

Abstract
Within the ovarian follicle, immature oocytes are surrounded and supported by granulosa cells (GCs). Stimulation of GCs by FSH leads to their proliferation and differentiation, events that are necessary for fertility. FSH activates multiple signaling pathways to regulate genes necessary for follicular maturation. Herein, we investigated the role of Y-box-binding protein-1 (YB-1) within GCs. YB-1 is a nucleic acid binding protein that regulates transcription and translation. Our results show that FSH promotes an increase in the phosphorylation of YB-1 on Ser102 within 15 min that is maintained at significantly increased levels until ∼8 h post treatment. FSH-stimulated phosphorylation of YB-1(Ser102) is prevented by pretreatment of GCs with the PKA-selective inhibitor PKA inhibitor (PKI), the MEK inhibitor PD98059, or the ribosomal S6 kinase-2 (RSK-2) inhibitor BI-D1870. Thus, phosphorylation of YB-1 on Ser102 is PKA-, ERK-, and RSK-2-dependent. However, pretreatment of GCs with the protein phosphatase 1 (PP1) inhibitor tautomycin increased phosphorylation of YB-1(Ser102) in the absence of FSH; FSH did not further increase YB-1(Ser102) phosphorylation. This result suggests that the major effect of RSK-2 is to inhibit PP1 rather than to directly phosphorylate YB-1 on Ser102. YB-1 coimmunoprecipitated with PP1β catalytic subunit and RSK-2. Transduction of GCs with the dephospho-adenoviral-YB-1(S102A) mutant prevented the induction by FSH of Egfr, Cyp19a1, Inha, Lhcgr, Cyp11a1, Hsd17b1, and Pappa mRNAs and estradiol-17β production. Collectively, our results reveal that phosphorylation of YB-1 on Ser102 via the ERK/RSK-2 signaling pathway is necessary for FSH-mediated expression of target genes required for maturation of follicles to a preovulatory phenotype.
Keywords: extracellular-signal-regulated kinase (ERK), follicle-stimulating hormone (FSH), gene expression, ovary, protein kinase A (PKA), Y-box-binding protein 1 (YB-1), granulosa cells (GCs)
Introduction
In the female, fertility requires maturation of the ovarian follicle that contains the oocyte surrounded by granulosa cells (GCs)2 and theca cells. Follicular maturation is a tightly regulated process that is initiated by FSH. FSH regulates at least 500 target genes within GCs whose expression drives development of the follicle, allowing it to respond to the surge of luteinizing hormone (LH) that promotes ovulation, oocyte maturation, and formation of the corpus luteum that serves to support the developing embryo after fertilization and implantation (for review, see Refs. 1 and 2).
The mechanism by which FSH signals to regulate gene and protein expression in GCs has been extensively investigated. FSH binds to its G protein-coupled receptor (GPCR) expressed exclusively on GCs to activate adenylyl cyclase, raise intracellular cAMP levels, and activate PKA (3–6). PKA then either directly phosphorylates proteins that regulate transcription or indirectly activates signaling cascades whose targets regulate primarily transcription and translation. Direct PKA targets in GCs include cAMP-response element binding protein (CREB) (Ser133) (7), histone H3 (Ser10) (8), and β-catenin (Ser552 and Ser675) (9). Upon phosphorylation, these direct PKA target proteins participate in the regulation of gene expression though interactions with DNA and additional transcription factors.
One of the signaling cascades activated by FSH is the MAPK/ERK pathway (8, 10), evidenced by the phosphorylation of ERK (Thr202/Tyr204) within 10 min of FSH treatment (11), and phosphorylation of its downstream target RSK-2 (Ser380) (8), shown schematically in Fig. 1. FSH also activates the PI3K/protein kinase B (AKT) pathway (12, 13). In a PKA-dependent manner, FSH enhances phosphorylation of the pivotal kinase AKT on both the proximal Thr308 and the distal Ser473 sites within 5 min after FSH treatment (12, 14) followed by the rapid phosphorylation of downstream AKT targets, tuberin and forkhead box-containing protein O1 (FOXO1) on Thr1462 and Ser256, respectively (12, 15, 16).
FIGURE 1.
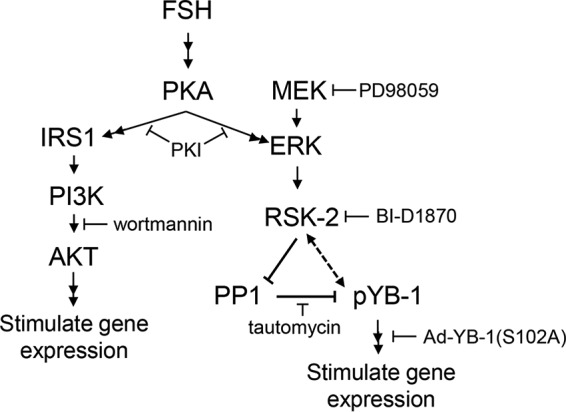
Proposed signaling pathway by which FSH promotes the PKA-dependent phosphorylation of YB-1 on Ser102 to enhance expression of select FSH target genes. Results support the schematic model in which FSH via PKA and ERK leads to an increase in phosphorylation of YB-1 on Ser102. This increase in YB-1 phosphorylation on Ser102 is dependent on ERK activation of RSK-2. RSK-2 appears to enhance phosphorylation of YB-1 on Ser102 primarily by inactivating PP1. YB-1 phosphorylated on Ser102 is necessary for FSH-dependent induction of target genes. IRS1 refers to insulin receptor substrate 1. Sites of actions of various inhibitors are indicated.
However, the mechanisms by which these signaling pathways regulate gene expression are less well understood. One of the more important pathways activated by FSH is the PI3K/AKT pathway, based on evidence that this pathway is necessary for GC differentiation (9, 15, 17, 18). Yet the only transcriptional factor regulated by this pathway that has been shown to modulate gene expression in GCs is FOXO1 (15, 18). Our recent results, however, suggest the PI3K/AKT pathway is likely regulating additional transcriptional modulators, based on the ability of exogenous insulin-like growth factor 1 (IGF1) at concentrations >1 ng/ml to synergize with FSH to enhance gene expression without further increasing the phosphorylation of FOXO1 (Ser256) (19). Strong evidence in cancer cells identified YB-1 as an AKT target (20–23) that, upon phosphorylation on Ser102, acts as a transcription factor to promote expression of genes such as Egfr (21, 24–26), a recognized FSH gene target (27).
YB-1 was first described as a major component of the inactive mRNA-protein complex (mRNP) (28, 29). Further studies demonstrated that YB-1 interacts with both RNA and DNA specifically through its cold shock domain (30, 31) and nonspecifically via basic and acidic clusters in its C terminus (for review, see Ref. 32). YB-1 functions in the cytosol to regulate mRNA translation and to stabilize mRNA; in the nucleus, YB-1 can regulate DNA repair, pre-mRNA splicing, and transcription (for review, see Ref. 32). Transcriptional targets for YB-1, identified mostly using cancer cell lines, in addition to Egfr, include multidrug resistance gene 1 (Mdr1) (33), protein-tyrosine phosphatase 1B (PTP1B) (34), cyclin A (Ccna) (35), and vascular endothelial growth factor (Vegf) genes (36).
Herein, we show that FSH promotes the protracted phosphorylation of YB-1 on Ser102. This phosphorylation is mimicked by forskolin and inhibited by expression of the selective PKA inhibitor (PKI), indicating its dependence on PKA activity. Phosphorylation of YB-1 on Ser102 is unaffected by the PI3K inhibitor wortmannin but is blocked by both the MEK/ERK inhibitor PD98059 and the RSK-2 inhibitor BI-1870, showing dependence on ERK and downstream RSK-2 signaling (Fig. 1). Our results surprisingly suggest that the major RSK-2 target is not YB-1; rather, RSK-2 promotes inactivation of protein phosphatase 1 (PP1) to enhance YB-1(Ser102) phosphorylation. Using adenoviral YB-1 phosphorylation mutants, we show that phosphorylation of YB-1 on Ser102 is necessary for the expression of multiple gene targets, including Cyp19a1, Lhcgr, Cyp11a1, Egfr, Hsd17b1, Pappa, and Inha. Together these results suggest that the PKA-dependent activation of ERK/RSK-2 pathways in GCs in response to FSH, by promoting phosphorylation of YB-1 on Ser102, contributes to the expression of multiple FSH-dependent gene targets that are necessary for follicular maturation. These results also suggest a previously underappreciated role for the ERK signaling pathway in immature GCs.
Experimental Procedures
Materials
The following were purchased: ovine FSH (oFSH-19) was from the National Hormone and Pituitary Agency of the NIDDK, National Institutes of Health (Torrance, CA); recombinant human IGF1 was from Atlanta Biologicals; purified mouse EGF, anti-FLAG (F3165), and testosterone propionate were from Sigma; human fibronectin was from BD Biosciences; antiphospho-YB-1 (Ser102; CST 2900), anti-YB-1 (CST 4202), anti-phospho-AKT (Ser473; CST 9271), anti-phospho-ERK (Thr202/Tyr204; CST 9107), anti-phospho-RSK-2 (Ser380; CST 9335), and anti-GST (CST 2622) were from Cell Signaling Technologies; anti-SRC homology-2 (SH2) domain-containing tyrosine phosphatase (SHP2) (SC 7384) and Protein A/G PLUS-agarose (SC 2003) were from Santa Cruz Biotechnology; wortmannin, PD98059, tautomycin, okadaic acid, forskolin, BI-D1870, 8-chlorophenylthiol (CPT)-cAMP, and myristoylated PKI were from Calbiochem/EMD. 8-CPT-2′-O-methyl-cAMP was purchased from Life Sciences Institute; TRIzol-RNA Lysis Reagent and Fast SYBR Green Master Mix were from Life Technologies; qScript was from VWR.
Animals
Sprague-Dawley, CD-outbred rats (breeders from Charles River Laboratories) were from a colony maintained by our laboratory in a pathogen-free facility at Washington State University. The facility is maintained in accordance with the Guidelines for the Care and Use of Laboratory Animals using protocols approved by the Washington State University Animal Care and Use Committee.
GC Culture and Western Blotting
Immature female rats were primed with subcutaneous injections of 1.5 mg of estradiol-17β (E2) in propylene glycol on days 21–23 to promote growth of preantral follicles. GCs were collected from ovaries on day 24 by puncturing individual follicles using 27-gauge needles (37). Cells were plated on fibronectin-coated plates at a density of ∼1 × 106 cells/ml of serum-free media supplemented with 100 units/ml penicillin (P), 100 μg/ml streptomycin (S), and 1 nm E2 (6). For the experiments measuring secreted estradiol-17β levels, 10 ng/ml androstenedione was added instead of E2. GC cultures are reported to be 88.6 ± 6.4% pure, based on the presence of the FSHR as detected by flow cytometry (38), are free of contaminating theca cells, based on the absence of response to LH (39), and initially contain ∼0.02% oocytes (40) that are removed with the addition of fresh media. These GC cultures faithfully mimic differentiation responses to FSH (for review, see Ref. 3) but do not mimic proliferation response to FSH (15, 41, 42). Indicated treatments were added to cells ∼20 h after plating and terminated by aspirating media and washing once with PBS. For Western blotting, total cell extracts were collected by scraping cells in SDS sample buffer (43) followed by heat denaturation. Equal protein loading was accomplished by plating equal numbers of cells, collecting at 50 μl/1 × 106 cells in SDS sample buffer, and loading equal volumes of extract per gel lane. Equal loading was verified by probing for total SHP2 or total AKT, as indicated. Proteins were separated by SDS/PAGE and transferred onto either Hybond C-extra or Protran (Amersham Biosciences) nitrocellulose membrane (8). The membrane was incubated with primary antibody overnight at 4 °C, and antigen-antibody complexes were detected using enhanced chemiluminescence (ThermoFisher). Westerns were scanned using an Epson Perfection V500 scanner and Adobe Photoshop CS2 9.0 software with minimal processing and quantified using Quantity One software (Bio-Rad). Experimental densitometric values were divided by load control protein values and expressed relative to vehicle values. Results were analyzed using GraphPad Prism, and significance was determined using either a one way ANOVA with Tukey's Multiple Comparison Test or a one-tailed Student's t test.
Immunoprecipitation
Briefly, ∼107 cells were scraped into 0.5 ml of immunoprecipitation lysis buffer (14), sonicated twice on ice for 45 s, and clarified by centrifugation at 10,000 × g for 5 min at 4 °C. After removal of a 0.03-ml sample for input, soluble GC extracts were precleared using a species-matched matrix (ExactaCruz Preclearing Matrix, Santa Cruz Biotechnology) for 60 min at 4 °C. Samples were then incubated by rotating at 4 °C overnight with the specified antibodies and Protein A/G PLUS-agarose (Santa Cruz Biotechnology; SC 2003) or antibody-agarose conjugate along with a species matched IgG antibody control (Santa Cruz Biotechnology). After centrifugation at 10,000 × g for 5 min at 4 °C, 0.4 ml of the supernatant fraction, representing the unbound protein fraction, was collected in SDS sample buffer. Agarose beads were washed by rotating 5 min at 4 °C with 1 ml of immunoprecipitation wash buffer (14) 4 times. Bound proteins were collected in SDS sample buffer and subjected to SDS/PAGE and Western blotting. Inputs represent ∼2%, and bound sample was ∼98% of total sample.
Transfection of GCs and Luciferase Assay
Egfr (−1109 to −16 bp) (44), Inha (−2021 to +68 bp) (45), and Cyp19a1 (−294 to + 20 bp) (46) promoter-luciferase constructs were previously described. Cells were plated on fibronectin-coated plates at a density of 1 × 106 cells/ml in Opti-MEM + P/S + E2 with expression constructs (500 ng/1 × 106 cells) and transfected using Lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions. After 6 h of incubation, media were removed, and fresh DMEM +P/S + E2 was added. Cells were treated as indicated after an ∼16 h recovery period then lysed in 0.1 ml of luciferase assay buffer (25 mm Hepes, pH 7.8, 15 mm MgSO4, 4 mm EGTA, 1 mm DTT, 1% Triton X-100). Samples were analyzed for luciferase activity using a Burthold Technologies Lumat LB 9507 luminometer using the Promega Luciferase Assay System according to the manufacturer's instructions.
Adenoviral Production
Full-length YB-1-FLAG was purchased from OriGene (Rockville, MD). YB-1-FLAG mutants were obtained using QuikChange Lightning Site-Directed Mutagenesis kit from Agilent Technologies (Santa Clara, CA) according to the manufacturer's instructions. The following primers were used: YB-1(S102D) forward (5′-GTC TCT CCA TCT CCT ACA TCG CGA AGG TAC TTC CTG GG-3′) and reverse (5′-CCC AGG AAG TAC CTT CGC GAT GTA GGA GAT GGA GAG AC-3′); YB-1(S102A) forward (5′-CCA GGA AGT ACC TTC GCG CTG TAG GAG ATG GAG AGA-3′) and reverse (5′-TCT CTC CAT CTC CTA CAG CGC GAA GGT ACT TCC TGG-3′). YB-1-FLAG, YB-1 (S102D)-FLAG, and YB-1 (S102A)-FLAG were cloned into pShuttleX and then cloned into Adeno-X by Vector Development Laboratory (Baylor College of Medicine, Houston TX). Adenoviral (Ad)-YB-1(S102D)- FLAG was used at 1.4 × 108 pfu/ml, and Ad-YB-1(S102A)-FLAG was used at 1 × 109 pfu/ml.
Cellular Fractionation
Fractionation was performed using a modified protocol previously described (47). GCs were treated as indicated, washed once with cold PBS, and collected in 0.5 ml of cytoplasmic lysis buffer (10 mm Hepes, pH 7.4, 10 mm KCl, 0.01 mm EDTA, 0.1 mm EGTA, 2 mm DTT, 5 mm Na2VO4, 10 mm NaF, 20 mm sodium β- glycerophosphate, 0.1% Nonidet P-40, Halt protease inhibitor mixture) and incubated on ice for 20 min. Nuclei were pelleted though centrifugation at 5000 × g 4 °C for 10 min, and the supernatant containing the cytoplasmic fraction was removed. The cytoplasmic fraction was collected in SDS sample buffer and heat-denatured. The nuclear pellet was washed with 1.0 ml of cytoplasmic lysis buffer and repelleted. The supernatant was removed, and the nuclear pellet was resuspended in 0.1 ml of SDS sample buffer and heat-denatured. Samples were subjected to SDS/PAGE and Western blotting with the indicated antibodies. Cytoplasmic fraction represents 9% of total sample, and the nuclear fraction represents 50% of total sample.
Adenoviral Transduction of GCs
GCs were plated on fibronectin-coated 6-well plates at a density of 1.5 × 106 cells/well in DMEM/F12 + P/S + E2. After allowing the cells to adhere to the plate for ∼4 h, the indicated adenoviruses were added to the media (9). After an overnight treatment, cells were allowed ∼1 h recovery and then treated as indicated.
RNA Isolation and Real-time PCR
GCs were treated as indicated, rinsed once with PBS, and collected in 1.0 ml of TRIzol-RNA lysis reagent. RNA isolation was carried out per the manufacturer's instructions. RNA concentration was determined using a NanoDrop 2000. cDNA was generated using qScript, and real time-PCR was performed using Fast SYBR Green Master Mix, both per the manufacturer's instructions. Primers used were: Lhcgr forward (5′-TCC AGA ACA CCA AAA ACC TGC-3′) and reverse (5′-AAG GGT TCG GAT GCC TGT G-3′); Egfr forward (5′-AGA TTG CAA AGG GCA TGA ACT AC-3′) and reverse (5′-ACA TTC CTG GCT GCC AAG TC-3′); Cyp19a1 forward (5′-CAC GGA TGT TTG ATG GTC TGA-3′) and reverse (5′-CTC GGC TTG CTG ACA AAC C-3′); Cyp11a1 forward (5′-GGG TGG ACA CGA CCT CCA T-3′) and reverse (5′-ACC TTC AAG TTG TGT GCC ATT TC-3′); Hsd17b1 forward (5′-CCT GCA CTT GGC TGT TCG T-3′) and reverse (5′-CGC AAT GTG GCA TAA ACT TTG A-3′); Pappa forward (5′-CCG TGA TCA CAG GGC TGT ATG-3′) and reverse (5′-CCC ATG ACC CAT CCT CGA T-3′); Inha forward (5′-TGG GAC CGC TGG ATC GTA-3′) and reverse (5′-GCA TCC CGC AGC TAC CAT-3′); Star forward (5′-AAG GAC TGC CCA CCA CAT CTA C-3′) and reverse (5′-TCT CGT TGT CCT TGG CTG GAA-3′); Egr1 forward (5′-CCT TTT GCC TGT GAC ATT TGT G-3′) and reverse (5′-TGG TAT GCC TCT TGC GTT CA-3′); Ereg forward (5′-GCT CTG ACA TGG ACG GCT ACT-3′) and reverse (5′-CTC GCT CAT GTC CAC CAG GTA-3′); ribosome L19 forward (5′-GTG ACC TGG ATG CGA AGG A-3′) and reverse (5′-GCC TTG TCT GCC TTC AGT TTG-3′).
Estradiol-17β Radioimmunoassay (RIA)
GCs were treated as described above in the presence of 10 ng/ml androstenedione. After treatment, cellular media was collected. Estradiol-17β levels were measured by RIA at the UVA Center for Research in Reproduction Ligand Assay and Analysis Core (48).
Results
FSH Signals to Stimulate Phosphorylation of YB-1(Ser102) through a PKA-dependent Pathway
We initially determined whether YB-1 was expressed in GCs and whether its expression at the protein level was regulated by FSH. Results (Fig. 2, A and B) show that YB-1 total protein, which migrates on SDS-PAGE at 50 kDa, is readily detected in GCs, and its expression is not acutely regulated by FSH. We then determined whether YB-1 was phosphorylated on Ser102 in response to FSH. FSH promoted a relatively slow increase in YB-1(Ser102) phosphorylation first detected 15 min after treatment (Fig. 2A). YB-1(Ser102) phosphorylation peaked 1 h after FSH treatment; this peak level of phosphorylation was maintained until ∼8 h and then declined to undetectable levels by 24 h (Fig. 2B) (the identities of the faster and slower migrating bands in phospho-YB-1 blots are not known). Increased phosphorylation of YB-1(Ser102) was mimicked by the adenylyl cyclase activator forskolin (Fig. 3A). Phosphorylation of AKT(Ser473) served as a positive control. YB-1(Ser102) phosphorylation also increased in response to 8-CPT-cAMP (Fig. 3B), a cell-permeable cAMP analog that activates both exchange proteins activated by cAMP and PKA (49). But 8-CPT-2′-O-Me-cAMP, a cAMP analog that does not activate PKA (50), failed to increase YB-1(Ser102) phosphorylation (Fig. 3B). Treatment of GCs with Ad-PKI, a pseudosubstrate for the catalytic subunit of PKA (51), led to a loss of FSH-dependent YB-1(Ser102) phosphorylation (82% ± 17%) (Fig. 3C). Similarly, treatment with myristoylated-PKI peptide resulted in a loss of YB-1 phosphorylation mediated by FSH (data not shown). Phosphorylation of AKT(Ser473) has been shown to be PKA-dependent in GCs (14) and serves as a positive control. Taken together, these data indicate that FSH stimulation results in phosphorylation of YB-1 on Ser102 in a PKA-dependent manner in GCs.
FIGURE 2.

Treatment of GCs with FSH leads to prolonged phosphorylation of YB-1 on Ser102. A, GCs were treated for the indicated times without (veh) or with 50 ng/ml FSH. Samples were heat-denatured after collection in SDS sample buffer, and proteins were separated by SDS/PAGE. A blot of whole cell extracts was probed with indicated antibodies. Results are representative of three independent experiments. Quantification of results represents the mean ± S.E. of three independent experiments, with the (*) representing p < 0.05, based on one-way ANOVA followed by Tukey's post test. B, GCs were treated without (v) or with FSH (F) for the indicated times, and samples were collected and analyzed as described in A, with (*) representing p < 0.05 based on a 1-way ANOVA followed by Tukey's post test. Arrow refers to phospho-YB1(Ser102).
FIGURE 3.

Phosphorylation of YB-1 on Ser102 is mediated by cAMP and PKA and independent of exchange proteins activated by cAMP. A, GCs were treated for 1 h without (veh) or with FSH (lanes 1 and 2) or without (EtOH) or with 10 μm forskolin in 0.25% ethanol, DMEM (lanes 3 and 4). Samples were collected as described in Fig. 2A. Quantification of the results represents the mean ± S.E. of three independent experiments, with the (*) representing p < 0.01, based on one-way ANOVA followed by Tukey's post test. B, GCs were treated for 1 h without (veh) or with FSH or with 250 μm concentrations of either 8-pCPT-2′-O-Me-cAMP or 8-CPT-cAMP. Samples were collected as described in Fig. 2A. Dotted lines between lanes represent cropped images. Results are representative of three independent experiments. Quantification was as described in A, with (*) representing p < 0.01, based on one-way ANOVA followed by Tukey's post test. C, GCs were transduced overnight with Ad-Empty (E) (5.8 × 103 optical particles per unit (OPU)/cell) or Ad-PKI (1.8 × 104 optical particles per unit (OPU)/cell), which expresses the full-length PKI protein. After media replacement, cells were treated for 2 h without (veh) or with FSH. Samples were collected as described in Fig. 2A, and quantification was as described in A, with (*) representing p < 0.01, based on a one-way ANOVA followed by Tukey's post test. Results are representative of three independent experiments.
Phosphorylation of YB-1(Ser102) Is Dependent on the ERK Pathway and Not the PI3K/AKT Pathway
The phosphorylation of YB-1(Ser102), investigated in various cancer cell lines, is most commonly mediated by AKT (20–23), although there is evidence that this site can be phosphorylated by RSK-2 downstream of ERK (26, 52). Because FSH in a PKA-dependent manner activates both the PI3K/AKT (14) and ERK signaling pathways (8, 10, 11, 53) in GCs, we investigated both pathways as possible regulators of YB-1 phosphorylation. Pretreatment of GCs with the PI3K inhibitor wortmannin (54) did not decrease FSH-stimulated YB-1(Ser102) phosphorylation, whereas AKT(Ser473) phosphorylation was inhibited significantly (74.2% ± 13.0%) (Fig. 4A). However, pretreatment with the MEK inhibitor PD98059 (54) promoted a near complete inhibition of YB-1(Ser102) phosphorylation (94.2% ± 3.6%) in the presence of FSH (Fig. 4B), similar to the inhibition of ERK(Thr202/Tyr204) phosphorylation (98.1% ±5.9%) that is used as the positive control. Equivalent results were obtained with a different MEK inhibitor U0126 (54) (data not shown). Consistent with these results, treatment of GCs with EGF, a known activator of the MEK/ERK pathway, promoted increased levels of YB-1(Ser102) phosphorylation as well as RSK-2(Ser380) phosphorylation (Fig. 4C). When GCs were treated with the PI3K/AKT pathway activator IGF1 or FSH, only cells treated with FSH showed enhanced YB-1(Ser102) phosphorylation (Fig. 4C). Unlike many other cell models, IGF1 does not activate the ERK pathway in GCs (55). Additionally, GCs treated with the RSK-2 inhibitor BI-D1870 (56) exhibited a marked inhibition (89.4% ± 5.3%) of YB-1(Ser102) phosphorylation in the presence of FSH (Fig. 5). CREB(Ser133) phosphorylation served as a negative control. Taken together, these results indicate that FSH-stimulated phosphorylation of YB-1(Ser102) is mediated by RSK-2 downstream of ERK.
FIGURE 4.

FSH-stimulated phosphorylation of YB-1 on Ser102 is via ERK signaling, not PI3K/AKT. A, GCs were pretreated without (DMSO) or with 100 nm wortmannin for 1 h followed by treatment without (veh) or with FSH for 1 h. Results are representative of three independent experiments. Samples were collected and quantified as described in Fig. 2A with (*) representing p < 0.05, based on a one-way ANOVA followed by Tukey's post test. B, GCs were pretreated without (DMSO) or with 50 μm PD98059 for 1 h followed by treatment without (veh) or with FSH for 1 h. Samples were collected, and quantification was as described in Fig. 2A, with (*) representing p < 0.001, based on a one-way ANOVA followed by Tukey's post test. Results are representative of three independent experiments. ns, not significant. C, GCs were treated without (veh) or with FSH, 50 ng/ml EGF, or 50 ng/ml IGF1 as indicated. Samples were collected and quantified as described in Fig. 2A with * representing p < 0.05 and ** representing p < 0.01, based on a one-way ANOVA followed by Tukey's post test. Results are representative of three independent experiments.
FIGURE 5.

FSH-stimulated phosphorylation of YB-1 on Ser102 is RSK-2-dependent. GCs were pretreated without (DMSO) or with 10 μm BI-D1870 for 1 h followed by treatment without (veh) or with FSH for 1 h. Samples were collected and quantified as described in Fig. 2A, with (*) representing p < 0.001, based on a one-way ANOVA followed by Tukey's post test. Results are representative of three independent experiments. Dotted lines between lanes represent cropped images.
Phosphorylation of YB-1(Ser102) Is Dependent on PP1 Inhibition
Enhanced phosphorylation of YB-1(Ser102) is relatively slow compared with the phosphorylation of CREB, which is detected within 1 min (11) or of ERK, which is detected within 10 min (8, 11) of FSH treatment. Moreover, YB-1(Ser102) phosphorylation is detected up to 8–12 h (Fig. 2B), whereas phosphorylations of both ERK (11) and RSK-2 (8) are undetectable by 2 h post treatment with FSH. We, therefore, sought to determine if the inhibition of a Ser/Thr phosphatase contributed to the phosphorylation of YB-1(Ser102).
Pretreatment of GCs with the protein phosphatase 2 (PP2) inhibitor okadaic acid (at 200 nm; Refs. 57 and 58) resulted in an increase in YB-1(Ser102) phosphorylation in both vehicle- and FSH-treated GCs (Fig. 6A). Phosphorylation of RSK-2(Ser380) is indicative of RSK-2 activity (59) and provides insight into the regulation of YB-1(Ser102) phosphorylation in the presence of okadaic acid. RSK-2(Ser380) phosphorylation was enhanced both in the absence and presence of FSH in response to okadaic acid and appears to account for the increase in YB-1(Ser102) phosphorylation. Fold activation by FSH relative to EtOH vehicle in the absence and presence of okadaic acid for pYB-1(Ser102) was 6.2 and 16.0 and for pRSK-2(Ser380) was 7.2 and 21.2, respectively. These results suggest that okadaic acid inhibits a phosphatase that acts upstream of YB-1 rather than at the level of YB-1.
FIGURE 6.

Prolonged FSH-stimulated phosphorylation of YB-1 on Ser102 is dependent on inhibition of the Ser/Thr phosphatase PP1. A, GCs were pretreated without (EtOH) or with 200 nm okadaic acid for 1 h followed by treatment without (veh) or with FSH for 1 h. Samples were collected as described in Fig. 2A, and quantification represents the mean ± S.E. of three independent experiments, with (**) representing p < 0.01 and (*) representing p < 0.05 based on a one-tailed Student's t test. B, GCs were pretreated without (EtOH) or with 1 μm tautomycin for 5.5 h followed by treatment without (veh) or with FSH for 1 h. Samples were collected, and quantification was as described in Fig. 2A, with (**) representing p < 0.01 and (*) representing p < 0.05, based on a one-tailed Student's t test. EtOH FSH-, tautomycin vehicle-, and tautomycin FSH-treated samples were not statistically different from each other (ns). Results are representative of three independent experiments. C, GCs were transfected with 500 ng of YB-1-FLAG plasmid for 6 h as described under “Experimental Procedures.” After an overnight recovery, GCs were treated without (veh) or with FSH for 1 h and then collected in immunoprecipitation lysis buffer and sonicated. After removal of the insoluble particles, the soluble fraction was rotated overnight with 20 μg of anti-FLAG (Sigma F3165) as described under “Experimental Procedures.” Inputs, bound (IP), and unbound (flow-though) samples were separated by SDS/PAGE, and the Western blot was probed with the indicated antibodies. Arrows indicate the protein band of interest. Results are representative of two independent experiments.
Pretreatment of GCs with the PP1 inhibitor tautomycin (at 1 μm; Refs. 57 and 58) resulted in an increase in YB-1(Ser102) phosphorylation in vehicle-treated cells to levels equivalent to those seen in FSH-treated cells in the absence of tautomycin, whereas FSH did not further enhance YB-1(Ser102) phosphorylation (Fig. 6B). Tautomycin pretreatment did not enhance FSH-stimulated RSK-2(Ser380) phosphorylation. Fold activation by FSH relative to EtOH vehicle in the absence and presence of tautomycin for pYB-1(Ser102) was 3.4 and 4.6 and for pRSK-2(Ser380) was 3.7 and 3.2, respectively. These results suggest that PP1 promotes the dephosphorylation of YB-1 on Ser102 in the absence of FSH.
If PP1 maintains the dephosphorylation of YB-1(Ser102) in the absence of FSH, then a PP1 catalytic subunit should co-immunoprecipitate with YB-1. As total YB-1 antibody was not immunoprecipitating, we transiently transfected GCs with a full-length FLAG-tagged YB-1 plasmid, allowed ∼16 h for cells to recover, treated GCs with vehicle or FSH, and then immunoprecipitated FLAG-tagged YB-1 with anti-FLAG antibody. Results (Fig. 6C) show that anti-FLAG antibody selectively pulls down comparable levels of the catalytic subunit PP1β (PP1cβ) in both vehicle- and FSH-treated samples. Additionally, when the same experiment was performed and the presence of RSK-2 was investigated, results show that RSK-2 also interacted (directly or indirectly) with FLAG-tagged YB-1 in a FSH-independent manner (Fig. 6C). These results suggest that both catalytic subunit PP1β (PP1cβ) and RSK-2 are in a complex with YB-1 and that these associations are independent of FSH.
Taken together, these results suggest (i) that PP2 inhibits YB-1 phosphorylation on Ser102 indirectly through regulation of RSK-2 itself or at a site upstream of RSK-2 and (ii) that PP2 activity is not regulated by FSH. Additionally, these results suggest (iii) that PP1 maintains YB-1(Ser102) in a dephosphorylated state in the absence of FSH and (iv) that FSH signals to inhibit the activity of PP1, preventing the dephosphorylation of YB-1 at Ser102. The inability of FSH to significantly enhance YB-1(Ser102) phosphorylation in the presence of tautomycin suggests that the major effect of PKA via ERK/RSK-2 is to promote inactivation of PP1.
YB-1 Phosphorylated on Ser102 Is Readily Detected in the Nuclear Fraction
As the phosphorylation of YB-1 on Ser102 in breast cancer cells is most often linked to its function as a transcriptional activator (Refs. 21, 23, 24, 26, and 33; for review, see Ref. 60), we asked if phospho-YB-1 was preferentially localized to the nuclear fraction of GCs.
To address this question, GCs were transfected with FLAG-tagged YB-1 plasmids that express wild type (WT) YB-1, the phospho- YB-1(S102D) mimic, and dephospho-YB-1(S102A) mutant. After a 16-h recovery period, GCs were treated with vehicle or FSH and then fractionated into cytosolic and nuclear fractions; GAPDH and histone H3 serve as positive controls for the purity of the cytoplasmic and nuclear fractions, respectively (cytoplasmic and nuclear fractions represent 9% and 50% of total sample, respectively). All of the FLAG-tagged constructs were readily detected in the cytoplasmic fraction (Fig. 7A), consistent with the presence of both phosphorylated YB-1(Ser102) and non-phosphorylated YB-1(Ser102) in this fraction (as shown below). However, the phospho-YB-1(S102D) mimic was readily detected in the nuclear fraction at higher levels compared with both YB-1-WT and the dephospho-YB-1(S102A) mutant.
FIGURE 7.

YB-1 and phospho-YB-1(Ser102) are detected in the both the nuclear and cytosolic fractions. A, GCs were transfected with 500 ng of the indicated plasmid for 6 h as described under “Experimental Procedures.” After recovery, GCs were treated without (veh) or with FSH for 4 h, then collected and separated into cytoplasmic and nuclear fractions as described under “Experimental Procedures.” Quantification of results represents the mean ± S.E. of two independent experiments, with (*) representing p < 0.05 and (**) representing p < 0.001, based on a one-way ANOVA followed by Tukey's post test. B, GCs were treated without (veh) or with FSH for 4 h, then collected and separated into cytoplasmic and nuclear fractions as described under “Experimental Procedures.” Western blots were probed with the indicated antibodies. Results are representative of three independent experiments.
Phosphorylated YB-1(Ser102) was also readily detected using the phospho-YB-1(Ser102) antibody in both the nuclear and cytosolic fractions of non-transfected GCs (Fig. 7B, left panel). However, FSH did not promote a change in total levels of YB-1 within the nuclear or cytoplasmic fractions (Fig. 7B, right panel), suggesting either that the phosphorylation of YB-1 on Ser102 occurs within the nuclear fraction or that a relatively small fraction of total YB-1 phosphorylated in the cytoplasmic fraction translocates into the nuclear fraction. Although the majority (∼90%) of total and phosphorylated YB-1 in non-transfected GCs is localized to the cytoplasmic fraction, the presence of phosphorylated YB-1(Ser102) in the nuclear fraction suggests a potential role for this phosphoprotein in the regulation of gene expression.
Phospho-YB-1(Ser102) Is Necessary for Expression of Select FSH Target Genes
Based on the increased levels of the phospho-YB-1(S102D) mutant detected in the nuclear fraction compared with WT and dephospho-YB-1(S102A) mutant, we hypothesized that YB-1 could participate in the transcriptional regulation of FSH target genes. Although ERK signaling is critical to LH-induced ovulation and luteinization in preovulatory GCs (61), the importance of the ERK signaling pathway to FSH-stimulated differentiation of GCs is less well understood. We, therefore, initially sought to identify FSH target genes that were regulated by the ERK signaling pathway. Pretreatment of GCs with the MEK inhibitor U0126 resulted in inhibition of FSH-induced Egfr (85.7% ± 6.2%), Inha (82.5% ± 3.2%), Cyp19a1 (96.1% ± 2.65%), and Ereg (67.7% ± 4.4%) mRNA levels measured at 48 h post FSH treatment, whereas Star mRNA levels were not affected (Fig. 8). Treatment with the MEK inhibitor PD98059 showed significant inhibition of FSH-induced expression of the immediate early gene Egr1 (86.4% ± 3.2%) 1 h post FSH treatment (Fig. 8).
FIGURE 8.

ERK signaling is necessary for FSH-stimulated target gene mRNA expression. For Egfr, Ereg, Cyp19a1, Inha, and Star, GCs were pretreated without (DMSO) or with 10 μm U0126 for 1 h, then treated without (veh) or with FSH for 48 h. For Egr1, GCs were pretreated without (DMSO) or with 50 μm PD98059 for 1 h, then treated without (veh) or with FSH for 1 h. Cells were collected for RNA isolation, and real-time PCR was performed for the genes indicated as described under “Experimental Procedures.” Quantification of results represents the mean ± S.E. of three independent experiments for Egfr and Ereg, and two independent experiments for Cyp19a1, Inha, Egr1, and Star with (*) representing p < 0.05, (**) representing p < 0.01, and (***) representing p < 0.001, based on a one-way ANOVA followed by Tukey's post test.
We next transduced GCs with adenoviral phospho- and dephospho-mutants of YB-1 to determine if select ERK-regulated gene targets were activated by the phosphorylation of YB-1 on Ser102. We initially focused on the three ERK-regulated target genes that showed the greatest inhibition by the MEK inhibitor at the 48 h time point; Star expression was included as a negative control. Transduction of GCs with Ad-YB-1(S102D) phospho-mutant resulted in a statistically significant FSH-independent increase in Egfr and Cyp19a1 mRNAs compared with Ad-Empty (E), which was not significantly enhanced further by FSH (Fig. 9A). In contrast, Inha mRNA levels were not different between Ad-E and Ad-YB-1(S102D) treatments. The ability of the Ad-YB-1(S102D) phospho-mutant to enhance Egfr and Cyp19a1 mRNA levels in the absence of FSH suggests that phosphorylated YB-1(Ser102) is a major contributor to the induction of these genes.
FIGURE 9.

Phospho-YB-1 regulates FSH- and ERK-dependent target gene mRNA expression. A, GCs were transduced with Ad-YB-1(S102D)-FLAG or Ad-Empty (E) overnight. After media replacement and >1 h of recovery, cells were treated without (veh) or with FSH for 48 h. GCs were collected and analyzed as described in Fig. 8. Quantification of results represents the mean ± S.E. of three independent experiments with (*) representing p < 0.05 and (**) representing p < 0.01, based on a one-tailed Student's t test. B, GCs were transduced with Ad-YB-1(S102A)-FLAG or Ad-Empty overnight. After media replacement and >1 h recovery, cells were treated without (veh) or with FSH for 48 h. Cells were collected and analyzed as described in A. Quantification of results represents the mean ± S.E. of three independent experiments for Egfr, Cyp19a1, Inha Cyp11a1, and Hsd17b1 and the mean ± S.E. of two independent experiments for Lhcgr and Pappa, with (**) representing p < 0.01 and (***) representing p < 0.001, based on a one-tailed Student's t test.
Transduction of GCs with the dephospho-Ad-YB-1(S102A) mutant prevented the induction by FSH of Egfr (82.0% ± 9.5%) and Cyp19a1 (88.6% ± 5.5%) mRNAs (Fig. 9B), consistent with results in Fig. 9A. FSH-stimulated induction of Lhcgr (98.6 ± 0.75%), Inha (90.9% ± 2.9%), Cyp11a1 (85.3 ± 3.2%), Hsd17b1 (94.5 ± 3.5%), and Pappa (92.7 ± 6.3%) mRNAs were also abrogated by the dephospho-Ad-YB-1(S102A) mutant. In contrast, FSH-stimulated Star mRNA expression was not reduced by the dephospho-Ad-YB-1(S102A) mutant. The unexpected ability of the dephospho-YB-1(S102A) mutant to induce expression of Egfr in the absence of FSH may be a consequence of YB-1 overexpression, such that the phosphorylation requirement for YB-1 to promote expression of this gene was negated (62). Taken together, we interpret these results to suggest that YB-1 phosphorylated on Ser102 is necessary for the induction of Lhcgr, Cyp19a1, Cyp11a1, Inha, Egfr, Hsd17b1, and Pappa mRNA by FSH but does not participate in the induction of Star mRNA by FSH.
GCs transfected with the indicated promoter-luciferase constructs followed by transduction with Ad-YB-1(S102A) mutant promoted a significant (p < 0.05) inhibition of Egfr (64.9% ± 13.2%), Cyp19a1 (59.9% ± 10.0%), and Inha (72.5% ± 5.0%) promoter-luciferase activities (Fig. 10). These promoter-luciferase results suggest that phosphorylated YB-1 acts primarily at the level of the gene promoters and not at the level of mRNA stability. The lack of complete inhibition of FSH-stimulated signal by the dephospho-YB-1(S102A) mutant of Cyp19a1 and Inha promoter-luciferase constructs as opposed to the ∼90% inhibition of mRNA expression shown in Fig. 9 either reflects the nature of the promoter-luciferase constructs that may be lacking important regulatory elements and/or provides indirect evidence that phospho-YB-1 not only regulates gene transcription but also mRNA stability.
FIGURE 10.

Phospho-YB-1 regulates FSH dependent activity of Egfr, Cyp19a1, and Inha promoter-reporter luciferase constructs. GCs were transfected with the indicated promoter-luciferase constructs for 6 h as described under “Experimental Procedures” followed by overnight transduction with Ad-YB-1(S102A)-FLAG or Ad-Empty (E). After media replacement and >1 h of recovery, cells were treated without (veh) or with FSH for 6 h then collected. Samples were analyzed (see “Experimental Procedures”) and results are expressed as the relative luciferase units (RLU) mean ± S.E. of triplicates and are representative of three independent experiments. (*) represents p < 0.05, (**) represents p < 0.01, and (***) represents p < 0.001, based on a one-tailed Student's t test.
Finally, estradiol-17β levels were measured in culture media of GCs that were transduced with Ad-E versus Ad-YB-1(S102A) and treated with vehicle or FSH for 48 h. Consistent with the ability of the dephospho-Ad-YB-1(S102A) mutant to inhibit the induction by FSH of Cyp19a1, Ad-YB-1(S102A) blocked the ability of FSH to enhance estradiol-17β biosynthesis in the presence of exogenous androstenedione (95.3 ± 2.9%) (Fig. 11). Collectively, these results indicate that YB-1(Ser102) phosphorylation is necessary for FSH-mediated induction of Egfr, Cyp19a1, Inha, Lhcgr, Cyp11a1, Hsd17b1, and Pappa genes that characterize the preovulatory GC as well as for estradiol-17β biosynthesis.
FIGURE 11.
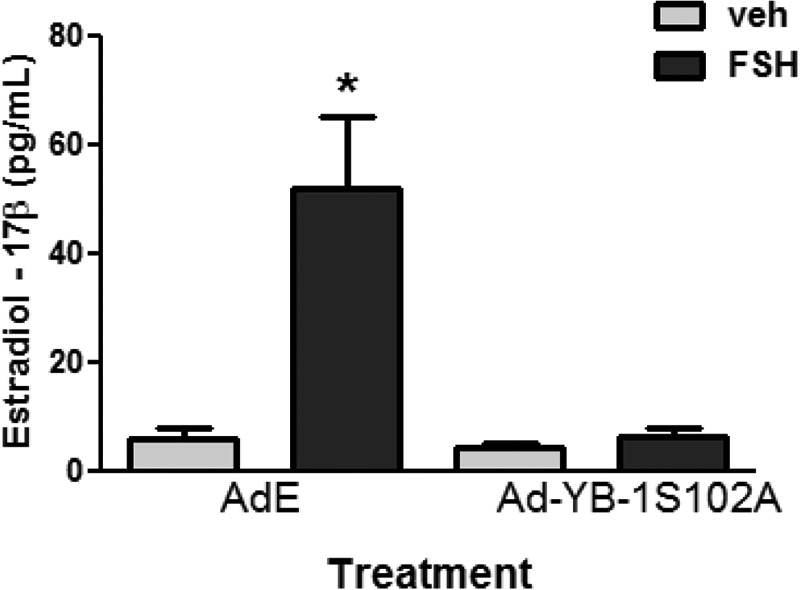
Phospho-YB-1 is necessary for FSH-dependent production of estradiol-17β by GCs. GCs plated in the presence of 10 ng/ml androstenedione were transduced with Ad-YB-1(S102A)-FLAG or Ad-Empty (E) overnight. After media replacement and >1 h of recovery, cells were treated without (veh) or with FSH for 48 h. Media was collected, and estradiol-17β levels were measured by RIA. Results represent the mean ± S.E. of three independent experiments, with (*) representing p < 0.05 based on a one-way ANOVA followed by Tukey's post test.
Discussion
Our results show that YB-1 is phosphorylated on Ser102 in response to FSH. However, in contrast to our original premise, YB-1 in GCs is not an AKT target. Rather, our results reveal that YB-1 is phosphorylated on Ser102 in response to FSH in a PKA- and ERK/RSK-2-dependent manner, consistent with previous reports that YB-1(Ser102) is a RSK-2 target (26, 52). Unexpectedly, our results suggest that the major effect of RSK-2 on YB-1(Ser102) phosphorylation is to inactivate PP1 rather than to promote the direct phosphorylation of YB-1, as discussed below. YB-1 phosphorylated on Ser102 is readily detected in the nuclear fraction and is required for the ability of FSH to induce key target genes required for follicular maturation. Together, our results also reveal a previously unidentified role for YB-1 and an unappreciated role for the ERK/RSK-2 signaling pathway in FSH-dependent GC maturation.
YB-1 is a multifunctional DNA and RNA-binding protein and consequently participates in most DNA- and RNA-dependent cellular functions (30, 31). In the cytoplasm YB-1 is one of two primary proteins (along with poly-A-binding protein) that compose the mRNP complex (for review, see Ref. 32). Hence, YB-1 is associated with both free nontranslated and polysomal mRNA and is a major regulator of translation (63–65). YB-1 also functions as a transcriptional factor to both repress and activate transcription by binding directly to gene promoters or in complex with other transcriptional factors (for review, see Refs. 22 and 32). YB-1 is generally viewed as an “oncogenic” transcription factor based on its overexpression in breast and lung cancer cells (21, 66) and on its gene targets, which include Egfr, Her2, Pi3kca, and Mdr1 (for review, see Ref. 32). Although YB-1 can be phosphorylated on a number of Ser and Tyr residues, phosphorylation of Ser102 within the cold shock domain is most commonly associated with the regulation of translation and transcription (for review, see Ref. 32). Phosphorylation of YB-1 on Ser102 promotes its nuclear localization in ovarian and breast cancer cells (20, 22, 26) and is required for Egfr expression in breast cancer cells (21, 23, 24, 26).
Our results show that YB-1 is also phosphorylated on Ser102 in ovarian GCs and that a phospho-YB-1(S102D) mutant is readily detected in the nuclear fraction. Unexpectedly, our results also reveal that YB-1 phosphorylated on Ser102 is necessary for the expression of select FSH gene targets that define a mature, preovulatory GC, such as Lhcgr, Cyp11a1, Egfr, Hsd17b1, Pappa, Cyp19a1, and Inha.
Lhcgr encodes the LH receptor that is required for ovulation, cumulus cell expansion, and the resumption of meiosis (67); Cyp11a1 encodes cytochrome P450 side chain cleavage, the rate-limiting enzyme for progesterone biosynthesis (18, 68); the EGFR contributes to LH-stimulated expansion of cumulus granulosa cells, resumption of meiosis, and ovulation (27, 69, 70); Cyp19a1 encodes aromatase, the rate-limiting enzyme in estrogen biosynthesis (71); Inha encodes for the inhibin hormone subunit inhibin-α that inhibits FSH expression by the anterior pituitary (72); Hsd17b1 encodes the enzyme that converts androstenedione and estrone to testosterone and estradiol-17β, respectively, and is required for normal ovulation and corpus luteum formation (73); and Pappa encodes a secreted metalloproteinase that cleaves IGF-binding proteins and contributes to estrogen and progesterone biosynthesis and to ovulation (74).
FSH-stimulated gene expression in GCs is complex and requires activation of a number of transcriptional factors coupled with inhibition of transcriptional repressors. These events are mediated by multiple signaling pathways activated by FSH and PKA (for review, see Ref. 2). CREB, SF-1(Nr5a1)/LRH-1(Nr5a2), and GATA-4/6 enhance expression of both Cyp19a1 and Inha (46, 75–77), and FOXO1 (15) and T-cell factor 3 (9) repress expression of both genes. Many transcriptional proteins that regulate expression of the Lhcgr (9, 78, 79) and Cyp11a1 (76, 80, 81) have been elucidated, although the mechanisms by which FSH enhances Egfr and Pappa expression have not been reported. However, a role for YB-1 and indeed for the ERK signaling pathway in the transcriptional activation of FSH target genes is novel.
FSH-dependent phosphorylation of YB-1 on Ser102 is mimicked by treatment of GCs with EGF. Although ERK phosphorylation is FSH-dependent, we previously reported that the canonical pathway upstream of ERK is constitutively active, yet necessary for ERK phosphorylation in immature GCs (8, 11). Consistent with this premise, FSH-stimulated ERK phosphorylation is blocked by the EGFR antagonist AG1478 (11). We expect that, in a similar manner, FSH-stimulated ERK/RSK-2-dependent YB-1 phosphorylation on Ser102 is dependent on an active upstream EGFR/MEK pathway that would be blocked by inhibition of upstream signaling proteins, such as the EGFR, RAS, and RAF.
Although YB-1 phosphorylated on Ser102 is readily detected in the nuclear fraction of GCs, the great majority of both phosphorylated YB-1(Ser102) and total YB-1 is present in the cytoplasmic fraction (see Fig. 7, where the cytoplasmic and nuclear fractions represent 9% and 50% of the total fraction). Cytoplasmic YB-1 has been linked with both translated polysomal mRNPs and free untranslated mRNPs (for review, see Ref. 32). YB-1 is recognized to stabilize both long- and short-lived mRNAs at high YB-1/mRNA ratios effectively by burying mRNAs within YB-1 protein complexes (for review, see Ref. 32). In NIH3T3 cells, AKT-dependent phosphorylation of YB-1 on Ser102 stimulated Cap-dependent translation (64). It is thus likely that cytosolic YB-1 is playing a major role in both stabilizing GC mRNA and, upon phosphorylation, enhancing translation.
FSH-stimulated phosphorylation of YB-1 on Ser102 in GCs is mediated by PKA, ERK, and its downstream kinase RSK-2. However, to our surprise, our results suggest that the major effect of RSK-2 is to inhibit the phosphatase activity of PP1. This conclusion is based largely on the inability of FSH to increase phospho-YB-1(Ser102) signal over that seen in vehicle-treated cells in the presence of the PP1 inhibitor tautomycin (see Fig. 6B). Moreover, the RSK-2 inhibitor BI-D1870 abrogates YB-1(Ser102) phosphorylation in vehicle- and FSH-treated cells even though PKA remains active (evidenced by CREB phosphorylation on Ser133; see Fig. 5). Additionally, the PP1β catalytic subunit, RSK-2, and YB-1 co-immunoprecipitate in a complex. The presence of phosphorylated YB-1(Ser102) in GCs pretreated with tautomycin and treated with vehicle (i.e. in the absence of FSH) further suggests that although RSK-2 may also directly phosphorylate YB-1 in the presence of FSH, RSK-2 is not the kinase that phosphorylates YB-1 on Ser102 in vehicle-treated cells. We hypothesize that Ser102 may be phosphorylated by the constitutively active kinase casein kinase 2, as the phosphorylation motif on YB-1 (KYLRSVGD, where is S is Ser102) partially aligns with that of casein kinase 2 (S(E/D)X(E/D)) (82).
We do not know the mechanism by which RSK-2 inactivates PP1 activity. The catalytic subunits of PP1 can associate with a large number of regulatory subunits (up to 200 have been identified), many of which inhibit the catalytic activity of PP1 (83). Unfortunately, we have not been able to identify the PP1 regulatory subunit inhibited by RSK-2. We hypothesize, however, that RSK-2-dependent phosphorylation of the PP1 regulatory subunit in GCs promotes its degradation, allowing for the prolonged phosphorylation of YB-1 that persists up to 8 h post FSH treatment despite the shorter-lived phosphorylation of RSK-2 that is undetectable within 2 h post FSH treatment (8).
The prolonged phosphorylation of YB-1 was not observed with other transcriptional targets in GCs, such as CREB and histone H3, whose phosphorylation has returned to the levels of untreated cells by 4 h post FSH treatment (8). We do not know the functional significance of this sustained phosphorylation of YB-1. It is unlikely to be required for transcriptional regulation. The prolonged YB-1 phosphorylation is more likely to participate either in mRNA stabilization and/or translation.
Although Ser102 is the most studied phosphorylation site on YB-1, multiple kinases have been shown to phosphorylate YB-1. ERK and glycogen synthase kinase 3β are reported to phosphorylate YB-1 on Thr38 and/or Ser41 and/or Ser45 (36). Indeed, mass spectrometric analysis has identified additional phosphorylations on YB-1, including Tyr162, Ser165 and/or Ser167, Ser174 and/or Ser176, and Ser313 and/or Ser314 (84, 85). Therefore, we cannot rule out the possibility that FSH promotes phosphorylation of YB-1 on additional sites by other kinases.
In conclusion, we have shown that FSH promotes YB-1(Ser102) phosphorylation through a PKA- and ERK/RSK-2-dependent signaling pathway. The major effect of RSK-2 appears to be to inhibit the Ser/Thr phosphatase PP1 rather than to directly phosphorylate YB-1 on Ser102. YB-1 phosphorylated on Ser102 is necessary for the expression of FSH-dependent gene targets that are essential for GC maturation. Taken together, our results not only establish a functional role for YB-1 as a transcriptional activator of FSH target genes in the GCs but also reveal an underappreciated role for ERK signaling pathway in the activation of gene expression by FSH in GCs.
Author Contributions
E. M. D. and M. E. H.-D. designed the experiments. E. M. D. performed the experiments and analyzed the data. E. M. D. and M. E. H.-D. wrote the manuscript.
Acknowledgments
We acknowledge the University of Virginia Center for Research in Reproduction Ligand Assay and Analysis Core, supported by National Institutes of Health Grant P50HD28934, for estradiol-17β RIAs. We thank Dr. Wenlong Bai, University of South Florida, Tampa, FL, for kindly providing the full-length human Egfr promoter-luciferase construct. We also thank Dr. John H. Nilson, Dr. Maria Herndon, and Nate Law for many helpful discussions.
This work was supported by National Institutes of Health Grants R01HD065859 and R01HD062053 (to M. E. H.-D.) and Training Grant T32GM083864 (NIGMS; to E. M. D). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- GC
- granulosa cell
- YB-1
- Y-box-binding protein-1
- FSH
- follicle stimulating hormone
- PKI
- PKA inhibitor
- RSK-2
- ribosomal S6 kinase-2
- PP1 and PP2
- protein phosphatase 1 or 2
- LH
- luteinizing hormone
- CREB
- cAMP-response element-binding protein
- AKT
- protein kinase B
- FOXO1
- forkhead box-containing protein O1
- SHP2
- SRC homology-2 (SH2) domain-containing tyrosine phosphatase
- E2
- estradiol-17β
- P
- penicillin
- S
- streptomycin
- mRNP
- mRNA-protein complex
- RIA
- radioimmunoassay
- EGFR
- epidermal growth factor receptor
- CPT
- 8-chlorophenylthiol
- ANOVA
- analysis of variance
- Ad-E
- adenoviral empty.
References
- 1. Richards J. S., Fitzpatrick S. L., Clemens J. W., Morris J. K., Alliston T., and Sirois J. (1995) Ovarian cell differentiation: a cascade of multiple hormones, cellular signals, and regulated genes. Recent Prog. Horm. Res. 50, 223–254 [DOI] [PubMed] [Google Scholar]
- 2. Hunzicker-Dunn M., and Mayo K. E. (2015) Gonadotropin Signaling in the Ovary. In Knobil and Neill's Physiology of Reproduction (Plant T. M., and Zeleznik A. J., eds.) 4th Ed., pp, 895–946, Elsevier Science Publishing Co., Inc., New York [Google Scholar]
- 3. Hsueh A. J., Adashi E. Y., Jones P. B., and Welsh T. H. Jr. (1984) Hormonal regulation of the differentiation of cultured ovarian granulosa cells. Endocr. Rev. 5, 76–127 [DOI] [PubMed] [Google Scholar]
- 4. Mukherjee A., Park-Sarge O. K., and Mayo K. E. (1996) Gonadotropins induce rapid phosphorylation of the 3′,5′-cyclic adenosine monophosphate response element binding protein in ovarian granulosa cells. Endocrinology 137, 3234–3245 [DOI] [PubMed] [Google Scholar]
- 5. Sprengel R., Braun T., Nikolics K., Segaloff D. L., and Seeburg P. H. (1990) The testicular receptor for follicle stimulating hormone: structure and functional expression of cloned cDNA. Mol. Endocrinol. 4, 525–530 [DOI] [PubMed] [Google Scholar]
- 6. DeManno D. A., Cottom J. E., Kline M. P., Peters C. A., Maizels E. T., and Hunzicker-Dunn M. (1999) Follicle-stimulating hormone promotes histone H3 phosphorylation on serine-10. Mol. Endocrinol. 13, 91–105 [DOI] [PubMed] [Google Scholar]
- 7. Hagiwara M., Brindle P., Harootunian A., Armstrong R., Rivier J., Vale W., Tsien R., and Montminy M. R. (1993) Coupling of hormonal stimulation and transcription via the cyclic AMP-responsive factor CREB is rate limited by nuclear entry of protein kinase A. Mol. Cell. Biol. 13, 4852–4859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Salvador L. M., Park Y., Cottom J., Maizels E. T., Jones J. C., Schillace R. V., Carr D. W., Cheung P., Allis C. D., Jameson J. L., and Hunzicker-Dunn M. (2001) Follicle-stimulating hormone stimulates protein kinase A-mediated histone H3 phosphorylation and acetylation leading to select gene activation in ovarian granulosa cells. J. Biol. Chem. 276, 40146–40155 [DOI] [PubMed] [Google Scholar]
- 9. Law N. C., Weck J., Kyriss B., Nilson J. H., and Hunzicker-Dunn M. (2013) Lhcgr expression in granulosa cells: roles for PKA-phosphorylated β-catenin, TCF3, and FOXO1. Mol. Endocrinol. 27, 1295–1310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Das S., Maizels E. T., DeManno D., St Clair E., Adam S. A., and Hunzicker-Dunn M. (1996) A stimulatory role of cyclic adenosine 3′,5′-monophosphate in follicle-stimulating hormone-activated mitogen-activated protein kinase signaling pathway in rat ovarian granulosa cells. Endocrinology 137, 967–974 [DOI] [PubMed] [Google Scholar]
- 11. Cottom J., Salvador L. M., Maizels E. T., Reierstad S., Park Y., Carr D. W., Davare M. A., Hell J. W., Palmer S. S., Dent P., Kawakatsu H., Ogata M., and Hunzicker-Dunn M. (2003) Follicle-stimulating hormone activates extracellular signal-regulated kinase but not extracellular signal-regulated kinase kinase through a 100-kDa phosphotyrosine phosphatase. J. Biol. Chem. 278, 7167–7179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Alam H., Maizels E. T., Park Y., Ghaey S., Feiger Z. J., Chandel N. S., and Hunzicker-Dunn M. (2004) Follicle-stimulating hormone activation of hypoxia-inducible factor-1 by the phosphatidylinositol 3-kinase/AKT/Ras homolog enriched in brain (Rheb)/mammalian target of rapamycin (mTOR) pathway is necessary for induction of select protein markers of follicular differentiation. J. Biol. Chem. 279, 19431–19440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Gonzalez-Robayna I. J., Falender A. E., Ochsner S., Firestone G. L., and Richards J. S. (2000) Follicle-stimulating hormone (FSH) stimulates phosphorylation and activation of protein kinase B (PKB/Akt) and serum- and glucocorticoid-induced kinase (Sgk): evidence for A kinase-independent signaling by FSH in granulosa cells. Mol. Endocrinol. 14, 1283–1300 [DOI] [PubMed] [Google Scholar]
- 14. Hunzicker-Dunn M. E., Lopez-Biladeau B., Law N. C., Fiedler S. E., Carr D. W., and Maizels E. T. (2012) PKA and GAB2 play central roles in the FSH signaling pathway to PI3K and AKT in ovarian granulosa cells. Proc. Natl. Acad. Sci. U.S.A. 109, E2979–E2988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Park Y., Maizels E. T., Feiger Z. J., Alam H., Peters C. A., Woodruff T. K., Unterman T. G., Lee E. J., Jameson J. L., and Hunzicker-Dunn M. (2005) Induction of cyclin D2 in rat granulosa cells requires FSH-dependent relief from FOXO1 repression coupled with positive signals from Smad. J. Biol. Chem. 280, 9135–9148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Richards J. S., Sharma S. C., Falender A. E., and Lo Y. H. (2002) Expression of FKHR, FKHRL1, and AFX genes in the rodent ovary: evidence for regulation by IGF-I, estrogen, and the gonadotropins. Mol. Endocrinol. 16, 580–599 [DOI] [PubMed] [Google Scholar]
- 17. Zeleznik A. J., Saxena D., and Little-Ihrig L. (2003) Protein kinase B is obligatory for follicle-stimulating hormone-induced granulosa cell differentiation. Endocrinology 144, 3985–3994 [DOI] [PubMed] [Google Scholar]
- 18. Liu Z., Rudd M. D., Hernandez-Gonzalez I., Gonzalez-Robayna I., Fan H. Y., Zeleznik A. J., and Richards J. S. (2009) FSH and FOXO1 regulate genes in the sterol/steroid and lipid biosynthetic pathways in granulosa cells. Mol. Endocrinol. 23, 649–661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Law N. C., and Hunzicker-Dunn M. E. (2016) Insulin receptor substrate 1, the hub linking follicle-stimulating hormone to phosphatidylinositol 3-kinase activation. J. Biol. Chem. 291, 4547–4560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Sutherland B. W., Kucab J., Wu J., Lee C., Cheang M. C., Yorida E., Turbin D., Dedhar S., Nelson C., Pollak M., Leighton Grimes H., Miller K., Badve S., Huntsman D., Blake-Gilks C., Chen M., Pallen C. J., and Dunn S. E. (2005) Akt phosphorylates the Y-box binding protein 1 at Ser-102 located in the cold shock domain and affects the anchorage-independent growth of breast cancer cells. Oncogene 24, 4281–4292 [DOI] [PubMed] [Google Scholar]
- 21. Stratford A. L., Habibi G., Astanehe A., Jiang H., Hu K., Park E., Shadeo A., Buys T. P., Lam W., Pugh T., Marra M., Nielsen T. O., Klinge U., Mertens P. R., Aparicio S., and Dunn S. E. (2007) Epidermal growth factor receptor (EGFR) is transcriptionally induced by the Y-box binding protein-1 (YB-1) and can be inhibited with Iressa in basal-like breast cancer, providing a potential target for therapy. Breast Cancer Res. 9, R61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Basaki Y., Hosoi F., Oda Y., Fotovati A., Maruyama Y., Oie S., Ono M., Izumi H., Kohno K., Sakai K., Shimoyama T., Nishio K., and Kuwano M. (2007) Akt-dependent nuclear localization of Y-box-binding protein 1 in acquisition of malignant characteristics by human ovarian cancer cells. Oncogene 26, 2736–2746 [DOI] [PubMed] [Google Scholar]
- 23. To K., Zhao Y., Jiang H., Hu K., Wang M., Wu J., Lee C., Yokom D. W., Stratford A. L., Klinge U., Mertens P. R., Chen C. S., Bally M., Yapp D., and Dunn S. E. (2007) The phosphoinositide-dependent kinase-1 inhibitor 2-amino-N-[4-[5-(2-phenanthrenyl)-3-(trifluoromethyl)-1H-pyrazol-1-yl]phenyl]-ace tamide (OSU-03012) prevents Y-box binding protein-1 from inducing epidermal growth factor receptor. Mol. Pharmacol. 72, 641–652 [DOI] [PubMed] [Google Scholar]
- 24. Wu J., Lee C., Yokom D., Jiang H., Cheang M. C., Yorida E., Turbin D., Berquin I. M., Mertens P. R., Iftner T., Gilks C. B., and Dunn S. E. (2006) Disruption of the Y-box binding protein-1 results in suppression of the epidermal growth factor receptor and HER-2. Cancer Res. 66, 4872–4879 [DOI] [PubMed] [Google Scholar]
- 25. Liu X., Su L., and Liu X. (2013) Loss of CDH1 up-regulates epidermal growth factor receptor via phosphorylation of YBX1 in non-small cell lung cancer cells. FEBS Lett. 587, 3995–4000 [DOI] [PubMed] [Google Scholar]
- 26. Stratford A. L., Fry C. J., Desilets C., Davies A. H., Cho Y. Y., Li Y., Dong Z., Berquin I. M., Roux P. P., and Dunn S. E. (2008) Y-box binding protein-1 serine 102 is a downstream target of p90 ribosomal S6 kinase in basal-like breast cancer cells. Breast Cancer Res. 10, R99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. El-Hayek S., Demeestere I., and Clarke H. J. (2014) Follicle-stimulating hormone regulates expression and activity of epidermal growth factor receptor in the murine ovarian follicle. Proc. Natl. Acad. Sci. U.S.A. 111, 16778–16783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Blobel G. (1972) Protein tightly bound to globin mRNA. Biochem. Biophys. Res. Commun. 47, 88–95 [DOI] [PubMed] [Google Scholar]
- 29. Morel C., Kayibanda B., and Scherrer K. (1971) Proteins associated with globin messenger RNA in avian erythroblasts: Isolation and comparison with the proteins bound to nuclear messenger-likie RNA. FEBS Lett. 18, 84–88 [DOI] [PubMed] [Google Scholar]
- 30. Tafuri S. R., and Wolffe A. P. (1992) DNA binding, multimerization, and transcription stimulation by the Xenopus Y box proteins in vitro. New Biol. 4, 349–359 [PubMed] [Google Scholar]
- 31. Bouvet P., Matsumoto K., and Wolffe A. P. (1995) Sequence-specific RNA recognition by the Xenopus Y-box proteins: an essential role for the cold shock domain. J. Biol. Chem. 270, 28297–28303 [DOI] [PubMed] [Google Scholar]
- 32. Eliseeva I. A., Kim E. R., Guryanov S. G., Ovchinnikov L. P., and Lyabin D. N. (2011) Y-box-binding protein 1 (YB-1) and its functions. Biochemistry 76, 1402–1433 [DOI] [PubMed] [Google Scholar]
- 33. Finkbeiner M. R., Astanehe A., To K., Fotovati A., Davies A. H., Zhao Y., Jiang H., Stratford A. L., Shadeo A., Boccaccio C., Comoglio P., Mertens P. R., Eirew P., Raouf A., Eaves C. J., and Dunn S. E. (2009) Profiling YB-1 target genes uncovers a new mechanism for MET receptor regulation in normal and malignant human mammary cells. Oncogene 28, 1421–1431 [DOI] [PubMed] [Google Scholar]
- 34. Fukada T., and Tonks N. K. (2003) Identification of YB-1 as a regulator of PTP1B expression: implications for regulation of insulin and cytokine signaling. EMBO J. 22, 479–493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Jurchott K., Bergmann S., Stein U., Walther W., Janz M., Manni I., Piaggio G., Fietze E., Dietel M., and Royer H. D. (2003) YB-1 as a cell cycle-regulated transcription factor facilitating cyclin A and cyclin B1 gene expression. J. Biol. Chem. 278, 27988–27996 [DOI] [PubMed] [Google Scholar]
- 36. Coles L. S., Lambrusco L., Burrows J., Hunter J., Diamond P., Bert A. G., Vadas M. A., and Goodall G. J. (2005) Phosphorylation of cold shock domain/Y-box proteins by ERK2 and GSK3β and repression of the human VEGF promoter. FEBS Lett. 579, 5372–5378 [DOI] [PubMed] [Google Scholar]
- 37. Carr D. W., DeManno D. A., Atwood A., Hunzicker-Dunn M., and Scott J. D. (1993) Follicle-stimulating hormone regulation of A-kinase anchoring proteins in granulosa cells. J. Biol. Chem. 268, 20729–20732 [PubMed] [Google Scholar]
- 38. Chen M. J., Chou C. H., Chen S. U., Yang W. S., Yang Y. S., and Ho H. N. (2015) The effect of androgens on ovarian follicle maturation: dihydrotestosterone suppress FSH-stimulated granulosa cell proliferation by upregulating PPARγ-dependent PTEN expression. Sci. Rep. 5, 18319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Erickson G. F., and Hsueh A. J. (1978) Stimulation of aromatase activity by follicle stimulating hormone in rat granulosa cells in vivo and in vitro. Endocrinology 102, 1275–1282 [DOI] [PubMed] [Google Scholar]
- 40. Otsuka F., and Shimasaki S. (2002) A negative feedback system between oocyte bone morphogenetic protein 15 and granulosa cell kit ligand: its role in regulating granulosa cell mitosis. Proc. Natl. Acad. Sci. U.S.A. 99, 8060–8065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. El-Hefnawy T., and Zeleznik A. J. (2001) Synergism between FSH and activin in the regulation of proliferating cell nuclear antigen (PCNA) and cyclin D2 expression in rat granulosa cells. Endocrinology 142, 4357–4362 [DOI] [PubMed] [Google Scholar]
- 42. Miró F., and Hillier S. G. (1996) Modulation of granulosa cell deoxyribonucleic acid synthesis and differentiation by activin. Endocrinology 137, 464–468 [DOI] [PubMed] [Google Scholar]
- 43. Hunzicker-Dunn M. (1981) Selective activation of rabbit ovarian protein kinase isozymes in rabbit ovarian follicles and corpora lutea. J. Biol. Chem. 256, 12185–12193 [PubMed] [Google Scholar]
- 44. Shen Z., Zhang X., Tang J., Kasiappan R., Jinwal U., Li P., Hann S., Nicosia S. V., Wu J., Zhang X., and Bai W. (2011) The coupling of epidermal growth factor receptor down regulation by 1α,25-dihydroxyvitamin D3 to the hormone-induced cell cycle arrest at the G1-S checkpoint in ovarian cancer cells. Mol. Cell Endocrinol. 338, 58–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Pei L., Dodson R., Schoderbek W. E., Maurer R. A., and Mayo K. E. (1991) Regulation of the α-inhibin gene by cyclic adenosine 3′,5′-monophosphate after transfection into rat granulosa cells. Mol. Endocrinol. 5, 521–534 [DOI] [PubMed] [Google Scholar]
- 46. Ito M., Park Y., Weck J., Mayo K. E., and Jameson J. L. (2000) Synergistic activation of the inhibin α-promoter by steroidogenic factor-1 and cyclic adenosine 3′,5′-monophosphate. Mol. Endocrinol. 14, 66–81 [DOI] [PubMed] [Google Scholar]
- 47. Sunters A., Armstrong V. J., Zaman G., Kypta R. M., Kawano Y., Lanyon L. E., and Price J. S. (2010) Mechano-transduction in osteoblastic cells involves strain-regulated estrogen receptor α-mediated control of insulin-like growth factor (IGF) I receptor sensitivity to ambient IGF, leading to phosphatidylinositol 3-kinase/AKT-dependent Wnt/LRP5 receptor-independent activation of β-catenin signaling. J. Biol. Chem. 285, 8743–8758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Gay V. L., Midgley A. R. Jr., and Niswender G. D. (1970) Patterns of gonadotrophin secretion associated with ovulation. Fed. Proc. 29, 1880–1887 [PubMed] [Google Scholar]
- 49. Kawasaki H., Springett G. M., Mochizuki N., Toki S., Nakaya M., Matsuda M., Housman D. E., and Graybiel A. M. (1998) A family of cAMP-binding proteins that directly activate Rap1. Science 282, 2275–2279 [DOI] [PubMed] [Google Scholar]
- 50. Enserink J. M., Christensen A. E., de Rooij J., van Triest M., Schwede F., Genieser H. G., Døskeland S. O., Blank J. L., and Bos J. L. (2002) A novel Epac-specific cAMP analogue demonstrates independent regulation of Rap1 and ERK. Nat. Cell Biol. 4, 901–906 [DOI] [PubMed] [Google Scholar]
- 51. Kemp B. E., Cheng H. C., and Walsh D. A. (1988) Peptide inhibitors of cAMP-dependent protein kinase. Methods Enzymol. 159, 173–183 [DOI] [PubMed] [Google Scholar]
- 52. Reipas K. M., Law J. H., Couto N., Islam S., Li Y., Li H., Cherkasov A., Jung K., Cheema A. S., Jones S. J., Hassell J. A., and Dunn S. E. (2013) Luteolin is a novel p90 ribosomal S6 kinase (RSK) inhibitor that suppresses Notch4 signaling by blocking the activation of Y-box binding protein-1 (YB-1). Oncotarget 4, 329–345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Andric N., and Ascoli M. (2006) A delayed gonadotropin-dependent and growth factor-mediated activation of the extracellular signal-regulated kinase 1/2 cascade negatively regulates aromatase expression in granulosa cells. Mol. Endocrinol. 20, 3308–3320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Bain J., Plater L., Elliott M., Shpiro N., Hastie C. J., McLauchlan H., Klevernic I., Arthur J. S., Alessi D. R., and Cohen P. (2007) The selectivity of protein kinase inhibitors: a further update. Biochem. J. 408, 297–315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Alam H., Weck J., Maizels E., Park Y., Lee E. J., Ashcroft M., and Hunzicker-Dunn M. (2009) Role of the phosphatidylinositol-3-kinase and extracellular-regulated kinase pathways in the induction of hypoxia-inducible factor (HIF)-1 activity and the HIF-1 target vascular endothelial growth factor in ovarian granulosa cells in response to follicle-stimulating hormone. Endocrinology 150, 915–928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Sapkota G. P., Cummings L., Newell F. S., Armstrong C., Bain J., Frodin M., Grauert M., Hoffmann M., Schnapp G., Steegmaier M., Cohen P., and Alessi D. R. (2007) BI-D1870 is a specific inhibitor of the p90 RSK (ribosomal S6 kinase) isoforms in vitro and in vivo. Biochem. J. 401, 29–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Chatfield K., and Eastman A. (2004) Inhibitors of protein phosphatases 1 and 2A differentially prevent intrinsic and extrinsic apoptosis pathways. Biochem. Biophys. Res. Commun. 323, 1313–1320 [DOI] [PubMed] [Google Scholar]
- 58. Favre B., Turowski P., and Hemmings B. A. (1997) Differential inhibition and posttranslational modification of protein phosphatase 1 and 2A in MCF7 cells treated with calyculin-A, okadaic acid, and tautomycin. J. Biol. Chem. 272, 13856–13863 [DOI] [PubMed] [Google Scholar]
- 59. Dalby K. N., Morrice N., Caudwell F. B., Avruch J., and Cohen P. (1998) Identification of regulatory phosphorylation sites in mitogen-activated protein kinase (MAPK)-activated protein kinase-1a/p90rsk that are inducible by MAPK. J. Biol. Chem. 273, 1496–1505 [DOI] [PubMed] [Google Scholar]
- 60. Wu J., Stratford A. L., Astanehe A., and Dunn S. E. (2007) YB-1 is a transcription/translation factor that orchestrates the oncogenome by hardwiring signal transduction to gene expression. Transl. Oncogenomics 2, 49–65 [PMC free article] [PubMed] [Google Scholar]
- 61. Fan H. Y., Liu Z., Shimada M., Sterneck E., Johnson P. F., Hedrick S. M., and Richards J. S. (2009) MAPK3/1 (ERK1/2) in ovarian granulosa cells are essential for female fertility. Science 324, 938–941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Hunter T., and Karin M. (1992) The regulation of transcription by phosphorylation. Cell 70, 375–387 [DOI] [PubMed] [Google Scholar]
- 63. Bader A. G., and Vogt P. K. (2005) Inhibition of protein synthesis by Y box-binding protein 1 blocks oncogenic cell transformation. Mol. Cell. Biol. 25, 2095–2106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Evdokimova V., Ruzanov P., Anglesio M. S., Sorokin A. V., Ovchinnikov L. P., Buckley J., Triche T. J., Sonenberg N., and Sorensen P. H. (2006) Akt-mediated YB-1 phosphorylation activates translation of silent mRNA species. Mol. Cell. Biol. 26, 277–292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Nekrasov M. P., Ivshina M. P., Chernov K. G., Kovrigina E. A., Evdokimova V. M., Thomas A. A., Hershey J. W., and Ovchinnikov L. P. (2003) The mRNA-binding protein YB-1 (p50) prevents association of the eukaryotic initiation factor eIF4G with mRNA and inhibits protein synthesis at the initiation stage. J. Biol. Chem. 278, 13936–13943 [DOI] [PubMed] [Google Scholar]
- 66. Gessner C., Woischwill C., Schumacher A., Liebers U., Kuhn H., Stiehl P., Jürchott K., Royer H. D., Witt C., and Wolff G. (2004) Nuclear YB-1 expression as a negative prognostic marker in nonsmall cell lung cancer. Eur. Respir. J. 23, 14–19 [DOI] [PubMed] [Google Scholar]
- 67. Zhang F. P., Poutanen M., Wilbertz J., and Huhtaniemi I. (2001) Normal prenatal but arrested postnatal sexual development of luteinizing hormone receptor knockout (LuRKO) mice. Mol. Endocrinol. 15, 172–183 [DOI] [PubMed] [Google Scholar]
- 68. Payne A. H., and Hales D. B. (2004) Overview of steroidogenic enzymes in the pathway from cholesterol to active steroid hormones. Endocr. Rev. 25, 947–970 [DOI] [PubMed] [Google Scholar]
- 69. Hsieh M., Lee D., Panigone S., Horner K., Chen R., Theologis A., Lee D. C., Threadgill D. W., and Conti M. (2007) Luteinizing hormone-dependent activation of the epidermal growth factor network is essential for ovulation. Mol. Cell. Biol. 27, 1914–1924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Hsieh M., Thao K., and Conti M. (2011) Genetic dissection of epidermal growth factor receptor signaling during luteinizing hormone-induced oocyte maturation. PloS ONE 6, e21574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Fisher C. R., Graves K. H., Parlow A. F., and Simpson E. R. (1998) Characterization of mice deficient in aromatase (ArKO) because of targeted disruption of the cyp19 gene. Proc. Natl. Acad. Sci. U.S.A. 95, 6965–6970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Woodruff T. K., and Mayo K. E. (1990) Regulation of inhibin synthesis in the rat ovary. Annu. Rev. Physiol. 52, 807–821 [DOI] [PubMed] [Google Scholar]
- 73. Hakkarainen J., Jokela H., Pakarinen P., Heikelä H., Kätkänaho L., Vandenput L., Ohlsson C., Zhang F. P., and Poutanen M. (2015) Hydroxysteroid (17β)-dehydrogenase 1-deficient female mice present with normal puberty onset but are severely subfertile due to a defect in luteinization and progesterone production. FASEB J. 29, 3806–3816 [DOI] [PubMed] [Google Scholar]
- 74. Nyegaard M., Overgaard M. T., Su Y. Q., Hamilton A. E., Kwintkiewicz J., Hsieh M., Nayak N. R., Conti M., Conover C. A., and Giudice L. C. (2010) Lack of functional pregnancy-associated plasma protein-A (PAPPA) compromises mouse ovarian steroidogenesis and female fertility. Biol. Reprod. 82, 1129–1138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Weck J., and Mayo K. E. (2006) Switching of NR5A proteins associated with the inhibin α-subunit gene promoter after activation of the gene in granulosa cells. Mol. Endocrinol. 20, 1090–1103 [DOI] [PubMed] [Google Scholar]
- 76. Bennett J., Wu Y. G., Gossen J., Zhou P., and Stocco C. (2012) Loss of GATA-6 and GATA-4 in granulosa cells blocks folliculogenesis, ovulation, and follicle stimulating hormone receptor expression leading to female infertility. Endocrinology 153, 2474–2485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Carlone D. L., and Richards J. S. (1997) Functional interactions, phosphorylation, and levels of 3′,5′-cyclic adenosine monophosphate-regulatory element binding protein and steroidogenic factor-1 mediate hormone-regulated and constitutive expression of aromatase in gonadal cells. Mol. Endocrinol. 11, 292–304 [DOI] [PubMed] [Google Scholar]
- 78. Chen S., Shi H., Liu X., and Segaloff D. L. (1999) Multiple elements and protein factors coordinate the basal and cyclic adenosine 3′,5′-monophosphate-induced transcription of the lutropin receptor gene in rat granulosa cells. Endocrinology 140, 2100–2109 [DOI] [PubMed] [Google Scholar]
- 79. Tsai-Morris C. H., Geng Y., Buczko E., and Dufau M. L. (1995) Characterization of diverse functional elements in the upstream Sp1 domain of the rat luteinizing hormone receptor gene promoter. J. Biol. Chem. 270, 7487–7494 [DOI] [PubMed] [Google Scholar]
- 80. Tremblay J. J., and Viger R. S. (2003) Novel roles for GATA transcription factors in the regulation of steroidogenesis. J. Steroid Biochem. Mol. Biol. 85, 291–298 [DOI] [PubMed] [Google Scholar]
- 81. Tremblay J. J., and Viger R. S. (2001) GATA factors differentially activate multiple gonadal promoters through conserved GATA regulatory elements. Endocrinology 142, 977–986 [DOI] [PubMed] [Google Scholar]
- 82. Ubersax J. A., and Ferrell J. E. Jr. (2007) Mechanisms of specificity in protein phosphorylation. Nat. Rev. Mol. Cell Biol. 8, 530–541 [DOI] [PubMed] [Google Scholar]
- 83. Bollen M., Peti W., Ragusa M. J., and Beullens M. (2010) The extended PP1 toolkit: designed to create specificity. Trends Biochem. Sci. 35, 450–458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Dephoure N., Zhou C., Villén J., Beausoleil S. A., Bakalarski C. E., Elledge S. J., and Gygi S. P. (2008) A quantitative atlas of mitotic phosphorylation. Proc. Natl. Acad. Sci. U.S.A. 105, 10762–10767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Olsen J. V., Blagoev B., Gnad F., Macek B., Kumar C., Mortensen P., and Mann M. (2006) Global, in vivo, and site-specific phosphorylation dynamics in signaling networks. Cell 127, 635–648 [DOI] [PubMed] [Google Scholar]