

Abstract
Depletion of the cartilage proteoglycan aggrecan is one of the earliest events that occurs in association with osteoarthritis. This loss is often accompanied by a coordinate loss in another glycosaminoglycan, hyaluronan. Chondrocytes experimentally depleted of cell-associated hyaluronan respond by switching to a pro-catabolic metabolism that includes enhanced production of endogenous inflammatory mediators and increased synthesis of matrix metalloproteinases. Hyaluronan turnover is also increased. Together, such a response provides for possible establishment of a self-perpetuating spiral of events that maintains or prolongs the pro-catabolic state. Chondrocytes or cartilage can also be activated by treatment with pro-inflammatory cytokines and mediators such as IL-1β, TNFα, LPS, fibronectin fragments, and hyaluronan oligosaccharides. To determine the mechanism of chondrocyte activation due to hyaluronan loss, a depletion method was required that did not include degrading the hyaluronan. In recent years, several laboratories have used the coumarin derivative, 4-methylumbelliferone, as a potent inhibitor of hyaluronan biosynthesis, due in part to its ability to sequester intracellular UDP-glucuronic acid and inhibition of hyaluronan synthase transcription. However, contrary to our expectation, although 4-methylumbelliferone was indeed an inhibitor of hyaluronan biosynthesis, this depletion did not give rise to an activation of chondrocytes or cartilage. Rather, 4-methylumbelliferone directly and selectively blocked gene products associated with the pro-catabolic metabolic state of chondrocytes and did so through a mechanism preceding and independent of hyaluronan inhibition. These data suggest that 4-methylumbelliferone has additional useful applications to block pro-inflammatory cell activation events but complicates how it is used for defining functions related to hyaluronan.
Keywords: cartilage biology, chondrocyte, hyaluronan, matrix metalloproteinase (MMP), osteoarthritis, 4-methylumbelliferone
Introduction
The pronounced loss of aggrecan from articular cartilage is an early critical event associated with osteoarthritis (OA)2 (1, 2). Aggrecan turnover within the extracellular matrix occurs due to the enhanced activity of endoproteinases such as a disintegrin and metalloproteinase with thrombospondin motifs (ADAMTS) 4 (3, 4) and 5 (5, 6) as well as other matrix metalloproteinases (MMPs) (7–11). In addition to aggrecan, a significant loss of hyaluronan (HA) is also observed in human OA cartilage as compared with normal human cartilage (12, 13). A marked depletion of HA was also observed in articular cartilage damaged experimentally, such as in the anterior cruciate ligament transection model of OA (14) and a reduced-loading, splint immobilization model (15) in dogs. In our studies, cultured explants of human articular cartilage treated with IL-1α displayed a loss of HA within the superficial and upper middle layers of cartilage, the same layers in which aggrecan loss occurred (16). Other studies on cytokine-stimulated cartilage explants suggested that HA is lost from the cartilage (17, 18), and other studies revealed that the HA is lost but via local catabolism of the resident chondrocytes (19, 20). What is clear from all the studies is that HA is lost from the articular cartilage in a coordinate fashion with the loss of aggrecan (16, 19, 20).
Experimentally, we have tried to mimic the loss of HA and proteoglycan via the use of HA oligosaccharides to displace the pericellular matrix (21–24) or with hyaluronidases to remove the HA-rich pericellular matrix (21, 22, 25). With both approaches we observe the induction of pro-catabolic state by the chondrocytes, one that includes the up-regulation of MMP2, -3, -9, and -13 (24–28) as well as ADAMTS4 and ADAMTS5 (28). Our approach was based on a working hypothesis originally developed by Toole (29, 30) and our laboratory (21, 22) that removal or displacement of HA from the surface of cells results in the unclustering of CD44, leading to cytoskeleton-dependent induction of cell signaling.
In this study, we explored an alternative approach to remove HA from the surface of chondrocytes, one that would not generate small HA oligosaccharides. In recent years, investigators have used the coumarin derivative 4-methylumbelliferone (4-MU) as a low toxicity, high potency inhibitor of HA biosynthesis (31). 4-MU blocks HA biosynthesis by its ability to sequester cytosolic UDP-glucuronic acid (UDP-GlcUA) (32). Additionally, 4-MU effects a pronounced reduction of HA synthase 2 (HAS2) transcription but by mechanisms that are currently unknown (32–34). As a potent inhibitor of HA, 4-MU has been used to examine the role of HA in epithelial-mesenchymal transitions (35), collagen-induced arthritis (36), invasion and metastasis of osteosarcoma (37) and breast cancer cells (38), inflammation and autoimmunity (31), chondrogenic differentiation (39), myofibroblast differentiation (40, 41), and keratinocyte activation (42). We have shown that 4-MU blocks the assembly of pericellular coats on bovine, rat, and mouse chondrocytes and diminishes the responsiveness of chondrocytes to bone morphogenetic protein-7 similar to results obtained by hyaluronidase treatment (43). In sum, 4-MU has been used as a loss-of-function approach to reveal or confirm biological events suggested to be dependent on HA.
In this study, we examined the comparative effect of Streptomyces hyaluronidase and 4-MU on bovine and human articular chondrocytes to generate HA-depleted cells. Although HA was depleted by both conditions, 4-MU did not mimic the biological response obtained by hyaluronidase treatment. We observed that 4-MU was a potent inhibitor of chondrocyte activation, an inhibition that was independent of its effects on HA biosynthesis. We have chosen a particular subset of outcome measures to define chondrocytes or cartilages that have become activated and switched to a pro-catabolic metabolism. Because we observe many of these outcome measures in human OA chondrocytes and explant cultures without the need for additional external stimulation, we consider these measures of an OA-like metabolism (27, 28). We have limited our study to MMP13 and ADAMTS4 as examples of cartilage-relevant proteinases, TNF-stimulated gene 6 protein (TSG6) as an early marker of inflammation and remodeling induced by a variety of stimuli (44, 45). In addition, when cartilage explants were examined, the release of sulfated glycosaminoglycan (GAG) into the medium was used as a measure of cartilage degradation.
Experimental Procedures
Materials
Ham's F-12 and DMEM were obtained from Mediatech; FBS was from HyClone, and IL-1β was from R&D Systems, Inc. 4-MU was from Sigma (M1381 or M1508) or Alfa Aesar (A10337) for comparison. Pronase (53702; EMD Millipore Calbiochem), collagenase P (11249002001; Roche Applied Science), and collagenase D (11088882001; Roche Applied Science) were used in dissociation of tissues. Cell Lysis Buffer was from Cell Signaling Technologies, and Clear Blue x-ray film was from Genesee Scientific.
Specific primers for real time RT-PCR were custom-made by Integrated DNA Technologies (Coralville, IA). HAS2, CD44, and control siRNAs were obtained from Thermo Scientific Dharmacon RNAi Technologies. iScriptTM cDNA synthesis kit was obtained from Bio-Rad, and RT2 Real TimeTM SYBR Green reagents were from SA Biosciences.
The DuoSet HA ELISA kit for hyaluronan (DY3614-05) was purchased from R&D Systems, Inc., and used following the manufacturer's instructions. Streptomyces hyaluronidase (H1136) was purchased from Sigma. Pharmaceutical grade high molecular mass HA (ARTZ) was a gift from Seikagaku Co. Hyaluronan oligosaccharides were generated from human rooster comb hyaluronan (Sigma), as described previously (27).
Specific antibodies used for analysis were goat anti-ADAMTS4 (sc-16533, clone K-20, lot I2010; Santa Cruz Biotechnology), rabbit anti-MMP13 (sc-30073, clone H-230, lot F1312; Santa Cruz Biotechnology), rabbit anti-TSG6 (sc-30140, clone FL-277, lot C0112; Santa Cruz Biotechnology), rabbit affinity-purified anti-CD44 cytoplasmic tail antisera (1:5,000) (46), and β-actin (A1978, clone AC-15, lot 065M4837V; Sigma). The following antibodies were all obtained from Cell Signaling Technology: mouse anti-Myc (catalog no. 2276, clone 9B11, lot 24); p-NF-κB p65 (Ser(P)536, catalog no. 3033, lot 14); p65 (catalog no. 8242, lot 4); p-p38 (catalog no. 9215, lot 7); p38 (catalog no. 9212, lot 16); p-ERK1/2 (catalog no. 4370S, lot 5); and ERK1/2 (catalog no. 4695, lot 8). Mouse anti-O-linked N-acetylglucosamine antibody (clone RL2, lot PB197682) as well as the secondary antibodies HRP-conjugated donkey anti-rabbit (SA1-200) and HRP-conjugated donkey anti-mouse (SA1-100) were from ThermoFisher. HRP-conjugated donkey anti-goat (sc-2020) was from Santa Cruz Biotechnology.
Cell Culture
Primary bovine articular chondrocytes were isolated from the metacarpophalangeal joints of 18–24-month-old adult steers as described previously (28, 46). Primary human articular chondrocytes were isolated from knee cartilage obtained following joint replacement surgery, within 24 h after surgery and with institutional approval. Human cartilage samples were from patients (∼60% female and 40% male) with an average age of 66.8 ± 10.0 years. Bovine and human chondrocytes were liberated from full thickness slices of articular cartilage by sequential Pronase/collagenase P digestion. The chondrocytes were plated as high density monolayers (0.5–1.0 × 106 cells/cm2) and cultured in a 1:1 mixture of DMEM/Ham's F-12 medium containing 10% FBS and 50 units/ml penicillin, l-glutamate, and ascorbic acid. Confluent cultures of bovine or human chondrocytes were incubated overnight with medium containing 1% FBS and then incubated in serum-free medium with or without 0.5 mm 4-MU for 1 h prior to treatment for varying times with 1 ng/ml IL-1β in fresh serum-free culture medium without or with varying concentrations of 4-MU. In all experiments, 4-MU was dissolved in DMSO and then added to the culture medium with a final concentration of 0.1% DMSO; DMSO only at the same concentration was used as a control.
The T84 (CCL-248; ATCC) human colon carcinoma cell line was a gift from Dr. Yan-Hua Chen (East Carolina University (47)). The HepG2 human liver hepatocellular carcinoma cell line was a gift from Dr. Elizabeth M. Wilson (University of North Carolina-Chapel Hill (48)).
For siRNA inhibition of chondrocyte CD44 or HAS2, human chondrocytes were released from monolayer culture using a 1.5-h incubation with 0.1% collagenase P and 0.1% Pronase. The released cells (2.0 × 106 cells) were next mixed with AmaxaTM human chondrocyte solution (Lonza) and 1 μg of siRNA (46, 49). The chondrocytes were then transfected by nucleofection using an Amaxa Nucleofector® device at setting U-28. The HAS2 siRNA 5′-GCCAGCUGCCUUAGAGGAAUU-3′ (sense strand) was a similar sequence as used by Tian et al. (50). The CD44 siRNA 5′-GAACGAAUCCUGAAGACAUCU-3′ (sense strand) was constructed using the human CD44 siRNA sequence originally described by Ghatak et al. (51). The control siRNA (D-001206-09-05, Dharmacon) was also as described previously (46, 49).
To study mRNA stability, bovine chondrocytes were pre-stimulated with 1 ng/ml IL-1β for 12 h and then co-treated with IL-1β and 5 μg/ml actinomycin D (A9415; Sigma) in the presence or absence of 0.5 mm 4-MU (52, 53). Total mRNA was extracted at the indicated time points, and levels of HAS2, MMP13, TSG-6, and GAPDH mRNA expression were determined by qRT-PCR.
Primary mouse chondrocytes were isolated from the knee and hip joints of 4-week-old Cd44−/− mice (43, 49). Briefly, the animals were euthanized; the hind limbs dissected from the acetabulum and freed of surrounding muscle, tendons, and connective tissue. Isolated femoral heads and condyles and tibial condyles were subjected to 3 mg/ml collagenase D with stirring at 37 °C, 5% CO2. Primary murine chondrocytes were collected every hour for the first 5 h and then plated into 35-mm dishes containing DMEM, 10% FBS, 50 units/ml penicillin, and 2 mm/ml l-glutamine. When the mouse chondrocytes reached confluence, the cells were passaged one time with 0.25% trypsin, 2.21 mm EDTA into 12-well plates for experiments.
In some experiments human chondrocytes were cultured in alginate beads, as described previously (54), for subsequent placement into bioengineered neocartilage disks. Chondrocytes released from alginate beads were allowed to form neocartilage by an additional 2 weeks in culture in 0.4-μm pore membrane cell culture inserts (BD Biosciences). For analysis, the treated neocartilage disks were fixed with 4% buffered paraformaldehyde overnight at 4 °C, rinsed in 30% sucrose, PBS, and embedded in OCT-freezing medium. Cryostat sections (8 μm) were prepared and stained with safranin O for the detection of proteoglycans and counterstained with Fast Green. Sections were visualized using a Nikon Eclipse E600 microscope equipped with images captured digitally using a Retiga 2000R digital camera and processed using NIS Elements BR 1.30 imaging software (Nikon).
Cartilage Explant Cultures
Full thickness 4-mm cores of bovine and human OA articular cartilage were cultured in 1.0 ml of DMEM/Ham's F-12 medium containing 10% FBS for 48 h. The medium was then replaced with serum-free DMEM/Ham's F-12, and the tissues were incubated for 1–3 days in the presence of various activators, including 1–10 ng/ml IL-1β; 250 μg/ml HA oligosaccharides; or 10 ng/ml LPS with or without 0.5–2.0 mm 4-MU. For analysis, the treated explants were fixed with 4% buffered paraformaldehyde overnight at 4 °C, rinsed in 30% sucrose, PBS, and embedded in paraffin. Sections (8 μm) were prepared and stained with safranin O for the detection of proteoglycans and counterstained with Fast Green. In some experiments, the culture medium was collected and processed for Western blotting. In other experiments, media fractions were analyzed for proteoglycan content by dimethylmethylene blue assay for sulfated GAG release (55).
Generation of Adeno-Tet-MycHas2
The murine Has2 coding sequence (NM_008216) and NH2-terminal 6× Myc tag in pCDNA3, kindly provided by Drs. Davide Vigetti and Alberto Passi (56) was PCR-amplified using AccuPrime Pfx DNA polymerase (Invitrogen). Primers were designed according to the manufacturer's protocol as detailed in the Adeno-X Adenoviral System 3 User Manual (Clontech). The upstream primer included the ATG start site for the Myc tag sequence, 5′-GTA ACT ATA ACG GTC GCT ATG GAG CAA AAG CTC ATT TCT-3′, and the downstream primer included the stop codon for murine Has2, 5′-ATT ACC TCT TTC TCC TCA TAC ATC AAG CAC CAT GTC ATA-3′. The PCR cycling conditions were as follows: initial denaturation of 95 °C for 2 min, followed by 30 cycles of 15 s denaturation at 95 °C, 30 s annealing at 57 °C, and a 2.15-min extension at 68 °C. The PCR-amplified product (MycHas2) was purified using the QIAquick gel extraction kit (Qiagen). MycHas2 was then subcloned into the pAdenoX-Tet3G-linearized vector using the Adeno-X adenoviral system 3 kit (Clontech) to form Adeno-Tet-MycHas2. DNA sequences were verified for all PCR-amplified regions. Upon DNA sequencing, it was noted that adeno-Tet-MycHas2 contained a 3× NH2-terminal Myc tag sequence. Adeno-Tet-MycHas2 was packaged and amplified in HEK293 cells (ATCC). Viral particles were purified using the Adeno-X purification kit (Clontech) and titered using the adeno-X rapid titer kit (Clontech) to obtain a 4.89 × 109 IFU/ml adeno-Tet-MycHas2-purified viral stock. Human chondrocytes at ∼70% confluence were incubated with 10 IFU/ml Ad-Tet-MycHas2 in serum-free medium for 24 h. Medium was replaced with medium containing 10% FBS. After an additional 24 h, the cells were then used for experiments in the absence or presence of IL-1β, 4-MU, and 100 ng/ml doxycycline (Clontech).
Western Blotting
Total protein was extracted using Cell-Lysis-Buffer containing protease and phosphatase inhibitor mixtures. Equivalent protein concentrations were loaded into 4–12% NuPAGE® Novex® Tris acetate gradient mini gels (Thermo Fisher). In some experiments, the conditioned culture medium was also collected and processed for Western blotting by loading aliquots of equivalent volume to mini gels. Following electrophoresis, proteins within the acrylamide gel were transferred to a nitrocellulose membrane using a Criterion blotter apparatus (Bio-Rad), and the nitrocellulose membrane was then blocked in TBS containing 0.1% Tween 20 and 5% nonfat dry milk (TBS-T/NFDM) for 1 h. Immunoblots were incubated overnight with primary antibody in TBS-T/NFDM at 4 °C, rinsed three times in TBS-T, and incubated with secondary antibody in TBS-T/NFDM for 1 h at room temperature. Detection of immunoreactive bands was performed using chemiluminescence (Novex ECL, Invitrogen). In some cases, the blots were stripped using Restore Plus Western Stripping Buffer (Thermo Fisher) for 30 min at room temperature and re-probed using another primary antibody.
Real Time Reverse Transcription-Polymerase Chain Reaction (RT-PCR)
Total RNA was isolated from the bovine and human chondrocyte cultures according to the manufacturer's instructions for the use of TRIzol® reagent (Thermo Fisher). Total RNA was reverse-transcribed to cDNA using the iScript cDNA synthesis kit (Bio-Rad). Quantitative PCR was performed using Sso-Advanced SYBR Green Supermix (Bio-Rad) and amplified on a StepOnePlus real time PCR system (Applied Biosystems) to obtain cycle threshold (Ct) values for target and internal reference cDNA levels.
The human-specific primer sequences were follows: ADAMTS4 (57), forward (5′-AGG CAC TGG GCT ACT ACT AT-3′) and reverse (5′-GGG ATA GTG ACC ACA TTG TT-3′); ACAN, forward (5′-TCT GTA ACC CAG GCT CCA AC-3′) and reverse (5′-CTG GCA AAA TCC CCA CTA AA-3′); COL2A1 (58), forward (5′-GGC AAT AGC AGG TTC ACG TAC A-3′) and reverse (5′-CGA TAA CAG TCT TGC CCC ACT T-3′); GAPDH, forward (5′-GAA TTT GGC TAC AGC AAC AGG-3′) and reverse (5′-AGT GAG GGT CTC TCT CTT CC-3′); MMP13, forward (5′-CAG TGG TGG TGA TGA AGA TGA T-3′) and reverse (5′-CGC GAG ATT TGT AGG ATG GTA G-3′); HAS2, forward (5′-CTG GAA GAA CAA CTT CCA CGA A-3′) and reverse (5′-GAC CAA TTG CGT TAC GTG TTGC-3′); TSG6, forward (5′-GTG GCG TCT TTA CAG ATC CAA AGC-3′) and reverse (5′-CAA CAT AAT CAG CCA AGC AAC-3′); UGT1, forward (5′-ATT CCT TGG ACG TGA TTG G-3′) and reverse (5′-ATG GGT CTT GGA TTT GTG G-3′); UGT2A1, 2, forward (5′-GGT GGA ACT AAT GGG ATC TAC G-3′) and reverse (5′-CCA CAC TTG TCA TTG TGT TTA GG-3′); UGT2A3, forward (5′-TTG CAG ATC AGT GTG TGA GG-3′) and reverse (5′-CAG GAC TTT CCC ACA GAA TCC-3′); UGT2B4, forward (5′-CAG CTT CCA TTT CTT TCG ATC C-3′) and reverse (5′-GGT TGA TCG GCA AAC AAT GG-3′); UGT2B7, forward (5′-GGT CAT CCA AAG ACC AGA GC-3′) and reverse (5′-TCA ATC ACT GTC TTG CCC CA-3′); UGT2B10, 11, 28, forward (5′-GGC ATC TAT GAG GCA ATC TAC C-3′) and reverse (5′-CAT TCA GCA GGT CTG TAC TCG-3′); UGT2B15, 17, forward (5′-ATT TCC TCG CCC ATT CTT ACC-3′) and reverse (5′-AGC TCT GCA CAA ACT CTT CC-3′); UGT3A1, 2, forward (5′-TCC TCA AAG CTC AGG AAA CC-3′) and reverse (5′-CAA AGG AGG TGC TCT CTA AGG-3′); and UGT8, forward (5′-TCA GCT TTC TGG TTC TTC CC-3′) and reverse (5′-CTA CGT CAG TAC ACA GCA TCC-3′).
The bovine-specific primer sequences were follows: ADAMTS4, forward (5′-TCA CTG ACT TCC TAG ACA ATG G-3′) and reverse (5′-ACT GGC GGT CAG CGT CGT AGT-3′); ACAN, forward (5′-AAA TAT CAC TGA GGG TGA AGC CCG-3′) and reverse (5′-ACT TCA GGG ACA AAC GTG AAA GGC-3′); COL2A1 (59), forward (5′-TGC AGG ACG GGC AGA GGT AT-3′) and reverse (5′-CAC AGA CAC AGA TCC GGC AG-3′); GAPDH, forward (5′-ATT CTG GCA A AG TGG ACA TCG TCG-3′) and reverse (5′-ATG GCC TTT CCA TTG ATG ACG AGC-3′); IL-1β, forward (5′-AAA TGA ACC GAG AAG TGG TGT T-3′) and reverse (5′-TTC CAT ATT CCT CTT GGG GTA GA-3′) (60); TNFα, forward (5′-CTG GTT CAG ACA CTC AGG TCC T-3′) and reverse (5′-GAG GTA AAG CCC GTC AGC A-3′) (60); and TSG6, forward (5′-GTG GTG TGT TTA CAG ATC CAA AGA G-3′) and reverse (5′-CTT CAA CAT AGT CAG CCA AGC AAG-3′). The mouse-specific primer sequences for Mmp13 were forward (5′-TTC CTG ATG ATG ACG TTC AAG G-3′) and reverse (5′-GGT AAT GGC ATC AAG GGA TAG G-3′); the mouse/human primer sequences for Has2 were forward (5′-AAG AAA GGA AAG AAT CCA GTG A-3′) and reverse (5′-AAC CAG CAG ACC CGT TG-3′); and the bovine/human primer sequences for Tsg6 were forward (5′-CTG GCA CAT TAG ACT CAA GTA TGG-3′) and reverse (5′-CCA CAG TAT CTT CCC ACA AAG C-3′).
Real time RT-PCR efficiency (E) was calculated as E = 10(−1/slope)(61). The fold increase in copy numbers of mRNA was calculated as a relative ratio of target gene to GAPDH (ΔΔCt), following the mathematical model introduced by Pfaffl (62) as described previously (28, 46).
Statistical Analysis
All data were obtained from at least three independent experiments performed in duplicate or triplicate. A two-tailed unpaired Student's t test was used for direct comparison of treatment group to control. For multiple comparisons of groups, one-way or two-way analyses of variance followed by Tukey-Kramer test or Dunnett's multiple comparison test were used. A p value of less than 0.05 was considered significant. One asterisk denotes a p value of less than 0.05, two asterisks denote a p value of less than 0.01.
Results
Streptomyces Hyaluronidase Removal of HA Activates Chondrocyte MMP13
To mimic the loss of HA and aggrecan observed in OA, we often treat chondrocytes in culture with a low concentration of Streptomyces hyaluronidase. We have found that Streptomyces hyaluronidase, an HA-specific lyase, provides an HA-depleted state around cells for extended time periods without detrimental effects on cell viability. Exposure of normal bovine articular chondrocytes (Fig. 1A) or human OA chondrocytes (Fig. 1B) to Streptomyces hyaluronidase resulted in a substantial up-regulation of MMP13, ADAMTS4, and TSG6 mRNA. Human OA chondrocyte cultures, derived from cartilage of patients of differing backgrounds, age, grade, and gender display higher variability of stimulation as compared with bovine chondrocytes. Antibodies to human epitopes were used to detect MMP13, ADAMTS4, and TSG6 protein, present in both cell lysates and/or conditioned medium (Fig. 1C). All three proteins displayed increases following hyaluronidase treatment. These results suggest that the removal of extracellular HA, and other HA-bound matrix macromolecules, results in the activation or further enhancement of a pro-catabolic state in these chondrocytes.
FIGURE 1.

Effects of HA depletion on chondrocyte metabolism. Following treatment without or with Streptomyces hyaluronidase (−/+) for 24 h, high density monolayers of bovine chondrocytes (A) or human OA chondrocytes (B) were lysed and analyzed by real time qRT-PCR. The fold changes (y axes) in mRNA copy number for MMP13, ADAMTS4, and TSG6 (A and B) were compared with control-untreated cultures (black bars) and quantified using the ΔΔCt approach with normalization to GAPDH. Values represent the fold increase and the average ± S.D. of data derived from three independent experiments. A two-tailed unpaired Student's t test was used for comparison of treatment group to control; *, p < 0.05; **, p < 0.01. Cell lysates and serum-free media from other human OA chondrocyte cultures were also processed for Western blotting analysis of MMP13, TSG6, and ADAMTS4 protein (C). Shown is a representative example of three replicated, independent experiments. Aliquots of equal protein were loaded onto gels from the cell lysate fractions, and the medium samples represent aliquots of equal volume. D–F, human OA chondrocytes were treated without or with 0.5 to 2.0 mm 4-MU for 24 h. Serum-free media were collected and processed for HA ELISA (D, values represent percent of control shown as 100%) or Western blot analysis of MMP13 protein (F). Cell lysates were collected either for quantitative real time RT-PCR analysis of MMP13 mRNA (E) or Western blot analysis of MMP13 protein (F); both using a similar approach as detailed for B and C. All blots with cell lysates were stripped and re-probed for β-actin. For statistical analysis of data shown in D and E, a one-way ANOVA followed by Dunnett's test was used; *, p < 0.05; **, p < 0.01.
Effect of 4-MU-mediated Depletion of HA
To determine whether the activation induced by Streptomyces hyaluronidase was due to the loss of high molecular mass HA or an activation due to the release of enzymatically derived HA oligosaccharides, a different approach was used to deplete HA in chondrocytes. The chemical inhibitor 4-MU is used by many investigators to block HA biosynthesis and lower HAS2 mRNA (31–33, 63). As expected, the human OA chondrocytes treated with 0.5 to 2.0 mm 4-MU displayed a marked reduction of HA as measured by ELISA (Fig. 1D). Surprisingly, however, treatment of these cells with 4-MU did not result in an increase in MMP13 mRNA but rather a dose-dependent decrease in its expression (Fig. 1E). This decrease was mirrored by a slight decrease in MMP13 protein, present in both in cell lysates and media fractions (Fig. 1F). The baseline values for MMP13 varied widely, and in some samples the drop in MMP13 protein due to 4-MU was less pronounced but certainly not increased as was originally expected. It was not possible to demonstrate similar effects of 4-MU in normal bovine chondrocytes as the baseline levels of the marker proteins are very low. Nonetheless, there was no stimulation of MMP13, ADAMTS4, or TSG6 mRNA due to 4-MU observed in bovine chondrocytes (data not shown) as was expected from the hyaluronidase results in Fig. 1A. These data suggest that the loss or absence of high molecular mass HA alone is not sufficient to affect the pro-catabolic activation of chondrocytes.
Effect of 4-MU on Chondrocytes Stimulated with IL-1β
As noted above, normal bovine chondrocytes exhibit very low levels of MMP13. To mimic OA, bovine are often treated with the inflammatory cytokine IL-1β. In human OA chondrocyte cultures, even though each patient population of cells exhibits a different baseline state of activation, most cells respond to subsequent treatment with IL-1β. Treatment of bovine (Fig. 2A) or human OA (Fig. 2B) chondrocytes with IL-1β resulted in a pronounced stimulation of MMP13, ADAMTS4, and TSG6 mRNA, similar to the level of activation observed following treatment with Streptomyces hyaluronidase. However, co-incubation of cells with IL-1β and 4-MU resulted in a dose-dependent decrease in all three transcripts (Fig. 2, A and B). Co-treatment of human OA cells with IL-1β and 4-MU also resulted in a dose-dependent inhibition of MMP13, ADAMTS4, and TSG6 protein, including decreases in MMP13 released into the medium (Fig. 2C). The 0.5 mm concentration of 4-MU was sufficient to affect a consistent 40–50-fold inhibition of MMP13 and ADAMTS4 expression without the potential toxicity issues of higher doses as noted by others (33, 63), and thus this concentration was used for most of the remaining experiments.
FIGURE 2.
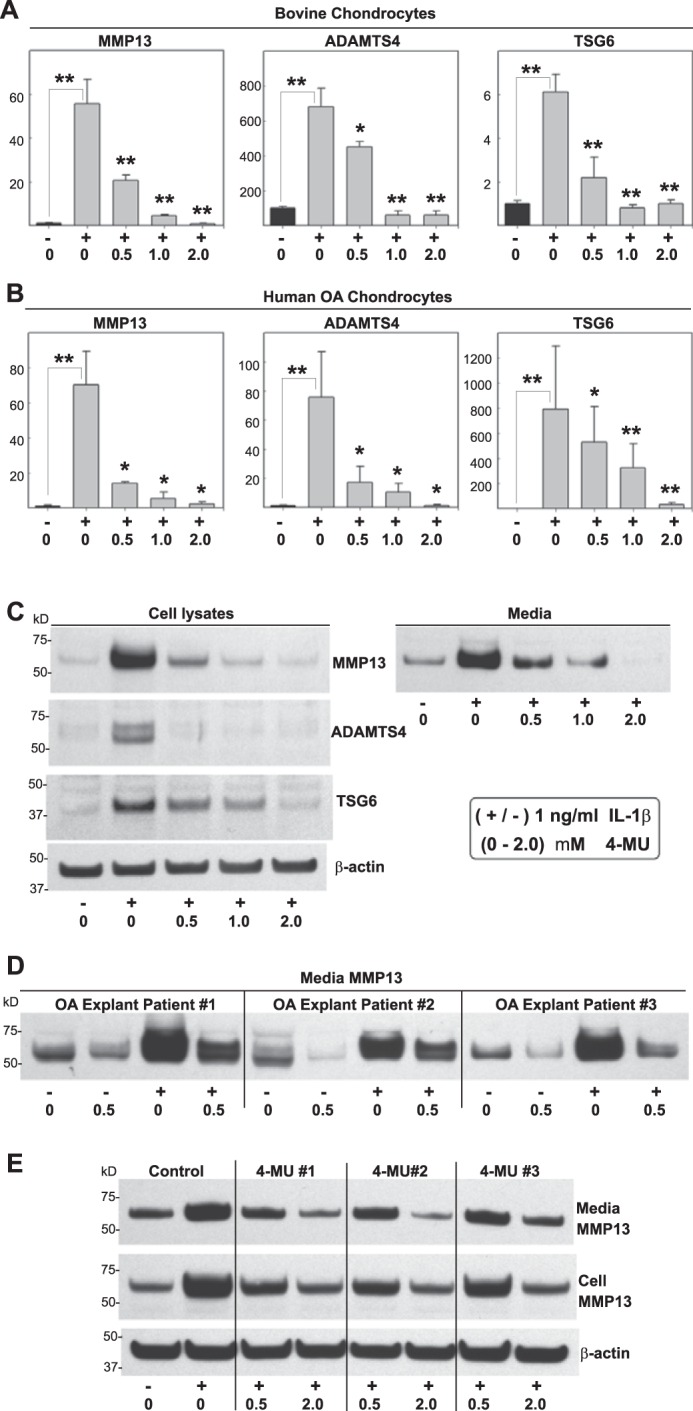
Effect of 4-MU on chondrocytes stimulated with IL-1β. High density monolayer cultures of bovine chondrocytes (A), human OA chondrocytes (B, C, and E), or explant cultures of human OA cartilage tissue cores (D) were treated without or with 1.0 ng/ml IL-1β (−/+) and co-treated with 0 to 2.0 mm concentrations of 4-MU as shown. After 24 h, the chondrocytes were lysed and analyzed by real time qRT-PCR (A and B). The fold changes (y axes) in mRNA copy number for MMP13, ADAMTS4, and TSG6 were compared with control (black bars) and quantified using the ΔΔCt approach with normalization to GAPDH. For statistical analysis, a two-way ANOVA followed by Tukey-Kramer test was used; *, p < 0.05; **, p < 0.01. Cell lysates and serum-free media from other human OA chondrocyte cultures were also processed for Western blotting for MMP13 protein (C). Shown is a representative example of three replicated, independent experiments. Aliquots of equal protein were loaded onto gels from the cell lysate fractions, and the medium samples represent aliquots of equal volume. Western blot analysis for MMP13 was also obtained from equal volume aliquots of medium samples from OA explants (D). E, human OA chondrocytes were co-treated without or with 4-MU that was obtained from three sources as follows: 1, Sigma M-1508; 2, Sigma M-1381; and 3, Aldrich M-1381. Shown is the Western blot analysis for MMP13 present in cell lysates and serum-free media. All blots with cell lysates were stripped and re-probed for β-actin.
The effects of 4-MU on pro-catabolically activated chondrocytes were not limited to primary cells in culture. Explant cultures of intact human OA cartilage release variable levels of MMP13 protein into their culture medium, and these are levels that could be enhanced by incubation of the tissues with IL-1β (Fig. 2D). However, even in cartilage explant cultures the inclusion of 0.5 mm 4-MU resulted in a striking diminution of MMP13 release and accumulation in the medium. It should be noted that both baseline and IL-1β-stimulated levels of MMP13 were inhibited.
As a quality control step, 4-MU obtained from three different commercial sources was evaluated. All three preparations inhibited MMP13 protein accumulation in human OA chondrocyte cell lysates and release into the media fractions as compared with controls (Fig. 2E).
Effects of 4-MU on Chondrocytes and Cartilage Are Not Limited to Stimulation with IL-1β
Like many cells types, mediators of other signaling pathways also induce a pro-catabolic state in chondrocytes. Treatment of bovine chondrocytes with HA oligosaccharides (Fig. 3A) or LPS (Fig. 3B) resulted in a stimulation of MMP13 protein present in both cell lysates and media fractions. As with IL-1β, co-treatment with LPS and 4-MU blocked the accumulation of MMP13 protein, including baseline levels of nontreated cells (Fig. 3, A and B).
FIGURE 3.
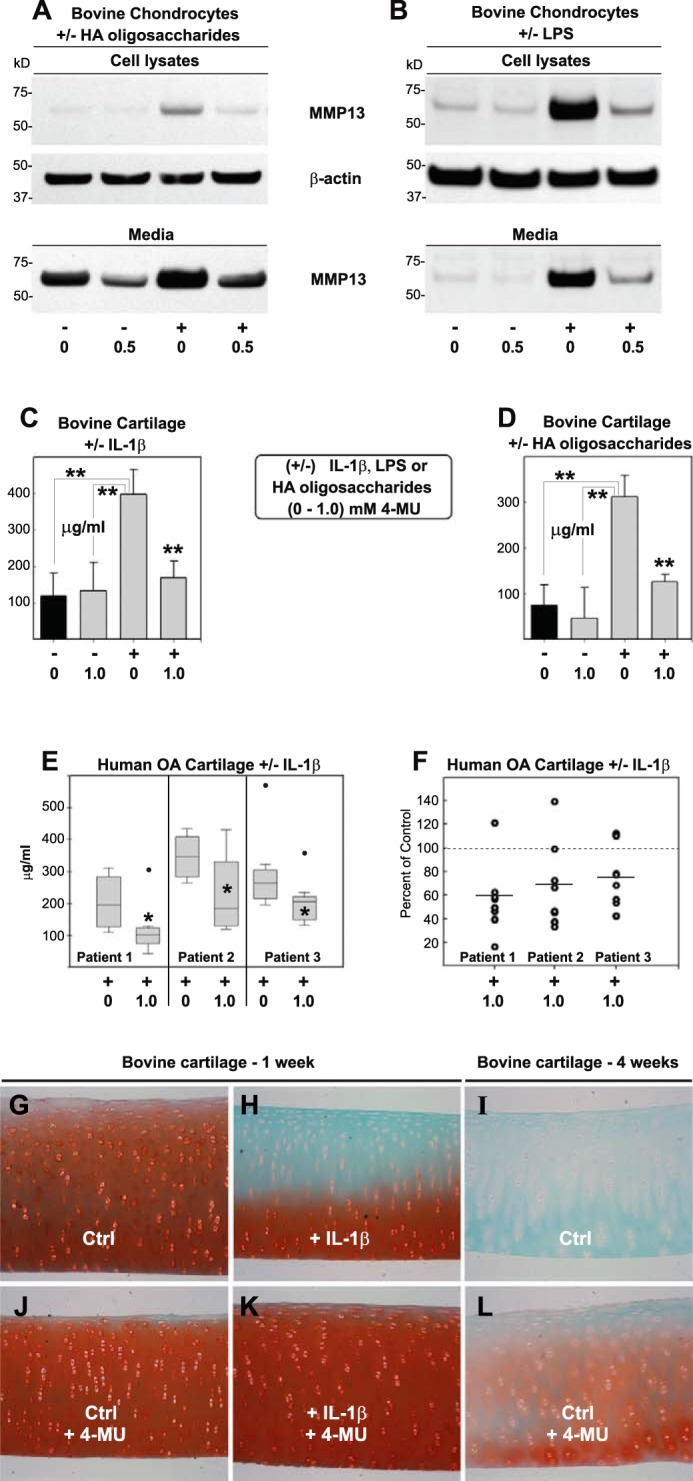
Effect of 4-MU on chondrocytes and cartilage explants induced by IL-1β, LPS, or HA oligosaccharides. To explore the effect of 4-MU on other reagents known to induce a pro-catabolic state in chondrocytes, high density monolayer cultures of bovine cells were treated without or with (−/+) 250 μg/ml HA oligosaccharides (A) or 10 ng/ml LPS (B) along with co-treatment with 0 or 1.0 mm 4-MU. After 24 h, cell lysates and serum-free media were isolated and processed for Western blotting for MMP13 protein. Shown are representative examples of three replicated, independent experiments. Aliquots of equal protein were loaded onto gels from the cell lysate fractions, and the medium samples represent aliquots of equal volume. All blots with cell lysates were stripped and re-probed for β-actin. To explore the effect of 4-MU on release of glycosaminoglycan, 6-mm cored disks of bovine cartilage (C, D, and G–L) or human OA cartilage (E and F) were treated without or with 1.0 ng/ml IL-1β or 250 μg/ml HA oligosaccharides as labeled (−/+) and including co-treatment with 1.0 nm 4-MU. In some experiments, serum-free medium was collected after 48 h and processed for glycosaminoglycan content using a dimethylmethylene blue colorimetric assay. Black bars depict values of control cultures. Values, shown in units of micrograms/ml, represent the average ± S.D. of data derived from five separate explant cultures of bovine cartilage for each condition (C and D) and seven separate explant cultures of human OA cartilage for each condition, performed on tissues derived from each of three patients (E and F). For statistical analysis, a two-way ANOVA followed by Tukey-Kramer test was used; *, p < 0.05; **, p < 0.01. F represents individual (non-averaged) data values that comprise the averaged data shown in E, wherein each individual IL-1β + 4-MU culture was compared with its control experiment (+IL-1β) set to a unit value of 100%. G–L depict 8-μm sections of fixed/paraffin-embedded bovine cartilage cores processed at the end of the experiment and stained with safranin O/fast green. G, H, J, and K show a representative experiment of explants cultured for 1 week in serum-free medium without (Ctrl) or with 1 ng/ml IL-1β and with 0 or 0.5 mm 4-MU as labeled. I and L represent bovine cartilage cultured without or with 0.5 mm 4-MU for 4 weeks. All images are of equivalent magnification and time exposure settings.
To determine whether the 4-MU inhibition of activated chondrocytes was of biological significance, GAG release from cartilage explants was examined. When bovine cartilage explants were treated with Il-1β (Fig. 3C) or HA oligosaccharides (Fig. 3D) for only 48 h, a substantial level of GAG was released into the medium. Co-incubation with 4-MU blocked the increase in GAG that accumulated in the medium (Fig. 3, C and D). In a similar fashion cartilage explants from OA patients, although highly variable from patient to patient, also exhibited a reduction of GAG released into the medium due to co-incubation with 4-MU (Fig. 3E, all samples treated with IL-1β). When the maximally stimulated GAG release values (in the absence of 4-MU) for each of the three human OA explant preparations were normalized to 100%, 4-MU treatment resulted in, on average, a 33% inhibition of GAG release. (Fig. 3F). Combined, these data suggest that 4-MU effectively blocks GAG release from cartilage likely by blocking the stimulation of proteases such as ADAMTS4 responsible for the degradation of aggrecan. After 1 week of treatment with IL-1β, a substantial loss of safranin O staining was observed in histological sections of bovine cartilage explants (Fig. 3H) as compared with untreated control tissues (Fig. 3G). Safranin O staining detects highly sulfated, GAG-rich proteoglycans such as aggrecan. The co-incubation of 4-MU and IL-1β blocked the loss of safranin O-stained extracellular matrix (Fig. 3K). The ability of 4-MU to block background loss of safranin O from untreated control explants was not discernable at the 1-week time point (Fig. 3, J compared with G) but was more apparent after 4 weeks in culture (Fig. 3, L compared with I). Thus, 4-MU appears to be chondroprotective of cartilage degradation.
Effects of 4-MU on Chondrocytes Is Not Because of Toxicity and Exerts Mild Pro-anabolic Effects
To determine whether the inhibition of pro-catabolic proteases and cytokines was due to a generalized inhibition of protein synthesis, the expressions of other genes important for cartilage biology were examined, namely aggrecan and type II collagen. Using similar culture conditions as the experiment shown in Fig. 2, 4-MU treatment did not decrease aggrecan or collagen II mRNA levels in either bovine (Fig. 4, A and B) or human OA chondrocytes (Fig. 4, C and D), respectively. It is well documented that IL-1β inhibits aggrecan and collagen II mRNA, although this inhibition was less prominent in these studies because only a low dose of IL-1β (1.0 ng/ml) was used. Nonetheless, co-treatment of bovine chondrocytes with 0.5 mm 4-MU and 1.0 ng/ml IL-1β resulted in a significant increase in aggrecan and collagen II. However, the stimulation of aggrecan or collagen apparent in bovine cells was not observed in similarly treated OA chondrocytes (Fig. 4, C and D), respectively. It should also be noted that no changes in β-actin, GAPDH mRNA, or total protein recovered in detergent cell lysates were observed in bovine or human chondrocytes treated with 4-MU (data not shown).
FIGURE 4.
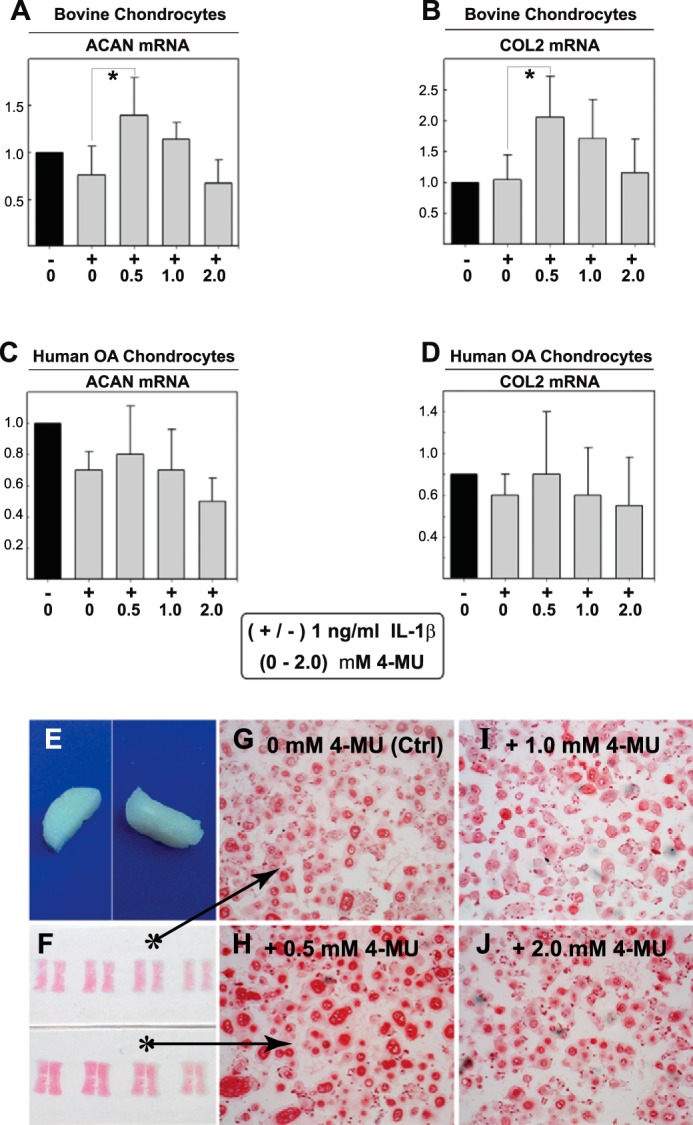
Effects to collagen II and aggrecan of 4-MU treatment of chondrocytes and bioengineered neocartilages. To demonstrate that 4-MU does not exhibit broadly based inhibitory effects on cell metabolism, high density monolayer cultures of bovine chondrocytes (A and B) or human OA chondrocytes (C and D) were treated without or with 1.0 ng/ml IL-1β (−/+) along with co-treatment with 0 to 2.0 mm concentrations of 4-MU as shown. After 24 h, the chondrocytes were lysed and analyzed by real time qRT-PCR. The fold changes (y axes) in mRNA copy number for aggrecan (ACAN, A and C) or collagen II (COL2, B and D) were compared with control (black bars) and quantified using the ΔΔCt approach with normalization to GAPDH. Values represent the average ± S.D. of data derived from three independent experiments. For statistical analysis, a two-way ANOVA followed by Tukey-Kramer test was used; *, p < 0.05; **, p < 0.01. In other experiments, bioengineered cartilage disks were generated from bovine chondrocytes (E) and treated for 72 h without (Ctrl) or with 0.5–2.0 nm 4-MU as labeled (F–J). After treatment, the neocartilage disks were fixed, sectioned, and stained using safranin O/fast green (actual slides shown in F). All images are of equivalent magnification and time exposure settings.
Next, bioengineered cartilage disks were established as a cartilage-like tissue model (Fig. 4E) in which newly synthesized aggrecan is being very actively deposited and organized. As compared with untreated control disks (Fig. 4, F, top, and G), the addition of 0.5–2.0 mm 4-MU (Fig. 4, F, bottom, and H–J) had no detrimental effect on the accumulation of safranin O-positive matrix. Moreover, at 0.5 mm 4-MU, the neocartilages were stained more richly with this proteoglycan-detecting reagent (Fig. 4H). Altogether, the data suggest that with 0.5–2.0 mm 4-MU, there is no generalized inhibition of protein synthesis or toxicity that might explain the diminution of MMP13, ADAMTS4, and TSG6. Again, 0.5 mm 4-MU may also be considered chondroprotective.
4MU Blocks Pre-initiated Pro-catabolic Events
To determine whether 4-MU blocked the initial steps in cytokine-mediated induction or subsequent signaling events downstream, chondrocytes were pre-treated for 4 h with IL-1β followed by a wash-out in the absence of cytokine (Fig. 5A). Under these conditions, MMP13 and ADAMTS4 mRNA remained elevated even after 20 h of cytokine wash-out (Fig. 5B). MMP13 in the cell lysates and media fractions also remained elevated (Fig. 5C). The addition of 4-MU during the wash-out phase was sufficient to block the IL-1β-induced stimulation (Fig. 5, B and C). Similar reductions by 4-MU of pre-stimulated MMP13 were observed in the medium from three additional OA patient chondrocyte cultures (Fig. 5D). These results suggest that 4-MU inhibition occurs downstream of the initial stimulatory signal. However, it remains unknown whether this downstream inhibition by 4-MU is coupled to the ability of 4-MU to block HA synthesis or is independent of effects on HA.
FIGURE 5.
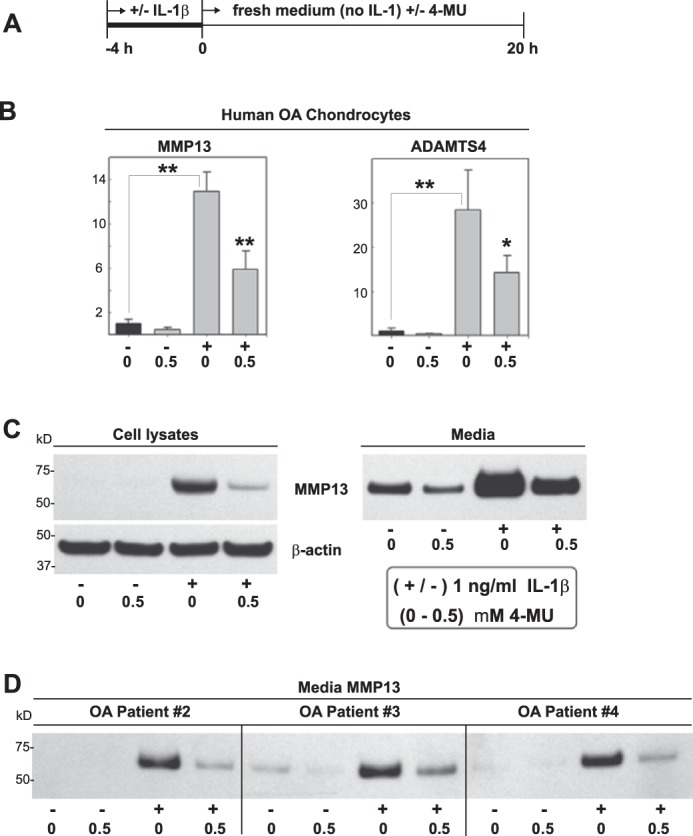
Effectiveness of 4-MU when added after induction of MMP13 and ADAMTS4. To determine whether 4-MU was affecting the initiation of signaling events (perhaps even extracellularly) or later phases of signal amplification, 4-MU was added after initiation by IL-1β. High density cultures of human OA chondrocytes were treated without or with 1.0 ng/ml IL-1β alone for 4 h (−/+) after which the medium was changed to fresh medium without or with 0.5 mm 4-MU, followed by incubation for an additional 20 h (A). The fold changes (y axes) in mRNA copy number for MMP13 and ADAMTS4 mRNA (B) were compared with control (black bars) and quantified using the ΔΔCt approach with normalization to GAPDH. For statistical analysis, a two-way ANOVA followed by Tukey-Kramer test was used; *, p < 0.05; **, p < 0.01. Cell lysates and serum-free media from other human OA chondrocyte cultures were also processed for Western blotting for MMP13 protein (C). Shown is a representative example of three replicated, independent experiments. Aliquots of equal protein were loaded onto gels from the cell lysate fractions, and the medium samples represent aliquots of equal volume. All blots with cell lysates were stripped and re-probed for β-actin. D depicts Western blot analysis for MMP13 accumulated in the media (equal volume aliquots) of experiments performed on chondrocyte cultures obtained from three additional OA patients.
Timing of 4-MU Inhibition of Pro-catabolic Events and Inhibition of HA Biosynthesis
When chondrocytes were treated with IL-1β for varying times (Fig. 6A), MMP13 mRNA was significantly elevated above untreated control after as little as 3 h of incubation (Fig. 6B). Although MMP13 mRNA was unchanged from control at 1 h (Fig. 6C), co-treatment with 4-MU (gray bars) significantly reduced MMP13 mRNA at this time point as well as all later time points (Fig. 6B). TSG6 mRNA was elevated above control as early as 1 h and was maximal at 6 h (Fig. 6, D and E). Like MMP13, 4-MU blocked the stimulation of TSG6 after 1 h of co-treatment. Interestingly, HA levels in the medium were unchanged at 1 h of co-treatment (Fig. 6, F and G). This suggested that reductions of medium HA (due to 4-MU) occurred later than inhibition of MMP13 or TSG6 mRNA.
FIGURE 6.
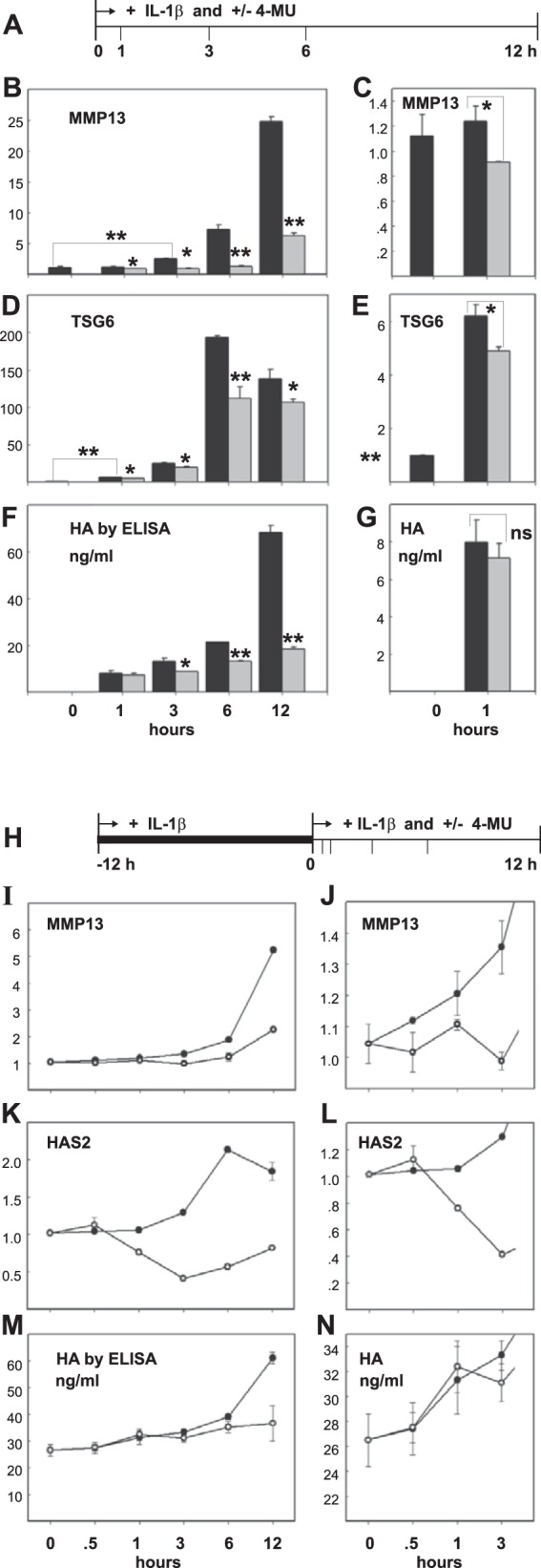
Timing of 4-MU inhibition of IL-1β induced changes to chondrocyte metabolism. A first approach (A) was used to determine when 4-MU was effective at blocking IL-1β-mediated changes in MMP13 and TSG6 mRNA levels in comparison with HA production. High density cultures of human OA chondrocytes were treated with 1.0 ng/ml IL-1β for 0–12 h (black bars) or with 1.0 ng/ml IL-1β and 0.5 mm 4-MU for 0–12 h (gray bars). The cells were lysed at each time point and analyzed by real time qRT-PCR. The fold changes (y axes) in mRNA copy number for MMP13 (B and C) and TSG6 (D and E) were compared with controls (0-h time points) and quantified using the ΔΔCt approach with normalization to GAPDH. For statistical analysis, a two-way ANOVA followed by Tukey-Kramer test was used; *, p < 0.05; **, p < 0.01. HA, present in the medium of each culture at the same time points was quantified by HA ELISA (F and G). Values represent average ± S.D. of data, shown in units of nanograms/ml. C, E, and G represent the 0- and 1-h data points shown in B, D, and F but using an expanded y axis scale. A second approach (H) was used to determine when 4-MU was effective at blocking IL-1β-mediated effects when chondrocytes were at a near-maximal state of IL-1β stimulation. High density cultures of human OA chondrocytes were first pre-treated with 1.0 ng/ml IL-1β for 12 h when 0.5 mm 4-MU was added directly into the culture medium (open circles) or left untreated (closed circles). Cells were lysed at each time point shown (I–N) and analyzed by real time qRT-PCR. The fold changes (y axes) in mRNA copy number for MMP13 (I and J) and HAS2 (K and H) were compared with controls (0-h time points) and quantified using the ΔΔCt approach with normalization to GAPDH. HA, present in the media of each culture at the same time points, was quantified by HA ELISA (M and N). Values represent average ± S.D. of data, shown in units of ng/ml. J, L, and N represent the 0–3-h data points shown in B, D, and F but using an expanded y axis scale.
In a different approach, after a 12-h pre-incubation of chondrocytes with IL-1β, cells were incubated for an additional 12 h with IL-1β in the presence or absence of 4-MU (Fig. 6H). Under these conditions, stimulation of pro-catabolic markers is at a maximum at the start of the experimental addition of 4-MU (value set to 1.0). As shown in Fig. 6I and the expanded scale in Fig. 6J, 0.5 h after the addition of 4-MU the relative copy number of MMP13 mRNA was inhibited due to the presence of 4-MU. HAS2 mRNA, which is also up-regulated by IL-1β treatment (data not shown), was unaffected by 4-MU at 0.5 h (Fig. 6, K and L) but displayed inhibition from 1 h onward. However, although HAS2 was down-regulated at 1 h, no change in HA production was observed until 6 h after exposure to 4-MU (Fig. 6, M and N). These data suggest that 4-MU reduces ongoing and maximally high levels of MMP13 mRNA in as little as 0.5 h (with IL-1β still present) and does so at a time point well before there is observable inhibition of HAS2 mRNA or the subsequent reduction of HA secreted into the medium. These results, although correlative in nature, suggest that reductions in HA due to 4-MU inhibition are separate from 4-MU-mediated inhibition of MMP13 and other pro-catabolic markers.
Participation of HA Is Not Required for the Inhibition of Pro-catabolic Events by 4-MU
To determine whether alterations in HA were required for 4-MU-mediated inhibition of MMP13, we explored mechanistic approaches to independently modify HA. As shown in Fig. 7A, exposure of chondrocytes to IL-1β resulted in a stimulation of MMP13 in both cell layer and media fractions. This enhanced production of MMP13 was blocked by the inclusion of 4-MU. Because HA levels were diminished by the 4-MU (as in Fig. 1D), 1.0 mg/ml exogenous pharmaceutical grade HA was added to the cultures to compensate for this loss. No rescue of MMP13 production was observed (Fig. 7A, 4th lane). Next, chondrocytes were transduced with an adeno-Tet-MycHas2 construct to re-populate the cells with HA synthases, as well as to enhance cell-associated and medium HA content. As shown in Fig. 7B, IL-1β stimulated MMP13 in cell lysates and media fractions, and both were blocked by co-incubation with 4-MU. In virally transduced human chondrocytes, prominent Myc epitope indicative of the transgene HAS2 protein was detected (Fig. 7B), and HA levels were elevated by 2.8-fold (Fig. 7C). However, even under these conditions, the presence of HAS2 and HA did not rescue MMP13 inhibition by 4-MU (Fig. 7B, 4th lane).
FIGURE 7.

Modulation of HA and the effect on 4-MU inhibition of IL-1β. To determine whether the 4-MU-mediated reduction in extracellular HA was responsible for the 4-MU-mediated inhibition of MMP13, HA rescue experiments were performed. In one approach, 1.0 mg/ml exogenous, high molecular mass, pharmaceutical grade HA was added to high density cultures of human OA chondrocytes during incubation without (−) or with (+) 1 ng/ml IL-1β and 0 or 0.5 mm 4-MU for 24 h. Cell lysates and serum-free media were processed for Western blotting for MMP13 protein (A). Shown is a representative example of three replicated, independent experiments. Aliquots of equal protein were loaded onto gels from the cell lysate fractions, and the medium samples represent aliquots of equal volume. All blots with cell lysates were stripped and re-probed for β-actin. In a second approach, human OA chondrocytes were transduced with an adeno-Tet-MycHas2 developed in our laboratory. Forty eight hours after transduction, the cultures were incubated without or with 200 ng/ml doxycycline (Dox), 1 ng/ml IL-1β, and 0.5 mm 4-MU as labeled. Cell lysates and serum-free media were again processed for Western blotting for MMP13 protein as well as MycHAS2 (B). The concentrations of HA present in cell lysates and media (in units of nanograms/ml) were determined by HA ELISA (C). Another series of experiments was developed to test the effects of HA depletion, specifically depletion that occurs independent of 4-MU. In one approach, human OA chondrocytes were treated without or with Streptomyces hyaluronidase (Streptomyces HA'ase) either during co-incubation (D) or as a 12-h pretreatment (E), without (−) or with (+) the addition of 1.0 ng/ml IL-1β and 0 or 0.5 mm 4-MU for 24 h as labeled. In a second approach, human chondrocytes were transfected with control, CD44, or HAS2-specific siRNA, 24 h before the addition of 1.0 ng/ml IL-1β and 0.5 mm 4-MU for 24 h. F depicts the effects of siRNA treatment on HAS2 mRNA; G and I, effects on HA concentration in cell lysates (black bars) and media fractions (gray bars), MMP13 mRNA (H), and MMP13 protein in cell lysates (J). The y axes for RT-PCR results depict fold change in mRNA copy number above control. For statistical analysis, a two-way ANOVA followed by Tukey-Kramer test was used; **, p < 0.01.
When human OA chondrocytes were co-treated with Streptomyces hyaluronidase along with IL-1β and 4-MU, the MMP13 levels remained reduced as compared with the positive control of IL-1β treatment alone (Fig. 7D). The hyaluronidase conditions were expected to stimulate MMP13 production as was observed earlier in Fig. 1C. However, hyaluronidase did not reverse or accentuate the inhibition of 4-MU. In a different experiment, chondrocytes were first pretreated with Streptomyces hyaluronidase, such that no HA would be available for inhibition by 4-MU at the start of the experiment. Again, the lack of HA due to hyaluronidase did not mimic 4-MU as cells treated only with IL-1β (no 4-MU) still exhibited strong MMP13 (Fig. 7E, 3rd lane). In addition, 4-MU continued to block IL-1β-stimulated MMP13 protein levels (Fig. 7E, 4th lane).
Next, human OA chondrocytes were incubated with HAS2 siRNA to selectively knock down endogenous HAS2. As shown in Fig. 7F, chondrocyte HAS2 mRNA was reduced to ∼20% the level of control siRNA and CD44 siRNA-treated cells. HA levels in both the medium and cell layer were reduced proportionally by HAS2 siRNA treatment (Fig. 7G). In control chondrocytes, IL-1β slightly up-regulated HA biosynthesis, whereas co-incubation with 4-MU reduced HA levels as expected (Fig. 7I). This trend was observed in HAS2 siRNA-treated chondrocytes but at much reduced levels of HA. Under these conditions in which HA was significantly reduced due to HAS2 knockdown, there was no inhibition of IL-1β-stimulated MMP13 mRNA (Fig. 7H, 5th bar) and MMP13 protein (Fig. 7J, 5th lane). Thus, simply affecting a reduction in HA is not sufficient to mimic the effects of 4-MU. In addition, HAS2 siRNA-mediated HA deficiency did not alter the ability of 4-MU to block IL-1β-stimulated MMP13 production (Fig. 7, H and J). 4-MU was just as effective an inhibitor in cells exposed to control siRNA as HAS2 siRNA. Together, these data suggest that the mechanism of 4-MU-mediated inhibition of the pro-catabolic state in chondrocytes is independent of its inhibition of HA synthesis and HAS2 expression.
Participation of CD44 Is Not Required for the Inhibition of Pro-catabolic Events by 4-MU
We often hypothesize that chondrocytes respond to changes in HA content by way of mechanisms involving the HA receptor CD44. To explore whether changes in CD44 were somehow masking or compensating for HA having little influence on 4-MU inhibitory activity, the effects of CD44 knockdown were examined. As shown in Fig. 8A, CD44 siRNA treatment substantially reduced CD44 protein expression even in the presence of IL-1β stimulation of CD44 (as compared with control siRNA treatment). CD44 mRNA was reduced to less than 10% of control siRNA or HAS2 siRNA values (Fig. 8B). However, even under these conditions wherein CD44 is largely absent, 4-MU treatment was still able to block IL-1β-activated MMP13 protein expression.
FIGURE 8.
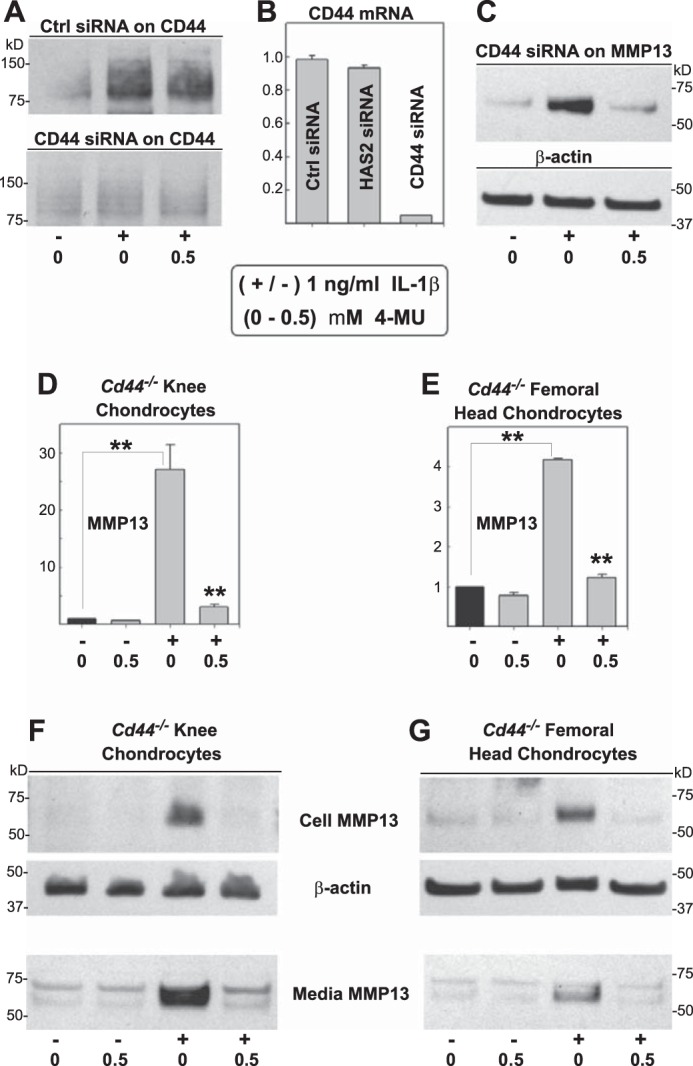
Modulation of CD44 and the effect on 4-MU inhibition of IL-1β. To determine whether CD44 was required for cells to detect changes in extracellular HA, such as changes in HA that occur due to 4-MU, CD44 depletion experiments were performed. In a first approach, human chondrocytes were transfected with control, HAS2-specific or CD44-specific siRNA, 24 h before the addition of 1.0 ng/ml IL-1β and 0 or 0.5 mm 4-MU for 24 h as indicated. A depicts the effects of siRNA treatment on CD44 protein as detected by Western blot analysis; B depicts the effects on siRNA on CD44 mRNA levels; and C depicts the effects of Cd44 siRNA on MMP13 protein in the medium of cells treated without or with IL-1β and 4-MU. In a second approach, murine chondrocytes derived from Cd44−/− knee condylar (D and F) or femoral head (E and G) cartilage were incubated without or with 1.0 ng/ml IL-1β and 0.5 mm 4-MU for 24 h. In some experiments, cell lysates were prepared for real time qRT-PCR, wherein the fold change (y axes) in mRNA copy number for MMP13 was compared with control conditions and quantified using the ΔΔCt approach with normalization to GAPDH (D and E). In other experiments, cell lysates and media fractions were processed for Western blotting for MMP13 and β-actin protein (F and G). For statistical analysis, a two-way ANOVA followed by Tukey-Kramer test was used; **, p < 0.01.
As a different approach, we next examined the effectiveness of 4-MU to block MMP13 in chondrocytes isolated from Cd44−/− murine knee joint articular cartilage (Fig. 8, D and F) as well as Cd44−/− femoral heads (Fig. 8, E and G). In both cell types, 4-MU continued to block IL-1β-stimulated Mmp13 mRNA (Fig. 8, D and E) and MMP13 protein in both cell and media fractions (Fig. 8, F and G). Altogether, these results indicate that 4-MU inhibition of pro-catabolic markers such as MMP13 is not dependent on changes to 4-MU-mediated inhibition of HA-dependent extracellular matrix structure, HAS2 organization of HA at the plasma membrane, or HA-mediated signaling events mediated via CD44.
Mechanism of 4-MU Inhibition of Pro-catabolic Events
As shown above, 4-MU inhibits the expression of a select group of pro-catabolic genes and does so in a relatively rapid time frame (as little as 1 h). In Fig. 9A, chondrocytes were pre-stimulated with IL-1β for 12 h (as in Fig. 6, H–N) prior to the addition of 4-MU at time 0. In the presence of actinomycin D, added to block ongoing transcription, mRNA for HAS2 dropped precipitously between 1 and 6 h of treatment (Fig. 9B, solid line) due to message decay. This drop was not affected by co-incubation with 4-MU (Fig. 9B, dotted line). Within the same time frame and conditions, little decay of MMP13 and TSG6 mRNA was observed (Fig. 9, C and D). Even so, the addition of 4-MU did not statistically alter the level of these transcripts. Thus, it is unlikely that the mechanism for inhibition by 4-MU is based on changes in message stability.
FIGURE 9.

Exploration of potential mechanisms responsible for 4-MU-mediated inhibition of pro-catabolic gene expression. To determine whether 4-MU inhibition was due to changes in message stability, bovine chondrocytes were first pre-stimulated with IL-1β for 12 h and then incubated in the absence or presence of actinomycin D (ActD, A). Under these conditions, the chondrocytes were at a maximum level of stimulation (as in Fig. 6, H–N) at time = 0. Fold change (y axes) in HAS2 mRNA (B), MMP13 mRNA (C), and TSG6 mRNA (D) were quantified at 0, 1, 3, and 6 h. Data collected from chondrocytes incubated in the absence of actinomycin D are depicted with solid lines and cells incubated with actinomycin D depicted with dotted lines. Data points represent the average ± S.D. of three independent experiments. For statistical analysis of data shown in B–D, a two-way ANOVA followed by Tukey-Kramer test was used; no statistical differences were observed. To explore whether 4-MU inhibition occurred earlier in the stimulation pathway, human OA (E) and bovine (F) chondrocytes were co-treated with IL-1β in the absence or presence of 0.5 mm 4-MU as labeled, and protein lysates were prepared after 0, 5, 15, and 60 min. Western blot analysis was used to detect activation of NF-κB (p-NF-κB), p38 MAPK (p-p38), and ERK1/2 (p-ERK1/2). Following detection, the blots were reprobed for detection of total NF-κB, p38 MAPK, ERK1/2, and β-actin. Aliquots of equal protein (20 μg) were loaded onto gels from the cell lysate fractions. Shown is a representative example of three replicated, independent experiments. E, results depict Western blot analysis of experiments from human OA chondrocytes derived from one patient. Similar results were obtained from chondrocyte cultures derived from the cartilage of two additional patients.
When chondrocytes are treated with IL-1β, investigators often observe the rapid activation of intracellular signaling proteins, including NF-κB, p38 MAPK, and ERK1/2 (27, 64, 65). In human OA chondrocytes, IL-1β treatment resulted in a prominent activation of these three proteins (p-NF-κB, p-p38 MAPK, and p-ERK1/2) in as little as 5 min (Fig. 9E); all three became reduced after 60 min. In bovine chondrocytes, the same prominent activation required 60 min (Fig. 9F). In the presence of 0.5 mm 4-MU, little change in the activation of these signaling proteins was observed in human or bovine chondrocytes. This suggests that the effect of 4-MU on inhibiting pro-catabolic gene mRNAs likely occurs downstream of these signaling pathway events.
Contribution of Protein O-Glucuronidation or O-GlcNAcylation to 4-MU-mediated Inhibition of Pro-catabolic Events
Given that 4-MU is thought to sequester UDP-glucuronic acid by driving the formation of 4-MU/glucuronosyl-glycoconjugates (32, 33), changes in the expression of UDP glucuronosyltransferases (UGTs) was examined. We hypothesized that 4-MU, either in the presence or absence of IL-1β, would promote or diminish the expression of one or more members of the transferase family. As shown in Fig. 10A, RT-PCR primers were designed to evaluate the expression of 13 members of the glucuronosyltransferase family selected to represent the major isoforms within the UGT1, UGT2A, UGT2B, UGT3, and UGT8 series. A study by Uchaipichat et al. (66) noted that isoforms except UGT1A4 had the capacity to glucuronidate 4-MU. All RT-PCR products were readily detected in lysates derived from either human Hep2A hepatocellular carcinoma or T84 colon carcinoma cells used as positive controls (Fig. 10A). However, in human OA chondrocytes, derived from three different human patients, UGT mRNA product detection was negligible as compared with the human liver or colon cells. When the agarose gel images were overexposed, the most prominent was UGT1 (overexposed, inverted image of UGT1 in Fig. 10A), and only small differences were observed in the expression of UGT1 following the addition of 4-MU, IL-1β, or both. Using real time qRT-PCR (Fig. 10B), no statistically significant changes in the expression of these genes was observed in the presence or absence of IL-1β or 4-MU as compared with control untreated cells. These data demonstrate that there are no changes in UGTs due to IL-1β or 4-MU but do not rule out the possibility that a reduction in glucuronidation of a regulatory protein could have occurred following the addition of 4-MU.
FIGURE 10.

Effect on 4-MU inhibition on glucuronidation transferases and O-GlcNAcylation of chondrocyte proteins. To determine whether the inhibitory effect of 4-MU was due to changes in UDP-glucuronic acid transferases (UGTs), human OA chondrocytes derived from the cartilage of three different patients were co-treated without or with 1.0 ng/ml IL-1β and without or with 0.5 mm 4-MU for 24 h. Lysates of total RNA were prepared, reverse-transcribed, and PCR-amplified using primer sets for various UGTs as labeled (A and B). One set of experiments was performed by conventional PCR with products identified by ethidium bromide staining following agarose gel electrophoresis (A). As positive controls, total RNA derived from human HepG2 liver hepatocellular carcinoma cells (Hep) and T84 colon carcinoma cells (T84) was analyzed in parallel. Data from analysis of UGT1 primer set is shown as a full size uncropped gel where gels from analysis of the other UGTs are depicted as cropped lanes. Base pair size of products are shown to the right of the panel; std, 100-bp ladder of standard markers. UGT1 data are also shown as an inverted (white field) digitally enhanced image. In another set of experiments, total RNA was amplified by real time qPCR using the same UGT primer sets as shown (B). Fold change (y axis) in products above control were compared by the ΔΔCt approach (values from untreated control chondrocytes set to 1.0) with normalization to GAPDH. For statistical analysis of data shown in B, a two-way ANOVA followed by Tukey-Kramer test was used; no statistical differences were observed. To determine whether the inhibitory effect of 4-MU was due to changes in UDP-GlcNAc transfer to chondrocyte proteins (GlcNAcylation), human OA chondrocytes (derived from the cartilage of three different patients, C) as well as bovine chondrocytes (derived from three different preparations, D) were co-treated with 1.0 ng/ml IL-1β and without or with 0.5 mm 4-MU for 0, 5, 15, and 60 min as labeled. Protein cell lysates were prepared, and Western blot analysis was used to detect O-GlcNAcylated cellular proteins. Following detection, the blots were stripped and re-probed for β-actin. C and D depict a representative example of the three replicated, independent experiments. In a different experiment, bovine chondrocytes were treated with varying concentrations of 4-MU for 24 h, protein lysates prepared, and Western blot analysis was used to detect O-GlcNAcylated cellular proteins and β-actin (E).
The addition of O-linked N-acetylglucosamine (GlcNAc) residues to a regulatory protein(s), is another potential mechanism that might be involved in 4-MU-mediated inhibition. An antibody is available that detects all O-GlcNAcylated proteins. An array of O-GlcNAcylated proteins, ranging in size from >250 to ∼60 kDa was observed in lysates from both human OA and bovine chondrocytes (Fig. 10, C and D, respectively). When these cells were treated with 1 ng/ml IL-1β for 0–60 min (as in Fig. 6), no apparent change in O-GlcNAcylated proteins was observed. Moreover, in the combined presence of IL-1β and 4-MU, again, no major change in the overall pattern of O-GlcNAcylated proteins was detected. In this study, we primarily focused on the lowest concentration of 4-MU (0.5 mm) that provided a significant inhibition in MMP13, ADAMTS4, TSG6, and HAS2 mRNA, in part to avoid cell toxicity that begins to be seen at higher 4-MU doses such as 5.0 mm (43). Although the addition of 4-MU alone to bovine chondrocytes shows little effect on the pattern of O-GlcNAcylated proteins at 0.5 mm after 1 h (Fig. 10D) or 24 h (Fig. 10E), increases in global O-GlcNAcylation of proteins does occur progressively after 24 h after incubation with 4-MU at higher concentrations (Fig. 10E).
Discussion
When human OA or normal bovine articular chondrocytes were stimulated with the inflammatory cytokine IL-1β, the elevated expression of MMP13, ADAMTS4, and TSG6 was inhibited by 4-MU treatment. 4-MU also blocked the expression of these genes when chondrocytes were activated by LPS or HA oligosaccharides. 4-MU is most often used by many investigators as a chemical inhibitor to block HA biosynthesis and to lower HAS2 mRNA. Indeed, in this study 4-MU did block HAS2 expression and HA biosynthesis in bovine and human articular chondrocytes. However, taken together, our data suggest that the mechanism of 4-MU-mediated inhibition of pro-catabolic metabolism in chondrocytes is independent of its inhibition of HA and HAS2. Our data also show that 4-MU is chondroprotective of cartilage degradation.
Yoshioka et al. (36) recently demonstrated that orally administered 4-MU substantially reduced the clinical symptoms of collagen-induced arthritis in DBA/1J mice. In addition to a reduction in footpad swelling, accumulation of MMP3 and MMP13 was reduced in knee joint cartilage, a reduction that matched the inhibition in cartilage damage observed by safranin O staining. The authors focused primarily on 4-MU inhibition of the synovial hyperplasia. Following 4-MU administration, they observed a reduction of hyperplasia to a mild synovitis with concurrent reduction in pannus formation and cytokine release. One interpretation of their data is that the 4-MU exerted a beneficial effect on preventing cartilage damage due to a reduction in synovial inflammation and the lowering serum and synovium HA levels. Our in vitro and cartilage explant data suggest that 4-MU may also have been exerting a direct inhibitory effect on MMPs in the knee cartilage of these mice as we observed in bovine cartilage (Fig. 3, K and L) as well as the promotion of aggrecan accumulation as in Fig. 4H.
In the study by Yoshioka et al. (36), hyaluronidase treatment mimicked the activity of 4-MU and successfully blocked TNF-α-stimulated MMP1 and MMP13 in primary cultures of human rheumatoid-like synovial cells. Additionally, the expression of the murine MMPs could be blocked by transfection of HAS siRNAs. Thus, these authors concluded that the inhibition of HA production by 4-MU was likely directly responsible for down-regulating MMPs in their synovial cells. Such was not the case in our study wherein the inhibitory effects of 4-MU could not be mimicked by hyaluronidase pre-treatment (Fig. 7E) or HAS2 siRNA knockdown (Fig. 7, H and J).
Inhibition of MMPs by 4-MU have been described in a few studies but appears to vary depending on the cell type. For example, 4-MU induces the expression of MT1-MMP mRNA, resulting in activation of MMP2 in human skin fibroblasts (67), but in human lymphoma cell lines, 4-MU decreased the level of mRNA for MMP9 (68). In primary human aortic smooth muscle cells 4-MU did not modify the activity of MMP2 but did cause a decrease in cellular migration, migration that could be rescued with the addition of HA (63). Whether this inhibition was dependent on HA inhibition or a direct effect remains unknown. Nakamura et al. (68) reported that 4-MU inhibited MMP9 in a human lymphoma cell line as well as other cultured human carcinoma cells, an inhibition that could not be mimicked by treatment of the cells with hyaluronidase. Given that elevated HA accumulation is often positively correlated with tumor growth and aggressiveness (31, 69), it is not surprising that 4-MU would provide inhibitory effects on tumor cell migration, invasion, or cell proliferation by way of blocking HA production. What must be considered is that 4-MU may, in addition, separately target MMPs and other pro-catabolic genes.
At present, the complete mechanism of action of 4-MU on chondrocytes and other cell types remains largely unknown. 4-MU is thought to act in large part by chelating UDP-GlcUA, one of two nucleotide sugars necessary for HA synthesis (32, 33), the end result being a inhibition of HA biosynthesis. Because UDP-sugar transporters in the endoplasmic reticulum and Golgi membranes have a low Km value, this diminution of cytosolic UDP-GlcUA has little to no effect on proteoglycan biosynthesis because the sugar residues for glycosaminoglycan chain elongation remain saturated in those compartments (70, 71). The more interesting aspect of 4-MU treatment is that it also has the capacity to down-regulate HAS2 mRNA by mechanisms that are far less clear or direct. To gain insight on this issue, several potential mechanisms were explored in this study. First, we observed that the addition of 4-MU to chondrocytes had little effect on reducing the activation of upstream signaling pathway intermediates (Fig. 9, E and F). Although other intermediates or cross-talk pathways may participate, the ones chosen, including activation of NF-κB/p65 (Ser(P)536), have been widely identified in IL-1β, HA oligosaccharide, or LPS-mediated stimulation of genes such as MMP13 (26, 27). We next examined the effect of 4-MU on altering mRNA stability as a potential post-transcriptional site of inhibitory action. Here also, 4-MU exerted little change and certainly no enhancement of mRNA decay (Fig. 9, B–D). Interestingly, in a recent transcriptome study performed on human OA chondrocytes exposed to actinomycin D by Tew et al. (72), HAS2 was identified as one of the transcripts with reduced stability. This was confirmed in our study (Fig. 9B) at least in comparison with the decay rate of MMP13 and TSG6 transcripts (Fig. 9, C and D); nevertheless, 4-MU co-treatment did not enhance further the rate of mRNA decay. In another study, co-treatment of PC3-ML prostate carcinoma cells with 4-MU and actinomycin D did not alter the decay of MMP2 and MMP9 mRNA (53). From these data we concluded that the inhibition by 4-MU on expression of this select group of pro-catabolic genes was most likely a transcriptional event.
Vigetti et al. (63) speculate that the reduction in UDP-GlcUA content might control gene transcription either directly or indirectly or that the slight decrease in UDP-GlcNAc they detected with 4-MU treatments of smooth muscle cells might alter the hexosamine biosynthesis pathway to impact gene regulation by the cellular function of O-GlcNAc glycosylation of proteins (74, 75). This opened up two possibilities, namely the possibility of a select, as yet unknown, transcriptional regulatory protein becomes differentially O-glucuronidated or differentially O-GlcNAcylated following the addition of 4-MU. We examined changes in the expression of a wide range of UGTs that participate in glucuronidation (Fig. 10, A and B). Although the expression of particular UGTs may be regulated by ligand-activated transcription factors in hepatic cells (76, 77), the expression of these genes was relatively low in human OA chondrocytes and unaffected by treatment of the cells with IL-1β and/or 4-MU. Although this does not rule out O-glucuronidation of a potential target protein, O-GlcNAcylation is thought to be a better modifier of protein function.
Therefore, we next examined global changes in O-GlcNAcylation of total cell proteins. However, neither 0.5 mm 4-MU nor 1 ng/ml IL-1β exerted detectable changes in O-GlcNAc transfer to cellular proteins of human OA or bovine chondrocytes (Fig. 10, C–E). Moreover, the ability of 4-MU to block IL-1β-stimulated MMP13 production was not changed by addition of exogenous glucosamine to the culture medium (data not shown). However, as the concentration of 4-MU was increased above 2.0 mm, the overall level of O-GlcNAcylated proteins increased. This opens the possibility that a particular regulatory protein or group of proteins could have been O-GlcNAcylated at 0.5 mm but below the limits of our detection. In a study by Kuroda et al. (78) using human skin fibroblasts, a moderate increase in O-GlcNAcylated proteins was observed at 0.1 and 1.0 mm 4-MU. The identity of this protein or factor remains unknown. Potential mediators are numerous (79). For example, Jokela et al. (80) found that conditions that enhanced O-GlcNAcylation of the transcription factor Yin Yang 1 (YY1) (in this case by the addition of glucosamine) was associated with an inhibition of HAS2 mRNA. YY1 is a downstream product of p65/NF-κB activation, and in fibroblast-like synoviocytes, it forms part of an NF-κB/YY1/miR10a regulatory circuit that promotes continued excessive secretion of inflammatory cytokines and MMPs, including MMP13 (81). Thus, one could speculate that 4-MU promotes the GlcNAcylation of YY1, reversing YY1 repression of miR10a, leading to an miR10a block of ongoing NF-κB activation.
The inhibitory effects of 4-MU on MMP13 in this study were observed in human OA chondrocytes, normal bovine articular chondrocytes, mouse chondrocytes, as well as cartilage explant cultures. 4-MU appeared to block pro-catabolic markers up-regulated by IL-1β, HA oligosaccharides, and LPS. One interpretation or mechanism for such a generalized effect might be that 4-MU was simply cytotoxic. However, we have previously demonstrated the viability of chondrocytes treated with 4-MU to be >98% at 0.1 and 1.0 mm 4-MU and greater than 85% at 5.0 mm 4-MU as determined by calcein AM/ethidium homodimer-1 live/dead cell assay (43). In Fig. 4, aggrecan and collagen II mRNA levels were relatively unaffected by 4-MU treatment, and GAPDH mRNA and β-actin protein remained unchanged. Moreover, 0.5 mm 4-MU appeared to be chondroprotective in part by up-regulating aggrecan mRNA and diminishing ADAMTS4 (Figs. 3 and 4). At 0.5 mm, 4-MU bovine chondrocytes still exhibit pericellular matrices or coats. In data not included, we observed the re-elevation of MMP13 release into the medium of explant cultures of human OA cartilage following a period of wash-out of 4-MU. Yoshioka et al. (36) also noted a return of MMP1 and MMP13 with wash-out of 4-MU from cultures of rheumatoid synovial cells activated with TNFα. Currently, 4-MU is used as a human dietary supplement (73), and although 4-MU is not approved for any indication in the United States, it has been used in several clinical trials (31). In animal models, 4-MU has been reported to have low toxicity in studies of the anti-tumor activity of 4-MU (73). Thus, it is unlikely that generalized cell toxicity is the mechanism responsible for 4-MU inhibition of MMPs.
In summary, 4-MU is an exciting small molecule as it provides for the selective inhibition of pro-inflammatory, pro-catabolic metabolism in many cell types, particularly activated chondrocytes within cartilage. 4-MU is also a useful tool to block the production of HA. Perhaps a common upstream element linking the stimulation of MMP13, ADAMTS4, TSG6, HAS2, and CD44 by IL-1β could be modulated by 4-MU. However, given these pleiotropic effects, caution and additional controls will be needed in the interpretation of results of future studies. Although inhibition of HA biosynthesis certainly occurs, our study demonstrates that 4-MU exerts effects that are independent of its effects on HA.
Author Contributions
W. K., C. B. K., and S. I. designed this study and wrote the paper and W. K. contributed to the preparation of the figures. E. B. A. contributed to the writing of the paper; designed, subcloned and constructed the adenoviral vectors for expression of HAS2, oversight of the Cd44−/− mouse colony, and development of methods to isolate murine chondrocytes from femoral hips and knees. S. I. and N. I. designed, performed, and analyzed the experiments shown in Figs. 1–8. All authors analyzed the results and approved the final version of the manuscript.
Acknowledgments
We thank Dr. Larry J. Dobbs, Jr. (Department of Pathology and Laboratory Medicine at East Carolina University), for assistance in the appropriation of human osteoarthritic tissue and Michelle Cobb and Joani Zary Oswald for their technical assistance with this project. We thank Dr. Tibor T. Glant and Dr. Katalin Mikecz (Rush University Medical Center) for graciously providing the Cd44−/− mice (BALB/c background); Drs. Alberto Passi and Davide Vigetti (Universita degli Studi dell'Insurbria) and Dr. Paraskevi (Uppsala University) for graciously providing the MycHAS2 plasmid.
The work was supported in part by National Institutes of Health Grants R21 AR066581 (to W. K.) and R01-AR039507 (to C. B. K.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- OA
- osteoarthritis
- ADAMTS
- a disintegrin and metalloproteinase with thrombospondin motif
- GAG
- sulfated glycosaminoglycan
- HA
- hyaluronan
- MMP
- matrix metalloproteinase
- 4-MU
- 4-methylumbelliferone
- ANOVA
- analysis of variance
- qRT
- quantitative RT
- NFDM
- nonfat dry milk
- UGT
- UDP glucuronosyltransferase.
References
- 1. Hardingham T. E., and Fosang A. J. (1992) Proteoglycans: many forms and many functions. FASEB J. 6, 861–870 [PubMed] [Google Scholar]
- 2. Rizkalla G., Reiner A., Bogoch E., and Poole A. R. (1992) Studies of the articular cartilage proteoglycan aggrecan in health and osteoarthritis. Evidence for molecular heterogeneity and extensive molecular changes in disease. J. Clin. Invest. 90, 2268–2277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Tortorella M. D., Burn T. C., Pratta M. A., Abbaszade I., Hollis J. M., Liu R., Rosenfeld S. A., Copeland R. A., Decicco C. P., Wynn R., Rockwell A., Yang F., Duke J. L., Solomon K., George H., et al. (1999) Purification and cloning of aggrecanase-1: a member of the ADAMTS family of proteins. Science 284, 1664–1666 [DOI] [PubMed] [Google Scholar]
- 4. Tortorella M. D., Pratta M., Liu R. Q., Austin J., Ross O. H., Abbaszade I., Burn T., and Arner E. (2000) Sites of aggrecan cleavage by recombinant human aggrecanase-1 (ADAMTS-4). J. Biol. Chem. 275, 18566–18573 [DOI] [PubMed] [Google Scholar]
- 5. Stanton H., Rogerson F. M., East C. J., Golub S. B., Lawlor K. E., Meeker C. T., Little C. B., Last K., Farmer P. J., Campbell I. K., Fourie A. M., and Fosang A. J. (2005) ADAMTS5 is the major aggrecanase in mouse cartilage in vivo and in vitro. Nature 434, 648–652 [DOI] [PubMed] [Google Scholar]
- 6. Glasson S. S., Askew R., Sheppard B., Carito B., Blanchet T., Ma H. L., Flannery C. R., Peluso D., Kanki K., Yang Z., Majumdar M. K., and Morris E. A. (2005) Deletion of active ADAMTS5 prevents cartilage degradation in a murine model of osteoarthritis. Nature 434, 644–648 [DOI] [PubMed] [Google Scholar]
- 7. Tortorella M. D., Malfait A. M., Deccico C., and Arner E. (2001) The role of ADAM-TS4 (aggrecanase-1) and ADAM-TS5 (aggrecanase-2) in a model of cartilage degradation. Osteoarthritis Cartilage 9, 539–552 [DOI] [PubMed] [Google Scholar]
- 8. Abbaszade I., Liu R. Q., Yang F., Rosenfeld S. A., Ross O. H., Link J. R., Ellis D. M., Tortorella M. D., Pratta M. A., Hollis J. M., Wynn R., Duke J. L., George H. J., Hillman M. C. Jr., Murphy K., et al. (1999) Cloning and characterization of ADAMTS11, an aggrecanase from the ADAMTS family. J. Biol. Chem. 274, 23443–23450 [DOI] [PubMed] [Google Scholar]
- 9. Sandy J. D., and Verscharen C. (2001) Analysis of aggrecan in human knee cartilage and synovial fluid indicates that aggrecanase (ADAMTS) activity is responsible for the catabolic turnover and loss of whole aggrecan whereas other protease activity is required for C-terminal processing in vivo. Biochem. J. 358, 615–626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Fosang A. J., Rogerson F. M., East C. J., and Stanton H. (2008) ADAMTS-5: the story so far. Eur. Cell Mater. 15, 11–26 [DOI] [PubMed] [Google Scholar]
- 11. Lark M. W., Bayne E. K., Flanagan J., Harper C. F., Hoerrner L. A., Hutchinson N. I., Singer I. I., Donatelli S. A., Weidner J. R., Williams H. R., Mumford R. A., and Lohmander L. S. (1997) Aggrecan degradation in human cartilage. J. Clin. Invest. 100, 93–106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Holmes M. W., Bayliss M. T., and Muir H. (1988) Hyaluronic acid in human articular cartilage. Age-related changes in content and size. Biochem. J. 250, 435–441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Roughley P. J., and Mort J. S. (2014) The role of aggrecan in normal and osteoarthritic cartilage. J. Exp. Orthop. 1, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Pita J. C., Muller F. J., Manicourt D. H., Buckwalter J. A., and Ratcliffe A. (1992) in Articular Cartilage and Osteoarthritis (Kuettner K. E., Schleyerbach R., Peyron J. G., and Hascall V. C., eds) pp. 455–470, Raven Press, New York [Google Scholar]
- 15. Haapala J., Lammi M. J., Inkinen R., Parkkinen J. J., Agren U. M., Arokoski J., Kiviranta I., Helminen H. J., and Tammi M. I. (1996) Coordinated regulation of hyaluronan and aggrecan content in the articular cartilage of immobilized and exercised dogs. J. Rheumatol. 23, 1586–1593 [PubMed] [Google Scholar]
- 16. Nishida Y., D'Souza A. L., Thonar E. J., and Knudson W. (2000) IL-1a stimulates hyaluronan metabolism in human articular cartilage. Arthritis Rheum. 43, 1315–1326 [DOI] [PubMed] [Google Scholar]
- 17. Sztrolovics R., Recklies A. D., Roughley P. J., and Mort J. S. (2002) Hyaluronate degradation as an alternative mechanism for proteoglycan release from cartilage during interleukin-1β-stimulated catabolism. Biochem. J. 362, 473–479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Durigova M., Roughley P. J., and Mort J. S. (2008) Mechanism of proteoglycan aggregate degradation in cartilage stimulated with oncostatin M. Osteoarthritis Cartilage 16, 98–104 [DOI] [PubMed] [Google Scholar]
- 19. Ng C. K., Handley C. J., Preston B. N., and Robinson H. C. (1992) The extracellular processing and catabolism of hyaluronan in cultured adult articular cartilage explants. Arch. Biochem. Biophys. 298, 70–79 [DOI] [PubMed] [Google Scholar]
- 20. Morales T. I., and Hascall V. C. (1988) Correlated metabolism of proteoglycans and hyaluronic acid in bovine cartilage organ cultures. J. Biol. Chem. 263, 3632–3638 [PubMed] [Google Scholar]
- 21. Knudson C. B. (1993) Hyaluronan receptor-directed assembly of chondrocyte pericellular matrix. J. Cell Biol. 120, 825–834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Knudson W., Aguiar D. J., Hua Q., and Knudson C. B. (1996) CD44-anchored hyaluronan-rich pericellular matrices: an ultrastructural and biochemical analysis. Exp. Cell Res. 228, 216–228 [DOI] [PubMed] [Google Scholar]
- 23. Knudson W., and Knudson C. B. (1991) Assembly of a chondrocyte-like pericellular matrix on non-chondrogenic cells. J. Cell Sci. 99, 227–235 [DOI] [PubMed] [Google Scholar]
- 24. Knudson W., Casey B., Nishida Y., Eger W., Kuettner K. E., and Knudson C. B. (2000) Hyaluronan oligosaccharides perturb cartilage matrix homeostasis and induce chondrogenic chondrolysis. Arthritis Rheum. 43, 1165–1174 [DOI] [PubMed] [Google Scholar]
- 25. Ohno-Nakahara M., Honda K., Tanimoto K., Tanaka N., Doi T., Suzuki A., Yoneno K., Nakatani Y., Ueki M., Ohno S., Knudson W., Knudson C. B., and Tanne K. (2004) Induction of CD44 and MMP expression by hyaluronidase treatment of articular chondrocytes. J. Biochem. 135, 567–575 [DOI] [PubMed] [Google Scholar]
- 26. Ohno S., Im H. J., Knudson C. B., and Knudson W. (2005) Hyaluronan oligosaccharide-induced activation of transcription factors in bovine articular chondrocytes. Arthritis Rheum. 52, 800–809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ohno S., Im H. J., Knudson C. B., and Knudson W. (2006) Hyaluronan oligosaccharides induce matrix metalloproteinase 13 via transcriptional activation of NFκB and p38 MAP kinase in articular chondrocytes. J. Biol. Chem. 281, 17952–17960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ariyoshi W., Takahashi N., Hida D., Knudson C. B., and Knudson W. (2012) Mechanisms involved in enhancement of the expression and function of aggrecanases by hyaluronan oligosaccharides. Arthritis Rheum. 64, 187–197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Toole B. P. (1991) in Cell Biology of Extracellular Matrix (Hay E. D., ed.) 2 Ed., pp. 305–339, Plenum Press, New York [Google Scholar]
- 30. Toole B. P. (1997) Hyaluronan in morphogenesis. J. Int. Med. 242, 35–40 [DOI] [PubMed] [Google Scholar]
- 31. Nagy N., Kuipers H. F., Frymoyer A. R., Ishak H. D., Bollyky J. B., Wight T. N., and Bollyky P. L. (2015) 4-Methylumbelliferone treatment and hyaluronan inhibition as a therapeutic strategy in inflammation, autoimmunity, and cancer. Front. Immunol. 6, 123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kakizaki I., Kojima K., Takagaki K., Endo M., Kannagi R., Ito M., Maruo Y., Sato H., Yasuda T., Mita S., Kimata K., and Itano N. (2004) A novel mechanism for the inhibition of hyaluronan biosynthesis by 4-methylumbelliferone. J. Biol. Chem. 279, 33281–33289 [DOI] [PubMed] [Google Scholar]
- 33. Kultti A., Pasonen-Seppänen S., Jauhiainen M., Rilla K. J., Kärnä R., Pyöriä E., Tammi R. H., and Tammi M. I. (2009) 4-Methylumbelliferone inhibits hyaluronan synthesis by depletion of cellular UDP-glucuronic acid and down-regulation of hyaluronan synthase 2 and 3. Exp. Cell Res. 315, 1914–1923 [DOI] [PubMed] [Google Scholar]
- 34. Vigetti D., Genasetti A., Karousou E., Viola M., Clerici M., Bartolini B., Moretto P., De Luca G., Hascall V. C., and Passi A. (2009) Modulation of hyaluronan synthase activity in cellular membrane fractions. J. Biol. Chem. 284, 30684–30694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Takahashi E., Nagano O., Ishimoto T., Yae T., Suzuki Y., Shinoda T., Nakamura S., Niwa S., Ikeda S., Koga H., Tanihara H., and Saya H. (2010) Tumor necrosis factor-α regulates transforming growth factor-beta-dependent epithelial-mesenchymal transition by promoting hyaluronan-CD44-moesin interaction. J. Biol. Chem. 285, 4060–4073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Yoshioka Y., Kozawa E., Urakawa H., Arai E., Futamura N., Zhuo L., Kimata K., Ishiguro N., and Nishida Y. (2013) Suppression of hyaluronan synthesis alleviates inflammatory responses in murine arthritis and in human rheumatoid synovial fibroblasts. Arthritis Rheum. 65, 1160–1170 [DOI] [PubMed] [Google Scholar]
- 37. Arai E., Nishida Y., Wasa J., Urakawa H., Zhuo L., Kimata K., Kozawa E., Futamura N., and Ishiguro N. (2011) Inhibition of hyaluronan retention by 4-methylumbelliferone suppresses osteosarcoma cells in vitro and lung metastasis in vivo. Br. J. Cancer 105, 1839–1849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Urakawa H., Nishida Y., Wasa J., Arai E., Zhuo L., Kimata K., Kozawa E., Futamura N., and Ishiguro N. (2012) Inhibition of hyaluronan synthesis in breast cancer cells by 4-methylumbelliferone suppresses tumorigenicity in vitro and metastatic lesions of bone in vivo. Int. J. Cancer 130, 454–466 [DOI] [PubMed] [Google Scholar]
- 39. Yoshioka Y., Kozawa E., Urakawa H., Arai E., Futamura N., Zhuo L., Kimata K., Ishiguro N., and Nishida Y. (2015) Inhibition of hyaluronan synthesis alters sulfated glycosaminoglycans deposition during chondrogenic differentiation in ATDC5 cells. Histochem. Cell Biol. 144, 167–177 [DOI] [PubMed] [Google Scholar]
- 40. Webber J., Jenkins R. H., Meran S., Phillips A., and Steadman R. (2009) Modulation of TGFβ1-dependent myofibroblast differentiation by hyaluronan. Am. J. Pathol. 175, 148–160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Webber J., Meran S., Steadman R., and Phillips A. (2009) Hyaluronan orchestrates transforming growth factor-β1-dependent maintenance of myofibroblast phenotype. J. Biol. Chem. 284, 9083–9092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Rilla K., Pasonen-Seppänen S., Rieppo J., Tammi M., and Tammi R. (2004) The hyaluronan synthesis inhibitor 4-methylumbelliferone prevents keratinocyte activation and epidermal hyperproliferation induced by epidermal growth factor. J. Invest. Dermatol. 123, 708–714 [DOI] [PubMed] [Google Scholar]
- 43. Luo N., Knudson W., Askew E. B., Veluci R., and Knudson C. B. (2014) CD44 and hyaluronan promote the BMP7 signaling response in chondrocytes. Arthritis Rheumatol. 66, 1547–1558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Milner C. M., Higman V. A., and Day A. J. (2006) TSG-6: a pluripotent inflammatory mediator? Biochem. Soc. Trans. 34, 446–450 [DOI] [PubMed] [Google Scholar]
- 45. Torihashi S., Ho M., Kawakubo Y., Komatsu K., Nagai M., Hirayama Y., Kawabata Y., Takenaka-Ninagawa N., Wanachewin O., Zhuo L., and Kimata K. (2015) Acute and temporal expression of tumor necrosis factor (TNF)-α-stimulated gene 6 product, TSG6, in mesenchymal stem cells creates microenvironments required for their successful transplantation into muscle tissue. J. Biol. Chem. 290, 22771–22781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Takahashi N., Knudson C. B., Thankamony S., Ariyoshi W., Mellor L., Im H. J., and Knudson W. (2010) Induction of CD44 cleavage in articular chondrocytes. Arthritis Rheum. 62, 1338–1348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Ding L., Lu Z., Foreman O., Tatum R., Lu Q., Renegar R., Cao J., and Chen Y.-H. (2012) Inflammation and disruption of the mucosal architecture in claudin-7-deficient mice. Gastroenterology 142, 305–315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. He B., Bai S., Hnat A. T., Kalman R. I., Minges J. T., Patterson C., and Wilson E. M. (2004) An androgen receptor NH2-terminal conserved motif interacts with the COOH terminus of the Hsp70-interacting protein. J. Biol. Chem. 279, 30643–30653 [DOI] [PubMed] [Google Scholar]
- 49. Ariyoshi W., Knudson C. B., Luo N., Fosang A. J., and Knudson W. (2010) Internalization of aggrecan G1 domain neoepitope ITEGE in chondrocytes requires CD44. J. Biol. Chem. 285, 36216–36224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Tian X., Azpurua J., Hine C., Vaidya A., Myakishev-Rempel M., Ablaeva J., Mao Z., Nevo E., Gorbunova V., and Seluanov A. (2013) High-molecular-mass hyaluronan mediates the cancer resistance of the naked mole rat. Nature 499, 346–349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Ghatak S., Misra S., and Toole B. P. (2005) Hyaluronan constitutively regulates ErbB2 phosphorylation and signaling complex formation in carcinoma cells. J. Biol. Chem. 280, 8875–8883 [DOI] [PubMed] [Google Scholar]
- 52. Fukui N., Ikeda Y., Ohnuki T., Hikita A., Tanaka S., Yamane S., Suzuki R., Sandell L. J., and Ochi T. (2006) Pro-inflammatory cytokine tumor necrosis factor-α induces bone morphogenetic protein-2 in chondrocytes via mRNA stabilization and transcriptional up-regulation. J. Biol. Chem. 281, 27229–27241 [DOI] [PubMed] [Google Scholar]
- 53. Lokeshwar V. B., Lopez L. E., Munoz D., Chi A., Shirodkar S. P., Lokeshwar S. D., Escudero D. O., Dhir N., and Altman N. (2010) Antitumor activity of hyaluronic acid synthesis inhibitor 4-methylumbelliferone in prostate cancer cells. Cancer Res. 70, 2613–2623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Masuda K., Sah R. L., Hejna M. J., and Thonar E. J. (2003) A novel two-step method for the formation of tissue-engineered cartilage by mature bovine chondrocytes: the alginate-recovered-chondrocyte (ARC) method. J. Orthop. Res. 21, 139–148 [DOI] [PubMed] [Google Scholar]
- 55. Ono Y., Ishizuka S., Knudson C. B., and Knudson W. (2014) Chondroprotective effect of kartogenin on CD44-mediated functions in articular cartilage and chondrocytes. Cartilage 5, 172–180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Karousou E., Kamiryo M., Skandalis S. S., Ruusala A., Asteriou T., Passi A., Yamashita H., Hellman U., Heldin C. H., and Heldin P. (2010) The activity of hyaluronan synthase 2 is regulated by dimerization and ubiquitination. J. Biol. Chem. 285, 23647–23654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Demircan K., Hirohata S., Nishida K., Hatipoglu O. F., Oohashi T., Yonezawa T., Apte S. S., and Ninomiya Y. (2005) ADAMTS-9 is synergistically induced by interleukin-1β and tumor necrosis factor α in OUMS-27 chondrosarcoma cells and in human chondrocytes. Arthritis Rheum. 52, 1451–1460 [DOI] [PubMed] [Google Scholar]
- 58. Indrawattana N., Chen G., Tadokoro M., Shann L. H., Ohgushi H., Tateishi T., Tanaka J., and Bunyaratvej A. (2004) Growth factor combination for chondrogenic induction from human mesenchymal stem cell. Biochem. Biophys. Res. Commun. 320, 914–919 [DOI] [PubMed] [Google Scholar]
- 59. Mehlhorn A. T., Niemeyer P., Kaiser S., Finkenzeller G., Stark G. B., Südkamp N. P., and Schmal H. (2006) Differential expression pattern of extracellular matrix molecules during chondrogenesis of mesenchymal stem cells from bone marrow and adipose tissue. Tissue Eng. 12, 2853–2862 [DOI] [PubMed] [Google Scholar]
- 60. Strandberg Y., Gray C., Vuocolo T., Donaldson L., Broadway M., and Tellam R. (2005) Lipopolysaccharide and lipoteichoic acid induce different innate immune responses in bovine mammary epithelial cells. Cytokine 31, 72–86 [DOI] [PubMed] [Google Scholar]
- 61. Rasmussen T. B., Uttenthal A., de Stricker K., Belák S., and Storgaard T. (2003) Development of a novel quantitative real-time RT-PCR assay for the simultaneous detection of all serotypes of foot-and-mouth disease virus. Arch. Virol. 148, 2005–2021 [DOI] [PubMed] [Google Scholar]
- 62. Pfaffl M. W. (2001) A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 29, e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Vigetti D., Rizzi M., Viola M., Karousou E., Genasetti A., Clerici M., Bartolini B., Hascall V. C., De Luca G., and Passi A. (2009) The effects of 4-methylumbelliferone on hyaluronan synthesis, MMP2 activity, proliferation, and motility of human aortic smooth muscle cells. Glycobiology 19, 537–546 [DOI] [PubMed] [Google Scholar]
- 64. Mengshol J. A., Vincenti M. P., and Brinckerhoff C. E. (2001) IL-1 induces collagenase-3 (MMP13) promoter activity in stably transfected chondrocytic cells: requirement for Runx-2 and activation by p38 MAPK and JNK pathways. Nucleic Acids Res. 29, 4361–4372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Liacini A., Sylvester J., Li W. Q., and Zafarullah M. (2002) Inhibition of interleukin-1-stimulated MAP kinases, activating protein-1 (AP-1) and nuclear factor κB (NF-κB) transcription factors down-regulates matrix metalloproteinase gene expression in articular chondrocytes. Matrix Biol. 21, 251–262 [DOI] [PubMed] [Google Scholar]
- 66. Uchaipichat V., Mackenzie P. I., Guo X. H., Gardner-Stephen D., Galetin A., Houston J. B., and Miners J. O. (2004) Human UDP-glucuronosyltransferases: isoform selectivity and kinetics of 4-methylumbelliferone and 1-naphthol glucuronidation, effects of organic solvents, and inhibition by diclofenac and probenecid. Drug Metab. Dispos. 32, 413–423 [DOI] [PubMed] [Google Scholar]
- 67. Nakamura T., Ishikawa T., Nanashima N., Miura T., Nozaka H., Nakaoka R., and Sato T. (2002) 4-Methylumbelliferone induces the expression of membrane type 1-matrix metalloproteinase in cultured human skin fibroblasts. Biochem. Biophys. Res. Commun. 298, 646–650 [DOI] [PubMed] [Google Scholar]
- 68. Nakamura R., Kuwabara H., Yoneda M., Yoshihara S., Ishikawa T., Miura T., Nozaka H., Nanashima N., Sato T., and Nakamura T. (2007) Suppression of matrix metalloproteinase-9 by 4-methylumbelliferone. Cell Biol. Int. 31, 1022–1026 [DOI] [PubMed] [Google Scholar]
- 69. Toole B. P., Wight T. N., and Tammi M. I. (2002) Hyaluronan-cell interactions in cancer and vascular disease. J. Biol. Chem. 277, 4593–4596 [DOI] [PubMed] [Google Scholar]
- 70. Vigetti D., Ori M., Viola M., Genasetti A., Karousou E., Rizzi M., Pallotti F., Nardi I., Hascall V. C., De Luca G., and Passi A. (2006) Molecular cloning and characterization of UDP-glucose dehydrogenase from the amphibian Xenopus laevis and its involvement in hyaluronan synthesis. J. Biol. Chem. 281, 8254–8263 [DOI] [PubMed] [Google Scholar]
- 71. Rowland A., Mackenzie P. I., and Miners J. O. (2015) Transporter-mediated uptake of UDP-glucuronic acid by human liver microsomes: assay conditions, kinetics, and inhibition. Drug Metab. Dispos. 43, 147–153 [DOI] [PubMed] [Google Scholar]
- 72. Tew S. R., McDermott B. T., Fentem R. B., Peffers M. J., and Clegg P. D. (2014) Transcriptome-wide analysis of messenger RNA decay in normal and osteoarthritic human articular chondrocytes. Arthritis Rheum. 66, 3052–3061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Yates T. J., Lopez L. E., Lokeshwar S. D., Ortiz N., Kallifatidis G., Jordan A., Hoye K., Altman N., and Lokeshwar V. B. (2015) Dietary supplement 4-methylumbelliferone: an effective chemopreventive and therapeutic agent for prostate cancer. J. Natl. Cancer Inst. 107, djv085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Hart G. W., Housley M. P., and Slawson C. (2007) Cycling of O-linked β-N-acetylglucosamine on nucleocytoplasmic proteins. Nature 446, 1017–1022 [DOI] [PubMed] [Google Scholar]
- 75. Hascall V. C., Wang A., Tammi M., Oikari S., Tammi R., Passi A., Vigetti D., Hanson R. W., and Hart G. W. (2014) The dynamic metabolism of hyaluronan regulates the cytosolic concentration of UDP-GlcNAc. Matrix Biol. 35, 14–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Bock K. W. (2010) Functions and transcriptional regulation of adult human hepatic UDP-glucuronosyl-transferases (UGTs): mechanisms responsible for interindividual variation of UGT levels. Biochem. Pharmacol. 80, 771–777 [DOI] [PubMed] [Google Scholar]
- 77. Hanioka N., Nonaka Y., Saito K., Negishi T., Okamoto K., Kataoka H., and Narimatsu S. (2012) Effect of aflatoxin B1 on UDP-glucuronosyltransferase mRNA expression in HepG2 cells. Chemosphere 89, 526–529 [DOI] [PubMed] [Google Scholar]
- 78. Kuroda Y., Kasai K., Nanashima N., Nozaka H., Nakano M., Chiba M., Yoneda M., and Nakamura T. (2013) 4-Methylumbelliferone inhibits the phosphorylation of hyaluronan synthase 2 induced by 12-O-tetradecanoyl-phorbol-13-acetate. Biomed. Res. 34, 97–103 [DOI] [PubMed] [Google Scholar]
- 79. Ozcan S., Andrali S. S., and Cantrell J. E. (2010) Modulation of transcription factor function by O-GlcNAc modification. Biochim. Biophys. Acta 1799, 353–364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Jokela T. A., Makkonen K. M., Oikari S., Kärnä R., Koli E., Hart G. W., Tammi R. H., Carlberg C., and Tammi M. I. (2011) Cellular content of UDP-N-acetylhexosamines controls hyaluronan synthase 2 expression and correlates with O-linked N-acetylglucosamine modification of transcription factors YY1 and SP1. J. Biol. Chem. 286, 33632–33640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Mu N., Gu J., Huang T., Zhang C., Shu Z., Li M., Hao Q., Li W., Zhang W., Zhao J., Zhang Y., Huang L., Wang S., Jin X., Xue X., et al. (2016) A novel NF-κB/YY1/microRNA-10a regulatory circuit in fibroblast-like synoviocytes regulates inflammation in rheumatoid arthritis. Sci. Rep. 6, 20059. [DOI] [PMC free article] [PubMed] [Google Scholar]