

Abstract
A quality control system in the endoplasmic reticulum (ER) efficiently discriminates polypeptides that are in the process of productive folding from conformers that are trapped in an aberrant state. Only the latter are transported into the cytoplasm and degraded in a process termed ER-associated protein degradation (ERAD). In the ER, an enzymatic cascade generates a specific N-glycan structure of seven mannosyl and two N-acetylglucosamine residues (Man7GlcNAc2) on misfolded glycoproteins to facilitate their disposal. We show that a complex encompassing the yeast lectin-like protein Htm1 and the oxidoreductase Pdi1 converts Man8GlcNAc2 on glycoproteins into the Man7GlcNAc2 signal. In vitro the Htm1-Pdi1 complex processes both unfolded and native proteins albeit with a preference for the former. In vivo, elevated expression of HTM1 causes glycan trimming on misfolded and folded proteins, but only degradation of the non-native species is accelerated. Thus, modification with a Man7GlcNAc2 structure does not inevitably commit a protein for ER-associated protein degradation. The function of Htm1 in ERAD relies on its association with Pdi1, which appears to regulate the access to substrates. Our data support a model in which the balanced activities of Pdi1 and Htm1 are crucial determinants for the efficient removal of misfolded secretory glycoproteins.
Keywords: endoplasmic-reticulum-associated protein degradation (ERAD), ER quality control, glycoprotein, protein degradation, protein disulfide isomerase, glycan processing
Introduction
In the endoplasmic reticulum (ER),5 most secretory proteins are modified with Glc3Man9GlcNAc2 oligosaccharides on specific asparagine residues (1, 2). The attachment of these so-called N-glycans supports the folding of polypeptides, as they define regions that are later exposed at the protein surface and promote the interaction with lectin-type chaperones. During protein maturation, a cascade of glucosidases followed by the mannosidase Mns1 trims the N-glycans by successive removal of terminal glucose and mannose moieties to generate a Man8GlcNAc2 structure (2, 3). In yeast this type of oligosaccharide is found on most mature glycoproteins by the time they are released from the ER (4). However, not all proteins successfully attain their native conformation. Mutations within the coding region, environmental stress as well as non-stoichiometric synthesis of protein subunits give rise to aberrantly folded polypeptides that are targeted by an ER protein quality control system. In a process referred to as ER-associated protein degradation (ERAD), aberrant conformers are transported back into the cytosol, where they are degraded by 26S proteasomes (5–7). Central to this system is the HRD ubiquitin ligase, a membrane-embedded protein complex that binds misfolded proteins via its ER-luminally exposed subunits and then ubiquitylates them in the cytosol (8–10).
N-Glycan processing plays an essential role in ERAD. Enzymes of the ER quality control system trim oligosaccharides on misfolded glycoproteins, thereby generating a unique Man7GlcNAc2 structure (Man5–7GlcNAc2 in mammalian cells) that displays a terminal α1,6-linked mannose on its C-branch (2). Proteins that expose both unstructured regions typically found on malfolded polypeptides and the Man7GlcNAc2 glycan are recognized by the Hrd3 and Yos9 (in mammals: Sel1L and OS9) subunits of the HRD ligase, which triggers their dislocation into the cytosol (11–16). Because glycan demannosylation appears to occur rather slow, this process is thought to delimitate the maturation phase, which protects folding intermediates from degradation (2, 3).
Sequence comparison identified the ER-luminal protein Htm1 (homologous to ER mannosidase 1) as a member of the glycosylhydrolase family 47. The deletion of HTM1 causes a significant delay in glycoprotein ERAD suggesting that this protein promotes the conversion of Man8GlcNAc2 glycans into Man7GlcNAc2 structures (17–19). In addition, three mammalian Htm1 orthologues, EDEM1–3, were implicated in supporting glycoprotein ERAD by demannosylation of N-glycans (20–28). A more recent study reports on the glycan-trimming activity of a purified protein complex containing Htm1 and the protein disulfide isomerase Pdi1 (29). However, due to experimental restraints, a conclusive proof of a mannosidase activity of Htm1 is still missing.
The mammalian EDEMs and Htm1 share mannosidase homology domains of high sequence similarity. Unique to Htm1 and other fungal homologues is a large carboxyl-terminal domain, which is required for the interaction to Pdi1 (19, 30). Pdi1 is one of the most abundant proteins in the yeast ER and is essential for cell viability (31–34). This oxidoreductase promotes the formation of native disulfide bonds in polypeptides but also mediates the reduction and isomerization of aberrant disulfide bridges (35, 36). Furthermore, mammalian orthologues of Pdi1 exhibit chaperone activity even toward proteins that do not form disulfide bridges (36–38). Notably, protein disulfide isomerases both from yeast and mammals directly assist in the unfolding as well as the export of ERAD-client proteins from the ER (39–41).
In this study we conclusively show both in vitro and in vivo that Htm1 in complex with Pdi1 trims Man8GlcNAc2 on glycoproteins to a Man7GlcNAc2 structure. This enzymatic activity relies on the association of Htm1 with Pdi1 and is per se not delimitated to misfolded clients. However, binding of Htm1 to Pdi1 seems to direct the demannosylating enzyme toward a defined set of polypeptides; the low amount of Htm1 relative to Pdi1 appears to restrict the activity of Htm1 toward misfolded proteins. Our findings suggest that processing by Htm1 does not ultimately induce ERAD of a glycoprotein but rather raises the efficiency of this process.
Experimental Procedures
Yeast Strains and Media
All Saccharomyces cerevisiae strains used were haploid descendants of DF5 (genotype: MATa/α trp1-1(am)/trp1-1(am) his3-Δ200/his3-Δ200 ura3-52/ura3-52 lys2-801/lys2-801 leu2-3,-112/leu2-3,-112) that were generated following standard protocols for transformation or crossing and are listed in Table 1. Polymerase chain reaction (PCR)-based techniques were employed to construct strains harboring deletions or expressing carboxyl-terminal epitope-tagged variants of the given genes from their chromosomal locus (42–44). Genomic mutations were introduced by replacing the gene of interest with the URA3 gene and subsequent exchange of the latter against a PCR product harboring the desired mutation (45).
TABLE 1.
Yeast strains used in this study
| Strain | Relevant genotype | Reference/source |
|---|---|---|
| YAK013 | TEF(NatNT2)-Htm1-D279N-TEV-ProtA-HIS6(HIS), prc1-1 | This study |
| YAK020 | Δhtm1::HIS, Pdi1–3xHA-HDEL(TRP), prc1-1 | This study |
| YAK051 | TEF(KanMX)-Htm1, Pdi-HA-HDEL(TRP), prc1-1 | This study |
| YAK052 | TEF(KanMX)-Htm1, Pdi-HA-HDEL(TRP), Δhrd1::hph, prc1-1 | This study |
| YAK054 | TEF(KanMX)-Htm1, Erg3–13Myc(TRP), prc1-1 | This study |
| YAK068 | TEF(KanMX)-Htm1, Erg3–13Myc(TRP), Δhrd1::LEU, prc1-1 | This study |
| YAK097 | TEF(KanMX)-Htm1, Δprc1::LEU | This study |
| YAK190 | Pdi1–3xHA-HDEL(TRP), Htm1-13xmyc(HIS), prc1-1 | This study |
| YAK206 | TEF2(natNT)-Htm1F632L-TEV-ProtA-7HIS(HIS), prc1-1 | This study |
| YAK207 | TEF2(natNT)-Htm1Y633S-TEV-ProtA-7HIS(HIS), prc1-1 | This study |
| YAK210 | Erg3–13xmyc: (TRP1), htm1Δ749::HIS, prc1-1 | This study |
| YAK211 | Erg3–13xmyc: (TRP1), htm1Δ760::HIS, prc1-1 | This study |
| YAK212 | Erg3–13xmyc: (TRP1), htm1Δ780::HIS, prc1-1 | This study |
| YAK213 | Erg3–13xmyc: (TRP1), htm1Δ792::HIS, prc1-1 | This study |
| YCH012 | Δhtm1::HIS3, prc1-1 | 57 |
| YCH030 | Htm1-13xmyc-(HIS), prc1-1 | 19 |
| YCH132 | Pdi1–3xHA-HDEL(TRP), prc1-1 | This study |
| YCH137 | Pdi1–3xHA-HDEL(TRP), htm1Δ749::13xmyc(HIS), prc1-1 | 19 |
| YCH175 | htm1Δ582::13xmyc (HIS3), Pdi1–3xHA-HDEL (TRP1), prc1-1 | 19 |
| YCH432 | Htm1-TEV-ProtA-HIS7(HIS), prc1-1 | This study |
| YCH444 | TEF(kanMX)-Htm1-TEV-ProtA-HIS7(HIS), prc1-1 | This study |
| YCH473 | Δhtm1::Trp, Δprc1::LEU2 | This study |
| YCH616 | htm1Δ780–13xmyc::HIS, Pdi1–3xHA-HDEL(TRP), prc1-1 | This study |
| YCH618 | htm1Δ760–13xmyc::HIS, Pdi1–3xHA-HDEL(TRP), prc1-1 | This study |
| YCH626 | htm1Δ792–13xmyc::HIS, Pdi1–3xHA-HDEL(TRP), prc1-1 | This study |
| YJU037 | Δhrd1::TRP1, prc1-1 | 58 |
| YJU047 | Δhrd1::TRP1, Δprc1::LEU2 | This study |
| YJU049 | Δprc1::LEU2 | This study |
| YLJ002 | Erg3–13xmyc(kanMX), prc1-1 | 57 |
| YLJ048 | Δhtm1::HIS3, Erg3–13xmyc(kanMx), prc1-1 | 57 |
| YTX140 | prc1-1 | 59 |
Plasmids
To generate plasmid pAK002, a fragment encoding Htm1–13xMyc from yeast strain YCH154 was amplified by PCR with primers 5′-GTACTCGAGGAGCGACCTCATACTATACCTG-3′ and 5′-TTGTCGACTGGCGACTCTATCTCACCATGG-3′ and cloned into pRS415 via the SalI/XhoI restriction sites. Plasmids pAK020 and pAK021 originate from site-directed mutagenesis of pAK002 with the primers 5′-CTAGAACCTGGATTGTATAACCGGTGGTCCAATC-3′ and5′-GATTGGACCACCGGTTATACAATCCAGGTTCTAG-3′ (pAK020) or 5′-CTAGAACCTGGATTTTCTAACCGGTGGTCCAATC-3′ and 5′-GATTGGACCACCGGTTAGAAAATCCAGGTTCTAG-3′ (pAK021). CPY* and unglycosylated CPY*0000 encoded by the prc1-1 gene on pRS316 were kind gifts from Dieter H. Wolf (46). To generate pDS030 (CPY*ΔCys), prc1-1 was PCR-amplified from genomic DNA of YTX140 and cloned into pRS413 via EcoRI/XhoI (pDS002) followed by site-directed mutagenesis of 11 codons encoding cysteines. Point mutations were introduced into the plasmid-encoded HTM1 gene with the Pfu UltraTM DNA-Polymerase (Agilent Technologies) according to the instructions of the QuikChange® site-directed mutagenesis kit (Agilent Technologies). Plasmid pCH016 (19) was used to modify the PDI1 genomic locus for the expression of HA epitope-tagged Pdi1 with an HDEL-ER retrieval signal (Pdi-HA).
Antibodies
Monoclonal anti-CPY antibodies were purchased from Life Technologies (A6428, clone 10A5B5) or Abcam (ab34636). Monoclonal anti-Pdi1 are commercially available from Pierce (MA1-10032, clone 38H8). The antibody against Sec61 is described elsewhere (47). Monoclonal antibodies specific for the HA epitope (H9658, clone HA-7) and the Myc epitope (M5546, clone 9E10) as well as the secondary HRP-coupled antibodies directed against mouse IgGs (A9044) or rabbit IgGs (A0545) were purchased from Sigma.
Protein Analysis
SDS-PAGE, silver staining, and immunoblotting was done following published protocols (48). Bulk microsomal glycoproteins were visualized with concanavalin A coupled to horseradish peroxidase (ConA-HRP; Sigma #L6397). Proteins were blotted onto a nitrocellulose membrane (Pall Gelman Laboratory), which was blocked overnight in PBS with 2% Tween 20 and then incubated with ConA-HRP in ConA buffer (50 mm Tris, pH 7.5, 0.1% SDS, 1 mm MgCl2, 2 mm CaCl2, 1 mm MnCl2, 0.5 m NaCl). The membrane was washed five times with ConA buffer, and proteins were detected via enhanced chemiluminescence.
Radioactive Pulse-Chase Analysis
Pulse-chase experiments were performed as described elsewhere (10). In brief, cells were radiolabeled with 35S Protein Labeling Mix (PerkinElmer Life Sciences #NEG072007) followed by a chase period with an excess of unlabeled amino acids. Aliquots were taken at the indicated time, and after cell lysis the proteins of interest were immunoprecipitated. CPY*-, CPY*ΔCys-, and Pdi-HA-containing samples were treated with N-glycosidase F (Roche Applied Science #11365177001) to simplify quantification. Bulk glycoproteins were isolated with ConA-coupled Sepharose (GE Healthcare). Samples were separated by SDS-PAGE, and quantification was done using phosphorimaging (Typhoon FLA9500; GE Healthcare).
Cycloheximide Decay Assay
In short, protein synthesis in cells was blocked by the addition of 0.33 mg/ml cycloheximide to the medium, and aliquots were taken at the indicated times. After cell lysis the samples were analyzed by SDS-PAGE and immunoblotting. A detailed description can be found in Bagola et al. (49).
Purification of Htm1
25,000 A600 yeast cells were disrupted in 400 ml of A200 buffer (0.2 m NaCl, 0.1 m Tris, pH 7.5, 2 mm CaCl2, 1 mm MgCl2, 1 mm PMSF) by vigorous shaking with glass beads. After low speed centrifugation (1000 × g, 10 min, 4 °C) the microsomes were recovered from the supernatant by high speed centrifugation (18,000 × g, 30 min, 4 °C). The pellet was suspended in lysis buffer (0.4 m NaCl, 50 mm Tris, pH 7.5, 2 mm CaCl2, 1 mm MgCl2, 1% CHAPS, 1 mm PMSF), and after 30 min of incubation the lysate cleared by centrifugation (18,000 × g, 30 min, 4 °C). Htm1-TPH7 was isolated from the supernatant with 4-ml Talon® beads (Clontech Laboratories, Inc.). After 3–4 h of incubation, beads were washed twice with lysis buffer, once with A200 supplemented with 0.1% SDS and once with A200 containing 1% CHAPS. Htm1-TPH7 was eluted with 10 ml elution buffer (0.2 m imidazole, pH 7.0, 0.2 m NaCl, 1% CHAPS). The eluate was diluted 1:3 with 20 mm Tris, pH 7.5, 0.2% CHAPS, and 2% glycerol and loaded on an ANX Sepharose 4 Fast Flow column (GE Healthcare). Bound material was eluted by a gradient (0.1–0.8 m NaCl) on an ÄKTA SMART FPLC (Amersham Biosciences). Fractions containing Htm1 were dialyzed overnight against GSH buffer (0.1 m Tris, pH 8.0, 65 mm NaCl, 1 mm CaCl2, 1 mm MgCl2, 0.1% CHAPS, 1 mm GSH, 0.2 mm GSSG). Protein concentrations were determined by measuring adsorption at 280 nm on a NanoDrop2000 (Thermo Scientific).
In Vitro Demannosylation of RNase B
0.5 mg of RNase B (Sigma, #R7884) was dissolved in 50 μl of 0.1 m Tris, pH 8.0 (native RNase B) or 50 μl of freshly prepared urea buffer (5 m urea, 0.1 m Tris, pH 8.0, 20 mm DTT, reductively denatured RNase B) and incubated for 1 h on ice and 37 °C, respectively. The buffer was exchanged to 0.1% acetic acid using desalting columns (Thermo Scientific #89882). Demannosylation was performed by incubating 60 μg of Htm1-Pdi1 with 0.5 mg of native or denatured RNase B in GSH buffer (0.1 m Tris, pH 8.0, 65 mm NaCl, 1 mm CaCl2, 1 mm MgCl2, 0.1% CHAPS, 1 mm GSH, 0.2 mm GSSG) at 28 °C for 15 h in a volume of 360 μl. 0.05% SDS was included in the reaction buffer in some experiments. 140 mm NaCl and 5.5 mm N-ethylmaleimide was added, and Htm1-TPH7/Pdi1 was removed by the addition of Talon® beads. After incubation for 4 h at room temperature, beads were removed, and the supernatant was dialyzed against 50 mm NH4HCO3, pH 8.0, overnight. Next, β-mercaptoethanol was added to a concentration of 10%, and samples were incubated at 95 °C for 20 min followed by 20 min at 20 °C. N-Glycans were cleaved from RNase B by treatment with 2.5 units of N-glycosidase F (Roche Applied Science #11365193001) for 6–8 h at 37 °C followed by evaporation of the solvent in a SpeedVac. Cleanup of neutral oligosaccharides was performed via SupelcleanTM ENVI-CarbTM SPE columns (Sigma #57109-U SUPELCO) as described (50), and samples were dried and dissolved in 10 μl of H2O.
Mass Spectrometric Analysis of Glycans
Quantification of glycans was performed on a matrix-assisted laser desorption/ionization time of flight mass spectrometry (MALDI-TOF-TOF) instrument (AB SCIEX TOF/TOF 5800; Applied Biosystems, Framingham, MA) equipped with an Nd:YLF laser (349 nm) operating at a frequency of 1000 Hz. 2,5-Dihydroxybenzoic acid (15 mg/ml in 0.3% TFA in acetonitrile/water (6:4, v/v)) served as the matrix. Samples were prepared by mixing 1 μl of the glycan solution with 1 μl of matrix solution. From the resulting mixture, 1 μl was applied to the sample plate, and samples were dried in air at 24 °C. Analysis was performed in the positive ion mode, and oligosaccharides were detected as cation adducts. For each spectrum 8000 consecutive laser shots were accumulated. Forty spectra (200 shots each) of one spot measured at different positions were averaged. Mass spectra were manually analyzed with the Data Explorer Software (Applied Biosystems). For each spectrum a baseline correction was conducted, and a noise filter smooth was performed using Gaussian Smooth with a filter width of 15.
Preparation of Microsomes
Depending on the experiment 50 or 100 A600 logarithmically growing cells were harvested, washed with water including 1 mm PMSF, and disrupted with glass beads in 0.4 ml or 1 ml of A200 (0.2 m NaCl, 0.1 m Tris, pH 7.5, 2 mm CaCl2, 1 mm MgCl2, 1 mm PMSF). After the addition of 1 ml of A200, debris was removed by centrifugation (1000 × g, 10 min, 4 °C), and microsomes were prepared by a further centrifugation step of the supernatant (20,000 × g, 20 min, 4 °C).
Preparation of Bulk Glycoproteins
Microsomes were prepared from 50 A600 logarithmically growing cells. The pellet was dissolved in 1 ml of lysis buffer (50 mm Tris, pH 7.5, 0.2 m NaCl, 2 mm CaCl2, 1 mm MgCl2, 0.5% Nonidet P-40, 1 mm PMSF) and incubated for 30 min on ice. Lysates were cleared by centrifugation (20,000 × g, 20 min, 4 °C), and glycoproteins were detected as described above.
Release of Oligosaccharides from Microsomes
Microsomes were prepared from 100 A600 logarithmically growing cells. The pellet was resuspended in 0.1 ml of denaturing buffer (50 mm Tris, pH 7.5, 1% SDS, 1 mm PMSF), incubated on ice for 10 min, diluted 1:10 with 50 mm Tris, pH 7.5, 1.1% Triton X-100, 165 mm NaCl, 5.5 mm EDTA, 1 mm PMSF, and incubated on ice again for 30 min. After centrifugation (20,000 × g, 20 min, 4 °C) 1% β-mercaptoethanol and 3 μl of N-glycosidase F (Roche Applied Science #11365177001) was added to the supernatant followed by incubation for 24 h at 37 °C. The samples were dried and purified as described elsewhere (50).
Fluorescent Labeling of Glycans
Free oligosaccharides were fluorescently labeled with 2-anthranilic acid as described (51).
Analysis of Glycans
2-Anthranilic acid-labeled oligosaccharides were separated over a 4.6 × 150 LudgerSepTM N2 amide column (Ludger, Abington, UK) via normal-phase high performance liquid chromatography (HPLC). Analysis was done on an Ultimate 3000 chromatography system (Dionex, Thermo Scientific) consisting of a FLM-3100 separation module and a FLD 3100 fluorescence detector (λextinction = 330 nm, λemission = 420 nm). Solvent A was composed of 50 mm ammonium formate, pH 4.4, in filtered double distilled H2O; solvent B was acetonitrile. The gradient started with 65% B, which was reduced over 22 min to 54% followed by a reduction over 2.5 min to 0%. After 2 min the column was brought to the starting condition of 65% B over 4 min before the next run was started. The column temperature was 40 °C, a flow rate of 1 ml/min was used, and samples were injected via an autosampler in a 65% acetonitrile solution. Data were collected and analyzed using the Chromeleon7-Software (Thermo Scientific).
Native Immunoprecipitation
Microsomes were prepared from 50 A600 logarithmically growing cells. The pellet was dissolved in 1 ml of lysis buffer (50 mm Tris, pH 7.5, 0.2 m NaCl, 2 mm CaCl2, 1 mm MgCl2, 0.5% Nonidet P-40, 1 mm PMSF) and incubated for 30 min on ice. Lysates were cleared by centrifugation at 20,000 × g for 20 min, and epitope-tagged proteins were precipitated with specific antibodies coupled to protein A Sepharose beads (GE Healthcare) at 4 °C overnight. Beads were washed 3 times with 1 ml of lysis buffer, and bound proteins were eluted with SDS sample buffer before analysis by SDS-PAGE and immunoblotting.
Results
Overexpression of HTM1 Promotes Processing of N-Linked Glycans
The forced expression of EDEM1 or -3 increases the electrophoretic mobility of aberrant glycoproteins, suggesting enhanced demannosylation of these polypeptides (21, 25, 28). We wanted to determine whether Htm1, the yeast homologue of the EDEMs, causes similar effects when overexpressed in cells. To this end we modified the chromosomal locus of the HTM1 gene by homologous recombination to allow the expression of Protein A-7His epitope-tagged Htm1 under the control of the strong, constitutive TEF promoter (42, 43). The corresponding protein product was termed Htm1-TPH7. Additionally, we generated a yeast strain expressing Htm1-D279N-TPH7, a variant of Htm1-TPH7 that is unable to support the turnover of misfolded glycoproteins (19). Immunoblotting confirmed that the TEF promoter-driven constructs dramatically elevates the cellular amounts of Htm1-TPH7 and Htm1-D279N-TPH7 (Fig. 1A).
FIGURE 1.

Overexpression of HTM1 accelerates the electrophoretic mobility of several glycoproteins. A, microsomes from wild-type or cells expressing the indicated Htm1 variants from the endogenous promoter or the TEF promoter (↑) were analyzed by immunoblotting (IB) with horseradish peroxidase-conjugated α-mouse antibody (α-mouse-HRP) to detect Protein A-tagged Htm1. Detection of Sec61 with a specific antibody served as loading control is shown. B, electrophoretic mobility of indicated glycoproteins was assayed as in A. C, Htm1-TPH7 and Htm1-D279N-TPH7 were purified from S. cerevisiae expressing either Htm1 variant from the TEF promotor. N-Linked glycans were released with PNGase F, fluorescently labeled with 2-anthranilic acid, and analyzed by normal-phase HPLC. The fluorescence intensity (arbitrary units) is plotted against the retention time on the column. Note the different scales of the ordinate. Man7, Man8, and Man9 refer to Man7GlcNAc2, Man8GlcNAc2 and Man9GlcNAc2, respectively. D, electrophoretic migration of bulk glycoproteins was determined as in B. Bulk glycoproteins were visualized with concanavalin A coupled to HRP (ConA-HRP). The outmost right lane shows a sample treated with PNGase F before the addition of sample buffer to verify detection of glycoproteins. < marks glycoproteins with apparent changes in electrophoretic mobility in Htm1-TPH7 overexpressing cells. WB, Western blot. E, microsomal proteins from the indicated yeast strains were treated with PNGase F to release N-linked glycans. The oligosaccharides were analyzed as in C. Man7, Man8, and Man9 refer to Man7GlcNAc2, Man8GlcNAc2, and Man9GlcNAc2, respectively. (↑) labels samples in which HTM1 was overexpressed from the TEF promoter. Note the different scales of the ordinate.
In extracts from cells that overexpressed Htm1-TPH7, we noticed that CPY*, a mutant variant of carboxypeptidase Y, which fails to adopt its native conformation and is, therefore, retarded in the ER, migrated significantly faster in denaturing polyacrylamide gels than CPY* from wild-type cells or cells that express Htm1-TPH7 from its endogenous promoter (Fig. 1B, upper part). By contrast, overexpression of Htm1-D279N-TPH7 did not increase the electrophoretic mobility of CPY*. Unexpectedly, however, we also observed faster migration of the ER-resident proteins Pdi1 and Htm1 when Htm1-TPH7 was expressed under the control of the TEF promoter (Fig. 1B, lower part). Analysis of the N-glycan profile of Htm1 variants isolated from S. cerevisiae confirmed that the gain in electrophoretic mobility was caused by an elevated amount of Man7GlcNAc2 glycans on overexpressed Htm1-TPH7 compared with overexpressed Htm1-D279N-TPH7, which predominantly carried Man9GlcNAc2 and Man8GlcNAc2 oligosaccharides (Fig. 1C). This indicates that elevated protein levels of Htm1-TPH7 enhance demannosylation of both aberrant and properly folded proteins in the ER. Concordantly, the mobility of other glycoproteins, which were detected by the lectin ConA-HRP, increased upon overexpression of Htm1-TPH7 (Fig. 1D). When we examined the spectra of N-glycans, cleaved from bulk microsomal proteins by HPLC analysis we observed higher amounts of Man7GlcNAc2 glycans in yeast cells with higher levels of Htm1-TPH7 but not of Htm1-D279N-TPH7 (Fig. 1E). Those Man7GlcNAc2 structures most likely originate from native proteins, as structurally defect polypeptides are rapidly degraded and, therefore, should account only for a small fraction of the cell's protein population. Although we cannot exclude that Htm1 demannosylates glycoproteins during folding when they expose similar structural features as terminally misfolded proteins, these data suggest that overexpressed Htm1 processes glycans on misfolded as well as native proteins.
Overexpression of HTM1 Accelerates Degradation of Misfolded Glycoproteins
Does the enhanced trimming of mannose residues change the stability of glycoproteins? We monitored the turnover of the ERAD substrates CPY* and Erg3-Myc in wild type and cells that were either deleted for HTM1 (Δhmt1) or that overexpressed Htm1-TPH7 or Htm1-D279N-TPH7. Radioactive pulse-chase analyses revealed a faster degradation of both proteins in cells containing elevated amounts of Htm1-TPH7 in comparison to wild type (Fig. 2, A and B). Forced expression of the catalytically inactive Htm1-D279N-TPH7 variant, on the other hand, significantly delayed CPY* breakdown. Notably, we did not observe stronger degradation of unglycosylated CPY*0000 (Fig. 2C) and thus were able to demonstrate that overexpressed HTM1 exclusively stimulates proteolysis of glycoproteins. The turnover of native, ER-resident glycoproteins like Htm1-TPH7 and Pdi-HA was not accelerated in Htm1-TPH7-overexpressing cells either, although these proteins displayed increased electrophoretic mobility on gels (Fig. 2, D and E). Furthermore, breakdown of bulk glycoproteins was only marginally enhanced upon forced expression of Htm1-TPH7, as determined in a radioactive pulse-chase assay and precipitation of glycoproteins with ConA-Sepharose beads (Fig. 2, F and G). Based on these results we concluded that oligosaccharide processing alone does not suffice to trigger glycoprotein disposal but, rather, facilitates this process.
FIGURE 2.

Overexpression of HTM1 increases degradation of misfolded glycoproteins. A and B, turnover of CPY* (A) and Erg3-Myc (B) was analyzed by pulse-chase experiments in the specified yeast strains. Representative autoradiographs are shown. * indicates an unspecifically precipitated protein. ♢ marks co-precipitated Protein A-tagged Htm1 variants. C, D, and E, the stability of unglycosylated CPY*0000 (C), overexpressed Htm1-TPH7 and Htm1-D279N-TPH7 (D), or Pdi-HA expressed from the endogenous promoter (E) was determined as in A. F, representative autoradiograph of radioactively labeled bulk glycoproteins that were precipitated from lysates of the indicated yeast strains with ConA-coupled Sepharose. G, quantification of F. A–E and G, except for B, where n = 2 graphs represent mean values of at least three independent experiments, and the S.D. is given. (↑) indicates the overexpression from the TEF promoter.
A Complex of Htm1 and Pdi1 Converts Man8GlcNAc2 Glycans to Man7GlcNAc2 in Vitro
So far an enzymatic activity of Htm1 has only been verified by using a crude mixture of proteins isolated from yeast cells as substrate (29). We, therefore, aimed to establish an in vitro assay with purified components to investigate the catalytic propensities of this enzyme in more detail. To this end we isolated Htm1-TPH7 and Htm1-D279N-TPH7 to near purity from yeast cell extracts employing metal affinity chromatography followed by separation on an anion exchange column (Fig. 3A). In silver-stained gels, an additional protein with a molecular mass of about 50 kDa was detectable in the Htm1-D279N-TPH7 sample, which was identified as UDP-glucose pyrophosphorylase by mass spectrometry. This protein is located in the yeast cytosol and hence represents most likely a contamination of the sample.
FIGURE 3.

A complex of Htm1 and Pdi1 converts Man8GlcNAc2 glycans to Man7GlcNAc2in vitro. A, Htm1-TPH7 and Htm1-D279N-TPH7 were isolated from S. cerevisiae by metal affinity chromatography followed by separation over an anion exchange column. The purified proteins were analyzed by SDS-PAGE and silver staining. The asterisk indicates UDP-glucose pyrophosphorylase that co-purified with Htm1-D279N-TPH7. B, representative mass spectra of the [M + Na]+ ions of N-linked glycans from RNase B; denatured RNase B was incubated without Htm1-TPH7/Pdi1, with Htm1-TPH7/Pdi1, or with Htm1-D279N-TPH7/Pdi1. After removal of Htm1-TPH7/Pdi1 or Htm1-D279N-TPH7/Pdi1, glycans of RNase B released by PNGase F were analyzed by MALDI-MS. The corresponding ManxGlcNAc2 oligosaccharide of each peak is indicated as Man5, Man6, Man7, Man8, and Man9, respectively. C, the amount of each glycan as determined by the peak areas in experiments (B) was set in relation to the total amount of oligosaccharides. Three independent experiments were quantified, and the S.D. is shown. D, MS spectrum obtained as in B after conducting the in vitro assay with Htm1-TPH7/Pdi1 only. Note that even with a highly reduced scale of the y-axis no glycans from Htm1 or any other source were detected.
Furthermore, we found a protein with an apparent molecular mass of 58 kDa in both purifications. Mass spectrometric analysis revealed that this signal originated from protein disulfide isomerase Pdi1, which forms a tight complex with Htm1 (19, 29, 30). Even when Htm1-TPH7 and Htm1-D279N-TPH7 were purified in a buffer containing 0.1% of the denaturing detergent sodium dodecyl sulfate (SDS) and 400 mm sodium chloride, significant amounts of Pdi1 were detected in the samples. Application of yet harsher buffer conditions, which may have facilitated dissociation of this protein complex, most likely would have affected the folding of Htm1 and rendered the protein inactive. For this reason, we used the Pdi1 containing preparations of Htm1-TPH7 and Htm1-D279N-TPH7 for our in vitro analysis.
First, we were interested if the purified Htm1 variants display mannosidase activity toward a misfolded glycoprotein. Therefore, we incubated chemically denatured RNase B in absence or presence of either Htm1-TPH7/Pdi1 or Htm1-D279N-TPH7/Pdi1. Because Htm1 and Pdi1 are glycosylated, Talon® beads were added to all samples at the end of the reaction to remove the Htm1-Pdi1 complexes. Next, we cleaved the N-glycans from the remaining RNase B with peptide:N-glycosidase F (PNGase F). Salt and proteinaceous material was subsequently withdrawn from the samples by passing them through a non-porous graphitized carbon column (52). The oligosaccharides were then analyzed by MALDI-Tof-MS (Fig. 3B), which allows the reliable quantification of neutral glycans (53). When performed in the absence of RNase B, the assay did not yield detectable amounts of N-glycans, which confirms that the samples were not contaminated with oligosaccharides derived from Pdi1, Htm1, or other sources (Fig. 3D). Only when RNase B was present in the assay, the MS spectrum showed five evident peaks that correspond to the [M+Na]+ ions of the following RNase B-derived oligosaccharides: Man5GlcNAc2 (m/z = 1257), Man6GlcNAc2 (m/z = 1419), Man7GlcNAc2 (m/z = 1582), Man8GlcNAc2 (m/z = 1744), and Man9GlcNAc2 (m/z = 1904). The relative amounts of all mannose forms have been calculated from the MS signals and represent the average of three independent experiments (Fig. 3C). Our approach allows the faithful quantification of the N-glycans on RNase B, as the relative glycan composition of the sample containing only RNase B perfectly matched published data (54). Compared with the control, the relative distribution of individual mannose forms remained unchanged after incubation of RNase B with Htm1-D279N-TPH7/Pdi1. Strikingly, the addition of Htm1-TPH7/Pdi1 to the reaction caused a significant reduction of Man8GlcNAc2 from 9.8% to 4.8%, whereas the fraction of Man7GlcNAc2 and Man6GlcNAc2 increased from 3.6% to 7.9% and 25.7% to 27.8%, respectively. These transformations in the glycan spectrum were exclusively observed in samples incubated with Htm1-TPH7/Pdi1. Therefore, fragmentation of the glycans during the MALDI-MS analysis or glycan processing by enzymes that were unspecifically co-purified does not contribute to the conversion of Man8GlcNAc2 to Man7GlcNAc2 and Man6GlcNAc2. The oligosaccharides on RNase B comprise three structurally diverse isomers of Man8GlcNAc2 (54). Most likely only one of those serves as a substrate for Htm1 (19), and therefore, only a subset of the Man8GlcNAc2 pool was converted to Man7GlcNAc2. Our results show that Htm1-TPH7/Pdi1 isolated from yeast cells exhibits mannosidase activity, which can be quantified by MALDI-Tof-MS.
Htm1-Pdi1 Preferentially Demannosylates Non-native Proteins
Our in vivo data suggest that Htm1-TPH7 targets not only misfolded polypeptides but also proteins that have obtained their native conformation. To assess if the complex of Htm1 and Pdi1 distinguishes between folded and non-native polypeptides, we compared Htm1-TPH7-dependent processing of native RNase B and its denatured form in the in vitro assay. The previous in vitro reactions contained 0.05% SDS to prevent refolding of the chemically misfolded RNase B. We now omitted this reagent and monitored the processing of native RNase B and RNase B that was pretreated with urea and a reducing agent to disrupt its folding. Also under these conditions Htm1-TPH7/Pdi1 processed the Man8GlcNAc2 glycan of denatured RNase B to the Man7GlcNAc2 form, albeit at a reduced rate (Fig. 4, A and B; compare the ratios of Man8GlcNAc2 and Man7GlcNAc2 in Figs. 3B and 4B). The ability of Htm1-TPH7/Pdi1 to trim glycans on native RNase B that had not been exposed to the denaturing reagents was even more compromised when compared with the denatured form (Fig. 4, A and B). Thus, Htm1-TPH7/Pdi1 appears to prefer glycoproteins that have not obtained their native conformation over fully folded polypeptides as demannosylation substrates.
FIGURE 4.

Htm1-Pdi1 preferentially processes non-native substrates. Demannosylation of native and reductively denatured RNase B was measured as in Fig. 3B but without the addition of SDS to the reaction buffer. A, representative MS spectra of the [M + Na]+ ions of N-linked glycans from either native RNase B incubated without or with Htm1-TPH7/Pdi1 or reductively denatured RNase B incubated with Htm1-TPH7/Pdi1p. B, relative quantification of RNase B-derived glycans from A. Three independent experiments were quantified, and the S.D. is given.
Binding of Htm1 to Pdi1 Is Required for ERAD
We wanted to illuminate the contribution of the protein disulfide isomerase to the demannosylation reaction. Htm1 forms a tight complex with Pdi1, which seems to support the structural integrity of the mannosidase (19, 29). Consistently, this complex did not dissociate during the purification procedure under near physiological conditions. The carboxyl-terminal domain of Htm1 comprises 278 amino acids that are required for the interaction with Pdi1 (19). Expression of an Htm1 variant lacking this domain delays degradation of CPY*, and its overexpression does not promote increased glycan processing (19). This indicates that the mannosidase homology domain alone does not suffice to support ERAD. To characterize the binding of Htm1 to Pdi1 in more detail, we generated yeast strains expressing chromosomally encoded carboxyl-terminal truncations of Htm1 (Htm1Δ582-Myc, Htm1Δ749-Myc, Htm1Δ760-Myc, Htm1Δ780-Myc, and Htm1Δ792-Myc) in addition to Pdi-HA.
We then immunoprecipitated Pdi-HA from microsomal extracts prepared under non-denaturing conditions and monitored the amount of co-precipitated Htm1-Myc by immunoblotting. Compared with the wild type, reduced amounts of the truncated Htm1 variants were co-purified with Pdi-HA (Fig. 5A). Even the deletion of only four amino acids at the very carboxyl terminus affected the ability of Htm1 to interact with the protein disulfide isomerase. In agreement with a former study, the association with Pdi-HA was completely lost in cells expressing Htm1Δ582-Myc (19) (Fig. 5A). Although some of the truncations still retained residual binding to Pdi-HA, all those variants of Htm1 completely failed to support turnover of the ERAD substrates CPY* and Erg3-Myc, as determined in pulse-chase experiments (Fig. 5, B and C). Thus, our data indicate that the function of the Htm1 carboxyl-terminal domain goes beyond the mere binding of Pdi1. However, the association with Pdi1 is absolutely required for the activity of Htm1 in ERAD.
FIGURE 5.
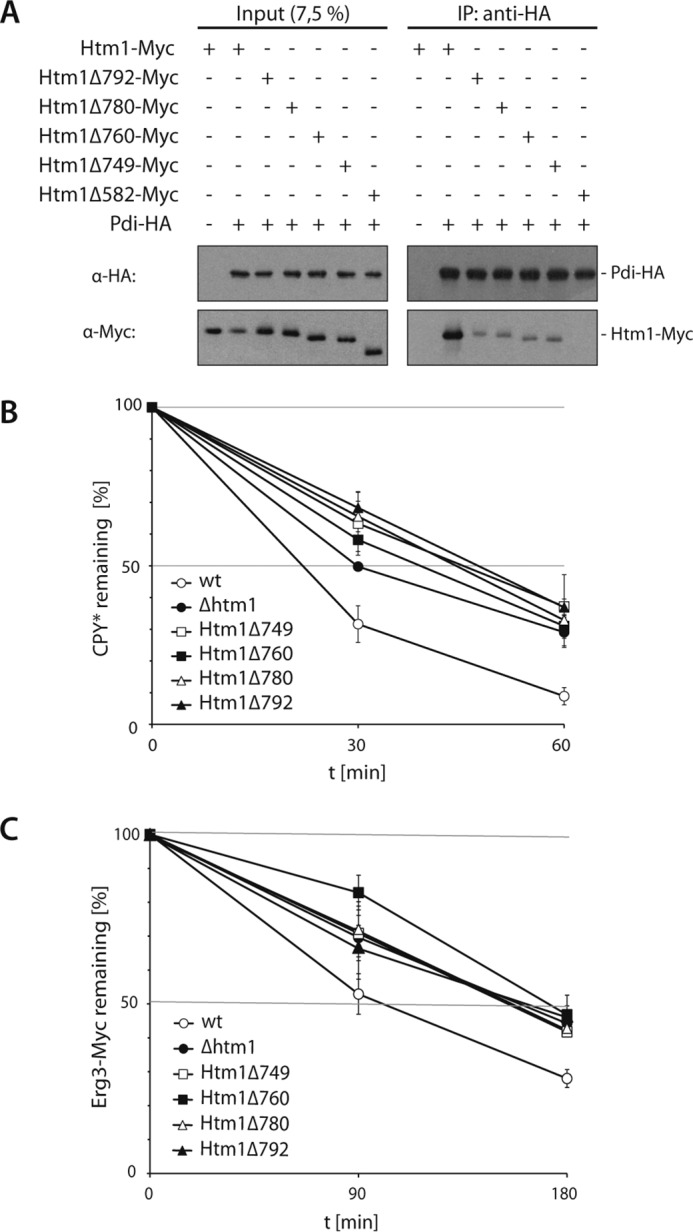
Binding of Htm1 to Pdi1 is required for ERAD. A, Pdi-HA was precipitated under non-denaturing conditions from cell lysates of yeast strains expressing Htm1-Myc or the indicated truncated variants. The proteins were detected by immunoblotting using the given antibodies. B and C, degradation of CPY* (B) or Erg3-Myc (C) was analyzed by pulse-chase experiments in the given yeast strains. Graphs represent the mean values of at least three independent experiments (CPY* and Erg3-Myc in Δhtm1 is n = 2), and the S.D. is given.
Pdi1 Binding-deficient Mutants of Htm1
As predicted by computational sequence analysis of Htm1, the region between Lys-582 and Pro-647, which is required for binding to Pdi1 (19), appears to be unstructured with the exception of an α-helix encompassing amino acids Pro-630 and Trp-636. To elucidate the contribution of this domain to the Htm1 function, we replaced phenylalanine 632 by leucine or tyrosine 633 by serine and monitored these Htm1 variants for their ability to interact with Pdi1. The Myc-tagged versions of both mutant proteins are stably expressed in yeast cells as determined in a cycloheximide decay assay (Fig. 6A). Compared with wild type we found substantially reduced amounts of Htm1-Y633S-Myc and even less Htm1-F632L-Myc in immunoprecipitates of Pdi-HA (Fig. 6B). Likewise, both Htm1 variants affected ERAD in a different manner. Cells expressing Htm1-Y633S-Myc displayed minor, yet significant defects in CPY* turnover, whereas degradation of the model substrate was impaired in Htm1-F632L-Myc-expressing yeasts to a similar extent as observed in an HTM1 deletion strain (Fig. 6C). These results confirm that binding of Htm1 to Pdi1 is crucial for the function of Htm1 in ERAD.
FIGURE 6.

The enzymatic activity of Htm1 requires binding to Pdi1. A, stability of chromosomally expressed Htm1-Myc or plasmid borne Htm1-Myc, Htm1-F632L-Myc and Htm1-Y633S-Myc was analyzed by cycloheximide decay assays. Sec61 visualized with specific antibodies served as loading control. B, Pdi-HA expressed from its endogenous locus was precipitated under non-denaturing conditions from lysates of the indicated yeast strains. Htm1-Myc variants were expressed chromosomally or from plasmids in Δhtm1 strains. Precipitates were subjected to immunoblotting with the indicated antibodies. C, turnover of CPY* was measured by radioactive pulse-chase analysis in wild-type or HTM1-deleted yeast strains harboring plasmid-borne Htm1-Myc, Htm1-F632L-Myc, or Htm1-Y633S-Myc. Graphs represent the mean values of at least four independent experiments, and the S.D. is given. D, microsomes prepared from the given yeast strains were subjected to immunoblot analysis to monitor the electrophoretic mobility of CPY*. E, microsomes from indicated yeast strains were solubilized and treated with PNGase F. The released glycans were fluorescently labeled with 2-anthranilic acid and analyzed via normal-phase HPLC. The fluorescence intensity in arbitrary units is plotted against the retention time on the column. Note the different scales of the ordinate. Man7, Man8, and Man9 represent Man7GlcNAc2, Man8GlcNAc2, and Man9GlcNAc2, respectively. D and E, (↑) indicates overexpression of HTM1 constructs from the TEF promoter.
Next, we were interested if Htm1-F632L-TPH7 and Htm1-Y633S-TPH7 exhibited demannosylation activity. To this end, we overexpressed Htm1-TPH7, Htm1-D279N-TPH7, Htm1-F632L-TPH7, and Htm1-Y633S-TPH7 in yeast cells and analyzed the migration pattern of CPY* in those strains on denaturing gels (Fig. 6D). The electrophoretic mobility of CPY* in cells overexpressing Htm1-Y633S-TPH7 was comparable with that of the Htm1-TPH7-overexpressing control, indicating that the Htm1-Y633S mutant is catalytically active. By contrast, elevated levels of Htm1-F632L-TPH7 or the catalytically inactive Htm1-D279N-TPH7 variant did not modify the mobility of CPY*. In accordance with this result, a slight but significant increase in the amount of Man7GlcNAc2 was detectable in the bulk glycan spectrum of cells overexpressing Htm1-Y633S-TPH7 but not in cells that contained higher levels of Htm1-F632L-TPH7 (Fig. 6E).
Obviously, the ability of Htm1 to support degradation of ERAD substrates is directly linked to its proficiency in glycan processing, which in turn depends on the binding to Pdi1. Because mutant versions of Htm1 that are impaired in their interaction with Pdi1 are stable, an association of Htm1 to the protein disulfide isomerase is most likely not required to maintain its structural integrity but, rather, regulates the catalytic activity of the enzyme.
Htm1 Affects the Turnover of a Cysteine-free Substrate
How does Pdi1 stimulate the function of Htm1 for glycan trimming? Because Pdi1 interacts with immature proteins to promote the formation of disulfide bridges, we wondered whether the isomerase presents polypeptides with unpaired cysteines to Htm1. In pulse-chase analyses we monitored the turnover of plasmid-encoded CPY*, a protein containing 11 cysteines and a version of this ERAD substrate in which all cysteine residues where substituted by serines (CPY*ΔCys) in wild-type-, Δhtm1-, or HTM1-overexpressing yeast strains. In cells deleted for HTM1 the degradation of CPY* and CPY*ΔCys was affected, suggesting that even cysteine-free proteins can be targeted by Htm1 (Fig. 7, A and B). Still, Pdi1 was shown to promote the folding of disulfide bond-free substrates in a chaperone-like manner (36–38). Notably, overexpression of HTM1 accelerated the degradation of CPY* but not of the cysteine-free variant. Thus, our data demonstrate that the impact of Htm1 on the turnover of cysteine-free substrates is lower than on proteins containing cysteines. Such polypeptides most likely interact more tightly or frequently with Pdi1 during disulfide bridge rearrangements, which in turn will increase their probability of being processed by Htm1.
FIGURE 7.
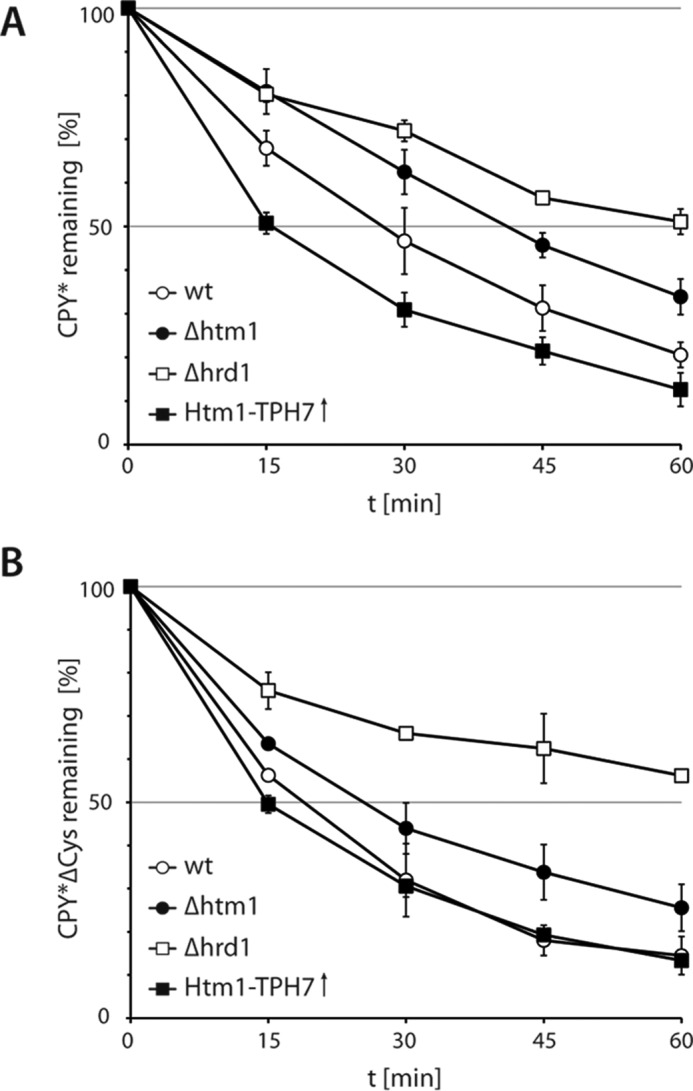
Htm1 affects the turnover of a cysteine-free substrate. A, turnover of plasmid-borne CPY* in cells deleted for PRC1 encoding endogenous CPY was monitored in radioactive pulse-chase experiments. B, as in A, except that cysteine-free CPY* (CPY*ΔCys) served as substrate. A and B, two independent experiments were quantified, and the S.D. is shown. (↑) indicates overexpression from the TEF promoter.
Discussion
A bipartite signal triggers ERAD of aberrantly folded glycoproteins (11–13, 55); Hrd3, a component of the HRD ligase, binds surface-exposed hydrophobic patches of unfolded proteins in the ER lumen and recruits such conformers to the ubiquitin ligase Hrd1. Associated with Hrd3 is the lectin Yos9, which identifies a terminal α1,6-linked mannosyl residue in the N-glycans of these polypeptides and thereby contributes to substrate recognition. Combined, both processes facilitate efficient degradation of malfolded glycoproteins. In the present study we demonstrate by in vitro as well as in vivo experiments that a protein complex comprising the mannosidase Htm1 and the protein disulfide isomerase Pdi1 generates the specific glycan structure recognized by Yos9.
By taking advantage of a well defined substrate in our in vitro assays we were able to conclusively show that Htm1 in complex with Pdi1 converts Man8GlcNAc2 on glycoproteins into Man7GlcNAc2 structures. Previous work investigated isolated Htm1 under less well defined conditions by studying glycan processing in total cell extracts (29). This approach did not exclude that the activity of Htm1 requires additional components or co-factors. Our experimental setup also enabled us to determine the performance of the mannosidase under varying conditions. Interestingly, the activity of Htm1-Pdi1 does not appear to be restricted to aberrantly folded glycoproteins; we observed that the mannosidase trims glycans on both denatured and native RNase B, albeit with a slight preference for the misfolded form. Consistently, increased glycan processing in cells overexpressing HTM1 was not confined to malfolded proteins; yet whereas the enhanced activity of Htm1 on aberrant glycoproteins accelerated their degradation, it had no impact on the stability of properly folded polypeptides or on non-glycosylated ERAD substrates.
Our results show that Htm1's function in ERAD depends on its interaction with Pdi1. By expressing Htm1 variants that harbored truncations or amino acid substitutions in the carboxyl-terminal part of Htm1, we were able to dissociate the Htm1-Pdi1 complex in yeast. This allowed us to monitor Htm1 activity in the absence of bound Pdi1 without expressing mutant forms of Pdi1 that may impair oxidative folding in general. Pdi1-L313P, which was previously used to separate Htm1 from Pdi1, not only affects the glycosylation status of Htm1, indicative of a mislocalization of this enzyme to the Golgi compartment, but also induces Hrd1-dependent ERAD of Htm1 (29). Despite recent findings that certain amino acid substitutions in the carboxyl-terminal part of Htm1 affect the general folding of the protein (30), all of our truncation constructs as well as the mutant variants Htm1-F632L and Htm1-Y633S turned out to be stable.
If an association with Pdi1 is not a prerequisite to maintain the structural integrity of Htm1 but is still required for the function of the mannosidase in ERAD, what is the role of the protein disulfide isomerase in the complex? Given the function of Pdi1 in the maturation of unfolded polypeptides, it is conceivable that the oxidoreductase confines the activity of Htm1 toward distinct clients. Thus, Htm1 may predominantly demannosylate glycoproteins that associate with Pdi1 during their folding attempts. Because the cellular amount of Pdi1 vastly exceeds that of Htm1 (31, 56), only a small number of the Pdi1 molecules are complexed with Htm1. Glycoproteins that rapidly obtain their native conformation associate only briefly with folding enzymes like Pdi1. In consequence, interactions of those proteins with Htm1 are unlikely to occur. By contrast, polypeptides that fold less efficient repeatedly bind to Pdi1 and are thereby more likely subjected to demannosylation by Htm1. Concordantly, we observed demannosylation of properly folded proteins only in cells overexpressing HTM1. Most probably these proteins were more often exposed to the now highly abundant Htm1-Pdi1 complexes during their maturation.
We also found that the turnover of a malfolded glycoprotein devoid of cysteines, CPY*ΔCys, in HTM1-deleted cells is affected to a much lesser degree than the degradation of cysteine containing CPY*. Accordingly, overexpression of HTM1 exclusively accelerated the turnover of CPY*. Obviously, CPY* is the preferred substrate of Pdi1 for disulfide bond rearrangements, which in turn increases its susceptibility to Htm1-mediated glycan processing. The shorter half-life of CPY*ΔCys in wild-type cells might result from a more loosely conformation due to a complete lack of disulfide bridges, which could facilitate recognition by the ERAD machinery or the translocation into the cytosol.
During their maturation polypeptides expose structurally disordered regions, which allows association not only with chaperones like BiP and Pdi1 but also with ERAD components such as Hrd3. Slowly folding proteins and polypeptides trapped in non-native conformations will engage more frequently in such interactions, thereby increasing the likelihood to be processed by the ERAD machinery. The Htm1-generated Man7GlcNAc2 glycan supports the interactions of proteins with Hrd3 by binding to the Hrd3-associated lectin Yos9. This enhances the chance of a protein to be subjected to ERAD. In line with this, we and others noticed that glycosylated ERAD model substrates are substantially faster processed than molecules that share the same polypeptide backbone but lack glycan residues (12, 46, 57). Importantly, an interaction with Yos9 alone does not suffice to trigger ERAD of a malfolded protein. Why should an ER quality control system based on glycan processing be required to proficiently detect aberrant conformers of glycoproteins? Disulfide bridge formation facilitates the partial folding of polypeptides, which probably renders the identification of malfolded species solely based on features of the peptide backbone fairly inefficient. Thus, we suggest that the stochastic demannosylation by Htm1 generates an additional signal that compensates the poor binding of aberrantly folded glycoproteins to ERAD receptors and thereby stimulates their degradation.
Author Contributions
A. P., C. V., C. H., and E. J. performed all experiments except glycan analysis by mass spectrometry (H. S.). A. P., E. K., C. H., E. J., and T. S. designed the experiments and discussed the results. A. P., C. H., E. J., and T. S. wrote the manuscript.
Acknowledgments
We thank all the members of the laboratory of Thomas Sommer for fruitful discussions and materials. CPY* and unglycosylated CPY*0000 encoded by the prc1−1 gene on pRS316 were kindly provided by Dieter H. Wolf (University of Stuttgart, Germany). We also thank Terry D. Butters (University of Oxford, UK) and his group as well as Gunnar Dittmar (Max-Delbrück-Center for Molecular Medicine, Germany) and the members of the Dittmar laboratory for excellent help in establishing glycan analysis by HPLC in the Sommer laboratory. Furthermore, we thank the laboratory of Gunnar Dittmar for support with mass spectrometry measurements. Regine Pfeiffer is acknowledged for carefully reading the manuscript.
This work was supported by Deutsche Forschungsgemeinschaft (DFG) (to T. S.). The authors declare that they have no conflicts of interest with the contents of this article.
- ER
- endoplasmic reticulum
- ERAD
- ER-associated protein degradation
- Htm1
- homologous to mannosidase 1
- Pdi1
- protein disulfide isomerase 1
- ConA-HRP
- concanavalin A coupled to horseradish peroxidase
- PNGase F
- peptide:N-glycosidase F
- CPY
- carboxypeptidase Y
- HRD
- HmgCoA reductade degradation
- TEF
- translation elongation factor.
References
- 1. Helenius A., and Aebi M. (2001) Intracellular functions of N-linked glycans. Science 291, 2364–2369 [DOI] [PubMed] [Google Scholar]
- 2. Aebi M., Bernasconi R., Clerc S., and Molinari M. (2010) N-glycan structures: recognition and processing in the ER. Trends Biochem. Sci. 35, 74–82 [DOI] [PubMed] [Google Scholar]
- 3. Hebert D. N., and Molinari M. (2012) Flagging and docking: dual roles for N-glycans in protein quality control and cellular proteostasis. Trends Biochem. Sci. 37, 404–410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Byrd J. C., Tarentino A. L., Maley F., Atkinson P. H., and Trimble R. B. (1982) Glycoprotein synthesis in yeast: identification of Man8GlcNAc2 as an essential intermediate in oligosaccharide processing. J. Biol. Chem. 257, 14657–14666 [PubMed] [Google Scholar]
- 5. Amm I., Sommer T., and Wolf D. H. (2014) Protein quality control and elimination of protein waste: the role of the ubiquitin-proteasome system. Biochim. Biophys. Acta 1843, 182–196 [DOI] [PubMed] [Google Scholar]
- 6. Hirsch C., Gauss R., Horn S. C., Neuber O., and Sommer T. (2009) The ubiquitylation machinery of the endoplasmic reticulum. Nature 458, 453–460 [DOI] [PubMed] [Google Scholar]
- 7. Ruggiano A., Foresti O., and Carvalho P. (2014) Quality control: ER-associated degradation: protein quality control and beyond. J. Cell Biol. 204, 869–879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bays N. W., Gardner R. G., Seelig L. P., Joazeiro C. A., and Hampton R. Y. (2001) Hrd1p/Der3p is a membrane-anchored ubiquitin ligase required for ER-associated degradation. Nat. Cell Biol. 3, 24–29 [DOI] [PubMed] [Google Scholar]
- 9. Deak P. M. (2001) Membrane topology and function of Der3/Hrd1p as a ubiquitin-protein ligase (E3) involved in endoplasmic reticulum degradation. J. Biol. Chem. 276, 10663–10669 [DOI] [PubMed] [Google Scholar]
- 10. Gauss R., Sommer T., and Jarosch E. (2006) The Hrd1p ligase complex forms a linchpin between ER-lumenal substrate selection and Cdc48p recruitment. EMBO J. 25, 1827–1835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Denic V., Quan E. M., and Weissman J. S. (2006) A luminal surveillance complex that selects misfolded glycoproteins for ER-associated degradation. Cell 126, 349–359 [DOI] [PubMed] [Google Scholar]
- 12. Quan E. M., Kamiya Y., Kamiya D., Denic V., Weibezahn J., Kato K., and Weissman J. S. (2008) Defining the glycan destruction signal for endoplasmic reticulum-associated degradation. Mol. Cell 32, 870–877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Gauss R., Jarosch E., Sommer T., and Hirsch C. (2006) A complex of Yos9p and the HRD ligase integrates endoplasmic reticulum quality control into the degradation machinery. Nat. Cell Biol. 8, 849–854 [DOI] [PubMed] [Google Scholar]
- 14. Mehnert M., Sommer T., and Jarosch E. (2014) Der1 promotes movement of misfolded proteins through the endoplasmic reticulum membrane. Nat. Cell Biol. 16, 77–86 [DOI] [PubMed] [Google Scholar]
- 15. Hosokawa N., Kamiya Y., Kamiya D., Kato K., and Nagata K. (2009) Human OS-9, a lectin required for glycoprotein endoplasmic reticulum-associated degradation, recognizes mannose-trimmed N-glycans. J. Biol. Chem. 284, 17061–17068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Satoh T., Chen Y., Hu D., Hanashima S., Yamamoto K., and Yamaguchi Y. (2010) Structural basis for oligosaccharide recognition of misfolded glycoproteins by OS-9in ER-associated degradation. Mol. Cell 40, 905–916 [DOI] [PubMed] [Google Scholar]
- 17. Nakatsukasa K., Nishikawa S., Hosokawa N., Nagata K., and Endo T. (2001) Mnl1p, an α-mannosidase-like protein in yeast Saccharomyces cerevisiae, is required for endoplasmic reticulum-associated degradation of glycoproteins. J. Biol. Chem. 276, 8635–8638 [DOI] [PubMed] [Google Scholar]
- 18. Jakob C. A., Bodmer D., Spirig U., Battig P., Marcil A., Dignard D., Bergeron J. J., Thomas D. Y., and Aebi M. (2001) Htm1p, a mannosidase-like protein, is involved in glycoprotein degradation in yeast. EMBO Rep. 2, 423–430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Clerc S., Hirsch C., Oggier D. M., Deprez P., Jakob C., Sommer T., and Aebi M. (2009) Htm1 protein generates the N-glycan signal for glycoprotein degradation in the endoplasmic reticulum. J. Cell Biol. 184, 159–172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hosokawa N., Tremblay L. O., Sleno B., Kamiya Y., Wada I., Nagata K., Kato K., and Herscovics A. (2010) EDEM1 accelerates the trimming of α1,2-linked mannose on the C branch of N-glycans. Glycobiology 20, 567–575 [DOI] [PubMed] [Google Scholar]
- 21. Hirao K., Natsuka Y., Tamura T., Wada I., Morito D., Natsuka S., Romero P., Sleno B., Tremblay L. O., Herscovics A., Nagata K., and Hosokawa N. (2006) EDEM3, a soluble EDEM homolog, enhances glycoprotein endoplasmic reticulum-associated degradation and mannose trimming. J. Biol. Chem. 281, 9650–9658 [DOI] [PubMed] [Google Scholar]
- 22. Ninagawa S., Okada T., Sumitomo Y., Kamiya Y., Kato K., Horimoto S., Ishikawa T., Takeda S., Sakuma T., Yamamoto T., and Mori K. (2014) EDEM2 initiates mammalian glycoprotein ERAD by catalyzing the first mannose trimming step. J. Cell Biol. 206, 347–356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hosokawa N., Wada I., Hasegawa K., Yorihuzi T., Tremblay L. O., Herscovics A., and Nagata K. (2001) A novel ER α-mannosidase-like protein accelerates ER-associated degradation. EMBO Rep. 2, 415–422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Oda Y., Hosokawa N., Wada I., and Nagata K. (2003) EDEM as an acceptor of terminally misfolded glycoproteins released from calnexin. Science 299, 1394–1397 [DOI] [PubMed] [Google Scholar]
- 25. Molinari M., Calanca V., Galli C., Lucca P., and Paganetti P. (2003) Role of EDEM in the release of misfolded glycoproteins from the calnexin cycle. Science 299, 1397–1400 [DOI] [PubMed] [Google Scholar]
- 26. Olivari S., Galli C., Alanen H., Ruddock L., and Molinari M. (2005) A novel stress-induced EDEM variant regulating endoplasmic reticulum-associated glycoprotein degradation. J. Biol. Chem. 280, 2424–2428 [DOI] [PubMed] [Google Scholar]
- 27. Mast S. W., Diekman K., Karaveg K., Davis A., Sifers R. N., and Moremen K. W. (2005) Human EDEM2, a novel homolog of family 47 glycosidases, is involved in ER-associated degradation of glycoproteins. Glycobiology 15, 421–436 [DOI] [PubMed] [Google Scholar]
- 28. Olivari S., Cali T., Salo K. E., Paganetti P., Ruddock L. W., and Molinari M. (2006) EDEM1 regulates ER-associated degradation by accelerating de-mannosylation of folding-defective polypeptides and by inhibiting their covalent aggregation. Biochem. Biophys. Res. Commun. 349, 1278–1284 [DOI] [PubMed] [Google Scholar]
- 29. Gauss R., Kanehara K., Carvalho P., Ng D. T., and Aebi M. (2011) A complex of Pdi1p and the mannosidase Htm1p initiates clearance of unfolded glycoproteins from the endoplasmic reticulum. Mol. Cell 42, 782–793 [DOI] [PubMed] [Google Scholar]
- 30. Sakoh-Nakatogawa M., Nishikawa S.-I., and Endo T. (2009) Roles of protein-disulfide isomerase-mediated disulfide bond formation of yeast Mnl1p in endoplasmic reticulum-associated degradation. J. Biol. Chem. 284, 11815–11825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Mizunaga T., Katakura Y., Miura T., and Maruyama Y. (1990) Purification and characterization of yeast protein disulfide isomerase. J. Biochem. 108, 846–851 [DOI] [PubMed] [Google Scholar]
- 32. Tachikawa H., Miura T., Katakura Y., and Mizunaga T. (1991) Molecular structure of a yeast gene, PDI1, encoding protein disulfide isomerase that is essential for cell growth. J. Biochem. 110, 306–313 [DOI] [PubMed] [Google Scholar]
- 33. Farquhar R., Honey N., Murant S. J., Bossier P., Schultz L., Montgomery D., Ellis R. W., Freedman R. B., and Tuite M. F. (1991) Protein disulfide isomerase is essential for viability in Saccharomyces cerevisiae. Gene 108, 81–89 [DOI] [PubMed] [Google Scholar]
- 34. Günther R., Bräuer C., Janetzky B., Förster H. H., Ehbrecht I. M., Lehle L., and Küntzel H. (1991) The Saccharomyces cerevisiae TRG1 gene is essential for growth and encodes a lumenal endoplasmic reticulum glycoprotein involved in the maturation of vacuolar carboxypeptidase. J. Biol. Chem. 266, 24557–24563 [PubMed] [Google Scholar]
- 35. Hatahet F., and Ruddock L. W. (2007) Substrate recognition by the protein disulfide isomerases. FEBS J. 274, 5223–5234 [DOI] [PubMed] [Google Scholar]
- 36. Gilbert H. F. (1997) Protein disulfide isomerase and assisted protein folding. J. Biol. Chem. 272, 29399–29402 [DOI] [PubMed] [Google Scholar]
- 37. Cai H., Wang C. C., and Tsou C. L. (1994) Chaperone-like activity of protein disulfide isomerase in the refolding of a protein with no disulfide bonds. J. Biol. Chem. 269, 24550–24552 [PubMed] [Google Scholar]
- 38. Song J. L., and Wang C. C. (1995) Chaperone-like activity of protein disulfide-isomerase in the refolding of rhodanese. Eur. J. Biochem. 231, 312–316 [DOI] [PubMed] [Google Scholar]
- 39. Weissman J. S., and Kim P. S. (1993) Efficient catalysis of disulphide bond rearrangements by protein disulphide isomerase. Nature 365, 185–188 [DOI] [PubMed] [Google Scholar]
- 40. Tsai B., Rodighiero C., Lencer W. I., and Rapoport T. A. (2001) Protein disulfide isomerase acts as a redox-dependent chaperone to unfold cholera toxin. Cell 104, 937–948 [DOI] [PubMed] [Google Scholar]
- 41. Gillece P., Luz J. M., Lennarz W. J., de La Cruz F. J., and Römisch K. (1999) Export of a cysteine-free misfolded secretory protein from the endoplasmic reticulum for degradation requires interaction with protein disulfide isomerase. J. Cell Biol. 147, 1443–1456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Janke C., Magiera M. M., Rathfelder N., Taxis C., Reber S., Maekawa H., Moreno-Borchart A., Doenges G., Schwob E., Schiebel E., and Knop M. (2004) A versatile toolbox for PCR-based tagging of yeast genes: new fluorescent proteins, more markers, and promoter substitution cassettes. Yeast 21, 947–962 [DOI] [PubMed] [Google Scholar]
- 43. Knop M., Siegers K., Pereira G., Zachariae W., Winsor B., Nasmyth K., and Schiebel E. (1999) Epitope tagging of yeast genes using a PCR-based strategy: more tags and improved practical routines. Yeast 15, 963–972 [DOI] [PubMed] [Google Scholar]
- 44. Longtine M. S., McKenzie A. 3rd, Demarini D. J., Shah N. G., Wach A., Brachat A., Philippsen P., and Pringle J. R. (1998) Additional modules for versatile and economical PCR-based gene deletion and modification in Saccharomyces cerevisiae. Yeast 14, 953–961 [DOI] [PubMed] [Google Scholar]
- 45. Gueldener U., Heinisch J., Koehler G. J., Voss D., and Hegemann J. H. (2002) A second set of loxP marker cassettes for Cre-mediated multiple gene knockouts in budding yeast. Nucleic Acids Res. 30, e23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Kostova Z., and Wolf D. H. (2005) Importance of carbohydrate positioning in the recognition of mutated CPY for ER-associated degradation. J. Cell Sci. 118, 1485–1492 [DOI] [PubMed] [Google Scholar]
- 47. Biederer T., Volkwein C., and Sommer T. (1996) Degradation of subunits of the Sec61p complex, an integral component of the ER membrane, by the ubiquitin-proteasome pathway. EMBO J. 15, 2069–2076 [PMC free article] [PubMed] [Google Scholar]
- 48. Ausubel F. M. (ed.) (1993–2006) Curr. Protoc. Mol. Biol., John Wiley & Sons, Inc., New York [Google Scholar]
- 49. Bagola K., von Delbrück M., Dittmar G., Scheffner M., Ziv I., Glickman M. H., Ciechanover A., and Sommer T. (2013) Ubiquitin binding by a CUE domain regulates ubiquitin chain formation by ERAD E3 ligases. Mol. Cell. 50, 528–539 [DOI] [PubMed] [Google Scholar]
- 50. Morelle W., and Michalski J.-C. (2007) Analysis of protein glycosylation by mass spectrometry. Nat. Protoc. 2, 1585–1602 [DOI] [PubMed] [Google Scholar]
- 51. Neville D. C., Coquard V., Priestman D. A., te Vruchte D. J., Sillence D. J., Dwek R. A., Platt F. M., and Butters T. D. (2004) Analysis of fluorescently labeled glycosphingolipid-derived oligosaccharides following ceramide glycanase digestion and anthranilic acid labeling. Anal. Biochem. 331, 275–282 [DOI] [PubMed] [Google Scholar]
- 52. Packer N. H., Lawson M. A., Jardine D. R., and Redmond J. W. (1998) A general approach to desalting oligosaccharides released from glycoproteins. Glycoconj. J. 15, 737–747 [DOI] [PubMed] [Google Scholar]
- 53. Harvey D. J., Royle L., Radcliffe C. M., Rudd P. M., and Dwek R. A. (2008) Structural and quantitative analysis of N-linked glycans by matrix-assisted laser desorption ionization and negative ion nanospray mass spectrometry. Anal. Biochem. 376, 44–60 [DOI] [PubMed] [Google Scholar]
- 54. Fu D., Chen L., and O'Neill R. A. (1994) A detailed structural characterization of ribonuclease B oligosaccharides by 1H NMR spectroscopy and mass spectrometry. Carbohydr. Res. 261, 173–186 [DOI] [PubMed] [Google Scholar]
- 55. Carvalho P., Goder V., and Rapoport T. A. (2006) Distinct ubiquitin-ligase complexes define convergent pathways for the degradation of ER proteins. Cell 126, 361–373 [DOI] [PubMed] [Google Scholar]
- 56. Ghaemmaghami S., Huh W.-K., Bower K., Howson R. W., Belle A., Dephoure N., O'Shea E. K., and Weissman J. S. (2003) Global analysis of protein expression in yeast. Nature 425, 737–741 [DOI] [PubMed] [Google Scholar]
- 57. Jaenicke L. A., Brendebach H., Selbach M., and Hirsch C. (2011) Yos9p assists in the degradation of certain nonglycosylated proteins from the endoplasmic reticulum. Mol. Biol. Cell 22, 2937–2945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Friedlander R., Jarosch E., Urban J., Volkwein C., and Sommer T. (2000) A regulatory link between ER-associated protein degradation and the unfolded-protein response. Nat. Cell Biol. 2, 379–384 [DOI] [PubMed] [Google Scholar]
- 59. Biederer T., Volkwein C., and Sommer T. (1997) Role of Cue1p in ubiquitination and degradation at the ER surface. Science 278, 1806–1809 [DOI] [PubMed] [Google Scholar]