

Abstract
Protein homeostasis is maintained by quality control mechanisms that detect and eliminate deficient translation products. Cytosolic defective proteins can arise from translation of aberrant mRNAs lacking a termination codon (NonStop) or containing a sequence that blocks translation elongation (No-Go), which results in translational arrest. Stalled ribosomes are dissociated, aberrant mRNAs are degraded by the cytoplasmic exosome, and the nascent peptides remaining in stalled 60S exit tunnels are detected by the ribosome-bound quality control complex (RQC) composed of Ltn1, Rqc1, Rqc2, and Cdc48. Whereas Ltn1 polyubiquitylates these nascent peptides, Rqc2 directs the addition of C-terminal alanine-threonine tails (CAT-tails), and a Cdc48 hexamer is recruited to extract the nascent peptides, which are addressed to the proteasome for degradation. Although the functions of most RQC components have been described, the role of Rqc1 in this quality control process remains undetermined. In this article we show that the absence of Rqc1 or Ltn1 results in the aggregation of aberrant proteins, a phenomenon that requires CAT-tail addition to the nascent peptides by Rqc2. Our results suggest that aberrant CAT-tailed protein aggregation results from a defect in Cdc48 recruitment to stalled 60S particles, a process that requires both Rqc1 and Ltn1. These protein aggregates contain Ltn1-dependent polyubiquitin chains and are degraded by the proteasome. Finally, aggregate characterization by proteomics revealed that they contain specific chaperones including Sis1, Sgt2, Ssa1/2, and Hsp82, suggesting that these protein aggregates may be addressed to aggresome-like structures when the RQC complex fails to deliver aberrant nascent peptides to the proteasome for degradation.
Keywords: protein aggregation, protein degradation, Saccharomyces cerevisiae, translation control, ubiquitin
Introduction
Protein homeostasis is maintained in eukaryotic cells by quality control pathways that ensure the recognition and the degradation of defective translation products. Whereas molecular chaperones perform unfolding and refolding of misfolded proteins, aberrant proteins are recognized and eliminated by the ubiquitin-proteasome system. These cellular responses prevent the toxicity generated by the accumulation of defective proteins that may be nonfunctional and are prone to aggregation. However, in the presence of a translational stress, the rapid increase of the amount of defective proteins often leads to the accumulation of protein aggregates (for review see Ref. 1). It has been shown that these protein aggregates can be transported via the microtubules network to a deposit called the aggresome (2), in which the coordinated action of chaperones and proteasome-recruiting factors enables aggregate processing, thus limiting their intracellular toxicity and maintaining the quality of the cellular protein pool (3). In the cytosol, accumulation of aberrant proteins may result from the translation of aberrant mRNAs that trigger ribosome stalling during translation elongation. These stalled ribosomes act as a signal for mRNA (4–6), nascent peptide (7, 8), and ribosome degradation and recycling (9, 10). Two types of aberrant transcripts causing translational arrest have been identified: mRNAs lacking a STOP codon (NonStop mRNAs) (4, 5), for which ribosome stalling is caused by the translation of the poly(A) tail into a positively charged polylysine (7), and mRNAs carrying a sequence or a structure that prevents elongating ribosomes from reaching the termination codon (no-go mRNAs) (6). In both cases, Dom34 (Pelota in mammals) and Hbs1 bind to the stalled ribosomes, which trigger the recruitment of the ATPase Rli1/ABCE1 (11). Together, they promote dissociation of the stalled ribosome. Following the dissociation, the aberrant transcripts undergo an endonucleolytic cleavage (6) and are degraded via the recruitment of the SKI complex, Ski7, the cytoplasmic exosome, and Xrn1 (5, 6, 12).
After ribosome dissociation, the exit tunnel of the 60S subunit still contains an aberrant nascent peptide bound to a tRNA that must be degraded to prevent the accumulation of defective translation products. Recent studies revealed that these aberrant substrates are detected by the ribosome-bound quality control complex (RQC),3 composed of the RING domain E3 ubiquitin ligase Ltn1, the factors Rqc1 and Rqc2, a hexamer of Cdc48 (Listerin, TCF25, NEMF, and p97 in mammals, respectively), and the Cdc48 co-factors Npl4 and Ufd1 (13–15) (see Fig. 1A). Rqc2 binds to the 60S interface surface and consequently prevents 40S reassociation (16). An additional function of Rqc2 is to sense the tRNA in the P-site of the stalled 60S and to coordinate the recruitment of alanyl- and threonyl-tRNAs in the A-site to add a nontemplated C-terminal alanine-threonine tail (CAT-tail) to the nascent peptide (17). This process is essential to trigger the Hsf1 response to translational stress (17). In parallel, Ltn1 performs the polyubiquitylation of the aberrant peptide (15–18). This enables the recruitment of a Cdc48 hexamer that hydrolyzes ATP to extract the aberrant substrate from the 60S exit tunnel and to escort it to the proteasome for degradation (14, 19). Whereas Rqc1 is not involved in the polyubiquitylation process, it is required for Cdc48 recruitment on the 60S subunit and therefore essential for efficient degradation of these aberrant proteins (13, 14).
FIGURE 1.
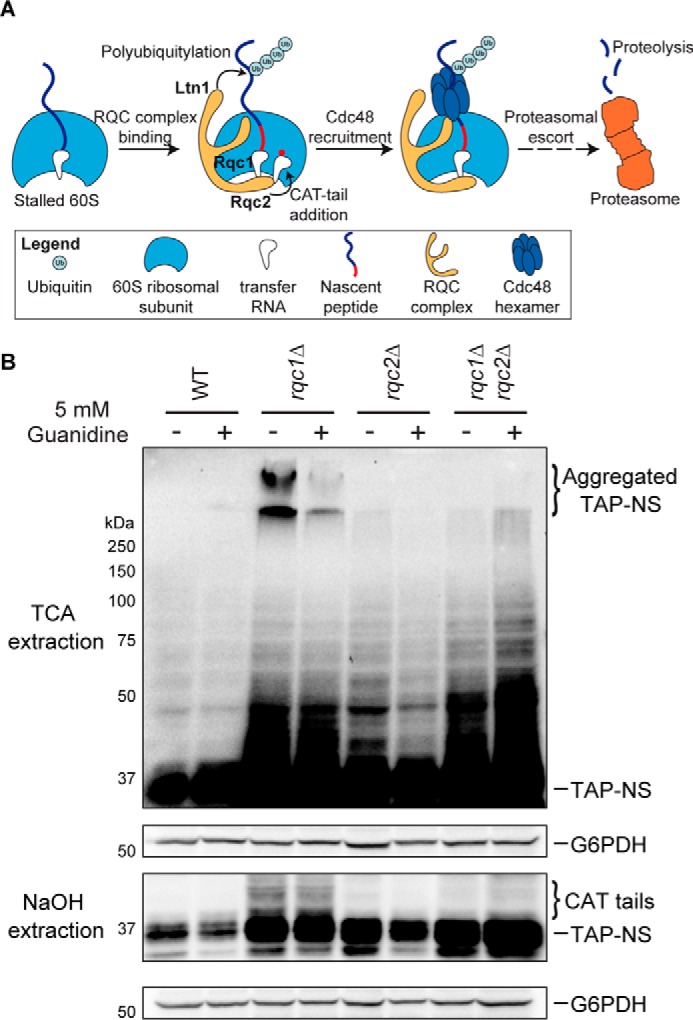
Rqc1 prevents the accumulation of Rqc2-dependent aberrant protein aggregates. A, schematic illustration of the ribosome-bound quality control pathway based on previous studies. Aberrant nascent peptides synthesized from mRNAs triggering translational arrest remain stalled in the 60S ribosomal subunit after ribosome dissociation. 60S particles stalled with a nascent peptide are recognized by the RQC complex composed of Rqc1, Rqc2, and Ltn1 that performs CAT-tail addition and polyubiquitylation of the nascent peptide. This polyubiquitylated substrate is then recognized by a Cdc48 hexamer that hydrolyzes ATP to extract it from the 60S exit tunnel and escorts it to the proteasome for degradation. B, total proteins extracted using TCA (top two panels) or NaOH (bottom two panels) prepared from wild-type, rqc1Δ, rqc2Δ, and rqc1Δrqc2Δ cells expressing the TAP-NonStop reporter and treated or not with 5 mm guanidine for 15 h were separated on 10% polyacrylamide gels. Analyses by Western blotting were performed using antibodies against the TAP-NonStop (peroxidase-anti-peroxidase (PAP) from Sigma-Aldrich) (1/10,000) and rabbit polyclonal against G6PDH (1/100,000) for loading control. Aggregated species of the TAP-NonStop in the stacking gel of TCA extracts and CAT-tailed forms of the TAP-NonStop are indicated. NS, NonStop.
Although the functions of Ltn1, Rqc2, and Cdc48 within this RQC-mediated translational quality control have been partly described (see review in Ref. 20), the function of Rqc1 remains widely unknown. Furthermore, rapid accumulation of defective proteins in case of stress often results in a saturation of quality control mechanisms (for review see in Ref. 21), and the fate of aberrant translation products in the absence of a fully functional RQC complex is yet to be determined.
In this study, we show that Rqc1, Ltn1, and Cdc48 prevent the aggregation of aberrant proteins that can only appear after CAT-tail addition by Rqc2. These protein aggregates contain polyubiquitin chains generated by Ltn1 and are degraded by the proteasome. Finally, these aggregates are recognized by specific chaperones including Sis1, Sgt2, Ssa1/2, and Hsp82, suggesting that they are targeted to aggresome-like structures. This work reveals the importance of Cdc48 recruitment on stalled 60S particles by Rqc1 and Ltn1 to prevent the accumulation of CAT-tail induced aggregates of aberrant translation products, thus participating in the maintenance of cellular protein homeostasis.
Experimental Procedures
Yeast Strains and Plasmids
The yeast strains used in this study are listed in Table 1. All of them were generated from either BY4741 or BY4742 by homologous recombination using PCR products. The plasmid pGFP-NonStop was generated by pTAP-NonStop (14) digestion with BamHI and BglII and yeast homologous recombination with a GFP sequence PCR fragment carrying overhangs that target 5′ and 3′ ends of the digested vector. Depletions of CDC48 were performed by generating a PrTetO2-CDC48 strain (14) where CDC48 is under the control of a tetracyclin-repressible promoter and by treating cell cultures with doxycyclin (5 μg·ml−1) for 13.5 h at 30 °C. To eliminate the aggregated forms of protein, the cells were treated for 15 h with 5 mm guanidine, and 5 A600 cells in the mid-log phase of treatment were harvested, and proteins were extracted as described below.
TABLE 1.
Yeast strains used in this study
| Strains | Genotypes | References |
|---|---|---|
| BY4741 | MATa, ura3Δ0, his3Δ1, leu2Δ0, met15Δ0 | |
| BY4742 | MATalpha, ura3Δ0, his3Δ1, leu2Δ0, lys2Δ0 | |
| LMA1967 | as BY4742, rqc1Δ::KANMX4 | Euroscarf |
| LMA843 | as BY4742, yel068cΔ::PralphaNATMX4, ydl242wΔ::KANMX4 | Ref. 14 |
| LMA2306 | as BY4742, yel068cΔ:: PralphaNATMX4, rqc1Δ::KANMX4 | Ref. 14 |
| LMA836 | as BY4742, rqc2Δ:: PralphaNATMX4,yel068cΔ::KANMX4 | Ref. 14 |
| LMA1921 | as BY4742, yel068cΔ:: PralphaNATMX4, ltn1Δ::KANMX4 | Ref. 14 |
| LMA1920 | as BY4742, rqc2Δ:: PralphaNATMX4, ltn1Δ::KANMX4 | Ref. 14 |
| LMA3768 | as BY4742, rqc1Δ:: PralphaNATMX4, ltn1Δ::KANMX4, | This study |
| LMA2135 | as BY4742, rqc1Δ::KANMX4, rqc2Δ:: PralphaNATMX4 | This study |
| LMA3124 | as BY4742, PralphaNATMX4:PrTetO2:CDC48, yel068cΔ::KANMX4 | This study |
| LMA3121 | as BY4742, PralphaNATMX4:PrTetO2:CDC48, rqc1Δ::KANMX4 | This study |
| LMA3041 | as BY4741, erg6Δ::LEU2 | This study |
| LMA3120 | as BY4741, erg6Δ::LEU2, rqc1Δ::KANMX4 | This study |
Polysome Gradients, Proteins Extraction, and Western Blotting
Polysome extracts were obtained from 120 A600 mid-log phase yeast cells were treated with 50 μg·ml−1 cycloheximide for 5 min and broken with glass beads using a MagNA lyser (three times for 60 s at 3000 rpm) in breaking buffer (20 mm Tris-HCl, pH 7.4, 50 mm KCl, 5 mm MgCl2, 50 μg·ml−1 cycloheximide, 10 mm N-ethylmaleimide, and EDTA-free protease inhibitors from Roche). For proteasome inhibition, the cells were treated for 2 h with 100 μm MG132 before lysis. They were deleted from ERG6 to increase the sensitivity to MG132. 7 A260 of clarified cell lysates were loaded on 10–50% sucrose gradients. Collected fractions were precipitated with 10% TCA and resuspended in 40 μl of sample buffer, and 10 μl were loaded on 10% SDS-PAGE. Total protein extracts were prepared from 5 A600 of exponential culture. Briefly, the cells were broken with glass beads using a MagNA Lyser (twice for 60 s at 4800 rpm) in 300 μl of TCA 20%. We let beads settle and transferred the supernatant into a new tube. 300 μl of 5% TCA were added to the beads, and after vortex for 10 s, we again let beads settle and transferred the supernatant into the collected tube. After centrifugation for 1 min at 16,000 × g, the samples were resuspended in 40 μl of sample buffer, and 10 μl were loaded on 10% SDS-PAGE. Alternatively, a fast method using alkaline treatment (22) was used as mentioned in the text. 5 A600 of exponential culture were resuspended in 200 μl of 0.1 n NaOH. After incubation for 5 min at room temperature, the cells were collected by centrifugation for 3 min at room temperature and resuspended in 40 μl of sample buffer. Proteins were denatured for 3 min at 95 °C, and cellular debris were pelleted by centrifugation. 10 μl of supernatant were loaded on 10% SDS-PAGE. After transfer to nitrocellulose membrane with a semidry system, proteins were detected by hybridization with the appropriate antibodies.
Mass Spectrometry Experiments and Data Analysis
For sample preparation from stacking gels, each lane was excised, cut in small cubes, and subjected to in-gel trypsin digestion after reduction and alkylation as described in Ref. 23. All steps from in-solution trypsin digestion, peptide sample analysis by LC-MS/MS on an Orbitrap Velos, protein identification, to comparative label-free quantification using MaxQuant suite were performed as previously described in Ref. 14.
Fluorescence Microscopy
Fluorescence microscopy was performed with GFP-NonStop-expressing yeast cells grown in liquid synthetic medium containing galactose without uracil to a final A600 of 4.0 and resuspended in sterile water. DNA was stained with Hoechst 33352 (5 ng·μl−1) for 5 min, and cells were washed twice in sterile water. Samples were imaged using a Leica DMRXA fluorescence microscope. Fluorescent signals were collected with single band pass filters for excitation of GFP (Leica) for 3500 ms, mCherry (Cy3; Leica) for 800 ms and Hoechst 33352 (A; Leica) for 40 ms. Images were acquired with a Hamamatsu ORCAII-ER cooled CCD camera controlled by the Openlab software (version 3.5; Improvision) and processed using Adobe Photoshop CS3 software (version 10; Adobe).
Results
Rqc1 Prevents the Accumulation of Rqc2-dependent Aberrant Protein Aggregates
Whereas the functions of the other components of the RQC complex have been described (13–15, 17), the role of Rqc1 in the translational quality control pathway remains poorly understood. To better understand the mechanism of action of Rqc1 in this quality control process, we analyzed by Western blotting the accumulation of an aberrant reporter containing a tandem affinity purification sequence (24) and lacking a translation termination codon (called “TAP-NonStop”) in the absence of RQC1 in the yeast Saccharomyces cerevisiae. In addition to the previously observed accumulation of the aberrant reporter protein in rqc1Δ cells compared with a wild type when protein extracts were prepared using NaOH (Fig. 1B, bottom two panels, and Ref. 14), extracting proteins using TCA and blotting the stacking part of the polyacrylamide gel revealed an additional form of the TAP-NonStop reporter stabilized in the absence of Rqc1 that was retained in the stacking gel (Fig. 1B, top two panels). Interestingly, these high molecular weight species of the TAP-NonStop were not visible in rqc2Δ cells, and the absence of Rqc2 suppressed their accumulation in the rqc1Δrqc2Δ double mutant, although the accumulation of the running gel-migrating TAP-NonStop was comparable between this double mutant and the rqc1Δ single mutant (Fig. 1B). Whereas these high molecular weight species were resistant to the SDS present in the protein sample buffer, treating the yeast cultures with 5 mm guanidine for 15 h significantly reduced high molecular weight species accumulation in the stacking gel of rqc1Δ cells (Fig. 1B), suggesting that they could correspond to aggregated forms of the TAP-NonStop aberrant protein that accumulate in the absence of Rqc1.
We addressed this hypothesis by replacing the TAP sequence in the TAP-NonStop reporter gene by a GFP sequence to monitor the aberrant “GFP-NonStop” reporter by fluorescence microscopy. Interestingly, whereas the fluorescent signal was weak and homogeneously distributed in the cytosol of wild-type cells, the GFP-NonStop aberrant protein accumulated to a higher extent in rqc1Δ cells and formed green foci corresponding to protein aggregates that were localized in the cytosol, according to a merge with nucleus staining (Fig. 2). In contrast, whereas the GFP-NonStop fluorescent signal in the cytosol of rqc2Δ cells was comparable with the intensity observed in rqc1Δ cells, no green foci were detected in the absence of Rqc2, whether in the presence or Rqc1 or not (Fig. 2). These observations corroborate those obtained in Fig. 1 and suggest that high molecular weight species observed with the TAP-NonStop correspond to the cytosolic aggregates observed with the GFP-NonStop reporter. Taken together, these results show that Rqc1 is essential to prevent the formation of cytosolic aggregates of aberrant peptides synthesized from NonStop mRNAs; however, aggregate formation strictly depends on the presence of Rqc2.
FIGURE 2.
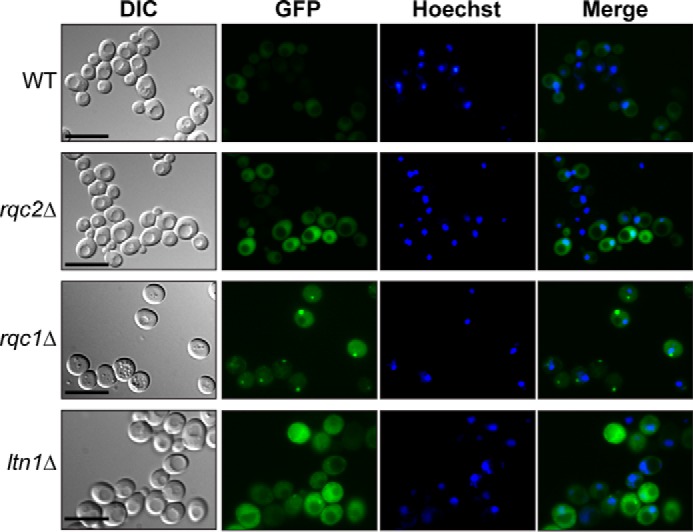
Aberrant nascent peptides form cytosolic aggregates in the absence of Rqc1. Wild-type, rqc2Δ, rqc1Δ, and ltn1Δ strains expressing the GFP-NonStop reporter gene were analyzed by fluorescence microscopy. Yeast cells are shown in the left column (Nomarski); GFP and Hoechst fluorescent signals are displayed in the middle columns; and a merge is displayed in the right column. Scale bars, 5 μm. DIC, differential interference contrast.
Addition of CAT-tails to Aberrant Peptides by Rqc2 Is Essential for Their Aggregation in the Absence of Rqc1
We then investigated why Rqc2 was essential for aggregate formation even in the absence of Rqc1. Because Rqc2 was shown to initiate CAT-tail addition to aberrant nascent peptides (17), we wondered whether the presence of CAT-tails on aberrant proteins could be responsible for their aggregation in the absence of Rqc1. To this end, we complemented the wild-type, rqc1Δ, rqc2Δ, and rqc1Δrqc2Δ strains with vectors coding the RQC2 gene or the RQC2aaa mutant, which is functional for aberrant protein degradation but not for CAT-tail addition, thus uncoupling these two distinct mechanisms (17). As expected, the CAT-tails addition to the soluble TAP-NonStop peptide was observed only when the entire RQC2 gene version is present but not with the RQC2aaa mutant (Fig. 3, bottom two panels). Interestingly, the accumulation of aggregated TAP-NonStop in the rqc1Δrqc2Δ double mutant could only be restored by complementation with the RQC2 gene but not with the RQC2aaa mutant (Fig. 3, top two panels, compare lanes 11 and 12), revealing that CAT-tail addition by Rqc2 is actually responsible for protein aggregation in the absence of Rqc1. Intriguingly, the quantity of protein aggregates was increased when two copies of RQC2 were expressed in the rqc1Δ mutant (Fig. 3, top two panels, compare lanes 7 and 8), which suggests that the overexpression of an Rqc2 version that is functional for CAT-tail addition has an aggravating effect on aberrant protein aggregation when Rqc1 is impaired.
FIGURE 3.

Addition of CAT-tails to aberrant peptides by Rqc2 is essential for their aggregation in the absence of Rqc1. Wild-type, rqc2Δ, rqc1Δ, and rqc2Δrqc1Δ strains expressing the TAP-NonStop reporter gene were transformed either with pRS315 empty vector or with pRS315-RQC2 or pRS315-RQC2aaa vectors (17). Total cellular proteins from each strain were extracted either by TCA (top two panels) or NaOH (bottom two panels) methods, separated on a 10% acrylamide gel, and analyzed by Western blotting using antibodies against the TAP-NonStop (PAP) and G6PDH as a loading control. Aggregated versions of the TAP-NonStop in the stacking gel of TCA extracts and CAT-tailed forms of the TAP-NonStop are indicated with curly brackets. NS, NonStop.
Recruitment of Cdc48 on Stalled 60S Particles by Rqc1 and Ltn1 Is Essential to Prevent Aberrant Protein Aggregation
We showed that the absence of Rqc1 leads to aberrant protein aggregation, a process that requires prior CAT-tail addition to aberrant proteins by Rqc2. Because the only documented role of Rqc1 is to optimize the recruitment of the Cdc48 hexamer on 60S particles stalled with an aberrant nascent peptide in their exit tunnel (13, 14), we suspected that the absence of efficient Cdc48 recruitment on stalled 60S particles in rqc1Δ cells, which leads to a defect in aberrant peptide extraction from the 60S and in escort to the proteasome for degradation (14, 19), could be the cause of aggregate formation in the absence of Rqc1. We therefore analyzed the accumulation of the TAP-NonStop reporter after CDC48 depletion by Western blotting after polysome fractionation using sucrose gradients. Interestingly, aberrant protein aggregates were also detected in the stacking gel upon CDC48 depletion as observed in the rqc1Δ mutant (Fig. 4). In addition, polysome fractionation revealed that these aggregates mainly co-sedimented with light nonribosomal fractions of the sucrose gradient, although a subset also co-sedimented with 60S particles (Fig. 4). We then addressed the effect of combining a Cdc48 depletion with an RQC1 deletion and observed a strong aggravating effect on aberrant protein aggregation (Fig. 4), indicating that the absence of both Rqc1 and Cdc48 leads to the aggregation of aberrant proteins in an additive or even synergistic manner. Remarkably, the proportion of aggregated proteins co-sedimenting in the 60S fractions was higher in the absence of both Rqc1 and Cdc48 compared with the rqc1Δ single mutant, which correlates with the function of the Cdc48 hexamer in nascent peptide extraction from the stalled 60S (13, 14) and suggests that aberrant proteins that are not efficiently extracted in the absence of Cdc48 can also form aggregates even when they are still bound to 60S particles (Fig. 4).
FIGURE 4.
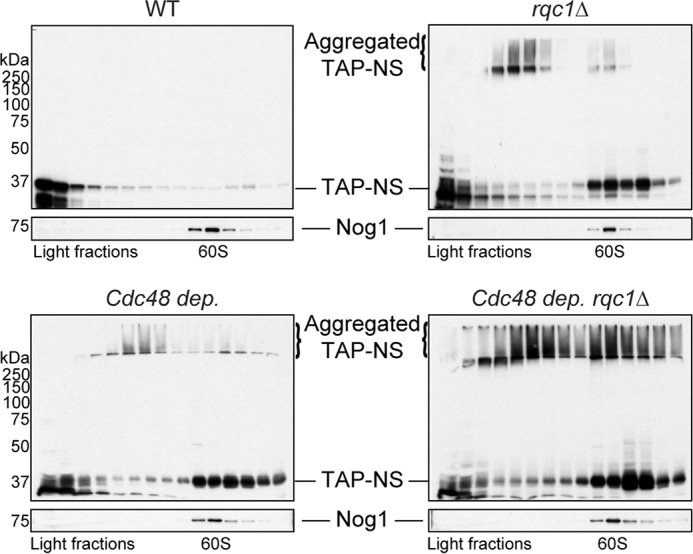
Recruitment of Cdc48 on stalled 60S particles by Rqc1 and Ltn1 is essential to prevent aberrant protein aggregation. Wild-type and rqc1Δ strains expressing the TAP-NonStop reporter were grown with or without Cdc48 depletion (13.5 h with doxycycline). Polysome extracts were separated on a sucrose gradient (10–50%), and TCA-precipitated fractions were migrated on polyacrylamide gels and analyzed by Western blotting using antibodies against the TAP-NonStop (PAP) and rabbit polyclonal against Nog1 (1/10,000) as a loading control and a marker of the 60S-sedimenting fractions. Aggregated species of the TAP-NonStop accumulating in the stacking gel are annotated. NS, NonStop.
In addition to the presence of Rqc1, Cdc48 recruitment to the stalled 60S also requires prior polyubiquitylation of the nascent peptide by the E3 ubiquitin ligase Ltn1 (13, 14). Therefore, one may expect that the absence of Ltn1 would also lead to an accumulation of aberrant protein aggregates. We thus analyzed the migration profile of the TAP-NonStop reporter in ltn1Δ mutant compared with rqc1Δ strain by Western blotting after polysome fractionation using sucrose gradients. As expected, aggregates of the TAP-NonStop reporter protein were also visible in the stacking gel and mainly co-sedimented with light fractions of the sucrose gradient from ltn1Δ cells, although the quantity of aggregates in this mutant was lower than in the rqc1Δ mutant (Fig. 5). We verified the presence of aggregates in the ltn1Δ mutant by fluorescence microscopy with the GFP-NonStop reporter and thus confirmed the presence of green cytosolic foci, although the fluorescence intensity of these aggregates was weaker than the one observed in the rqc1Δ (Fig. 2). We also analyzed the migration profile of the TAP-NonStop reporter in the ltn1Δrqc1Δ mutant. Surprisingly, the accumulation level of aggregated TAP-NonStop in the ltn1Δrqc1Δ mutant was comparable with the ltn1Δ and thus not as high as in the rqc1Δ (Fig. 5), indicating that the deletion of LTN1 is epistatic over the absence of RQC1 with regard to the level of accumulation of protein aggregates. One possibility to explain this epistatic phenotype is that aberrant peptides aggregating in the absence of RQC1 are polyubiquitylated in an Ltn1-dependent manner and that this polyubiquitylated state promotes protein aggregation, as previously reported in the case of α-synuclein (25).
FIGURE 5.
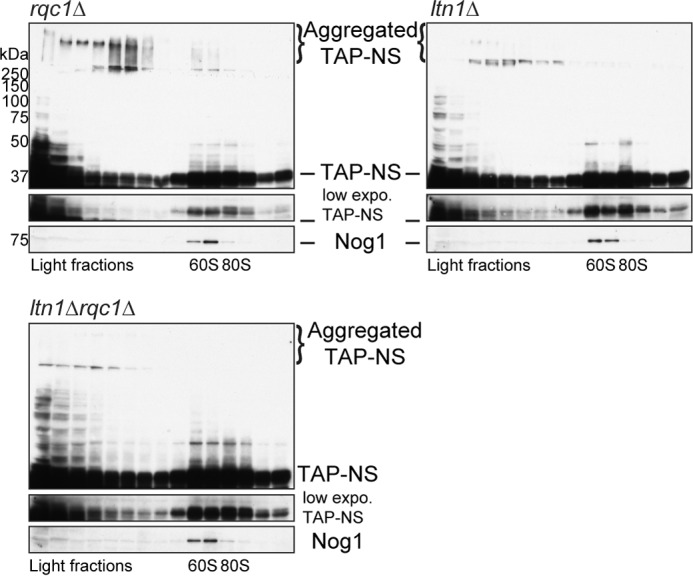
Ltn1 is also essential to prevent the accumulation of aberrant protein aggregates. Polysome extracts from rqc1Δ, ltn1Δ, and rqc1Δltn1Δ cells expressing the TAP-NonStop reporter were prepared and separated on a sucrose gradient (10–50%), and TCA-precipitated fractions were migrated on polyacrylamide gels and analyzed by Western blotting using antibodies against the TAP-NonStop (PAP) and Nog1 as a loading control and a marker of the 60S-sedimenting fractions. Aggregated species of the TAP-NonStop accumulating in the stacking gel are annotated. NS, NonStop.
We therefore analyzed the polyubiquitination level of protein aggregates by Western blotting using anti-ubiquitin antibodies on sucrose-fractionated protein extracts prepared from wild-type, rqc1Δ, ltn1Δ, and ltn1Δrqc1Δ reporter-free strains. Whereas the ubiquitinated proteins profiles migrating in the running gel were comparable among these four strains, a fraction of ubiquitinated proteins migrating in the stacking gel was observed specifically in the rqc1Δ mutant, demonstrating that the aggregates accumulating in the absence of RQC1 are constituted (at least in part) of polyubiquitinated proteins (Fig. 6). Notably, these species migrating in the stacking gel displayed a sedimentation pattern similar to the aggregated TAP-NonStop in the same rqc1Δ mutant (Fig. 4). Because the strains used for Fig. 4 do not express any aberrant reporter, this result shows that protein aggregates can spontaneously appear when Rqc1 is impaired and are not an artifact resulting from the overexpression of an aberrant NonStop reporter protein. In contrast, no polyubiquitin-containing aggregates were detected in the wild-type strain, the ltn1Δ mutant, or the ltn1Δrqc1Δ double mutant, which shows that the LTN1 deletion is also epistatic over the RQC1 deletion concerning the accumulation of polyubiquitinated protein aggregates (Fig. 6). Furthermore, these results show that aggregates accumulating in the absence of RQC1 contain aberrant proteins that have been polyubiquitylated in an Ltn1-dependent manner. Taken together, these results suggest that ubiquitylation of aberrant proteins by Ltn1 may contribute to their aggregation in the absence of Rqc1, which may explain the observed epistatic phenotypes of the LTN1 deletion over the RQC1 deletion (Figs. 5 and 6).
FIGURE 6.
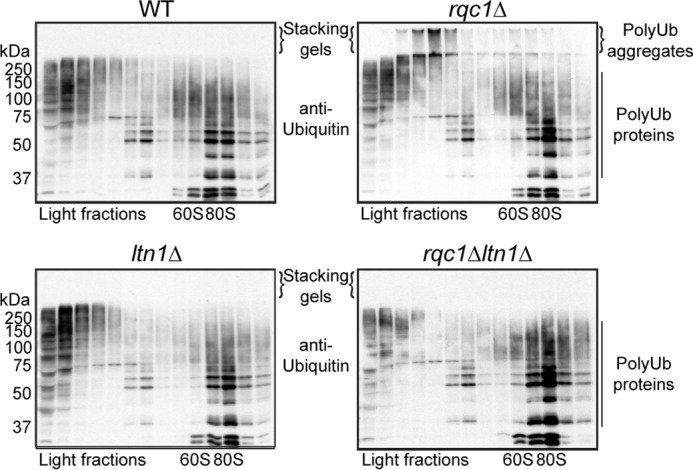
Aggregated aberrant proteins accumulating in the absence of Rqc1 are polyubiquitinated in an Ltn1-dependent manner. Polysome extracts prepared from wild-type, rqc1Δ, ltn1Δ, and ltn1Δrqc1Δ cells were separated on a sucrose gradient (10–50%). Fractions of the gradient were TCA-precipitated, separated on a 10% acrylamide gel, and analyzed by Western blotting using monoclonal antibodies against ubiquitin (P4D1 from Covance) (1/1,000) to visualize the ubiquitinated forms of soluble (running gel) and aggregated (stacking gel) endogenous proteins. Aggregates containing polyubiquitinated proteins and accumulating in stacking gels are indicated. Ub, ubiquitin.
Aberrant Protein Aggregates Are Eliminated by the Proteasome
Because aberrant peptides synthesized from NonStop mRNAs are known to be degraded by the proteasome (15), we wondered whether the aggregated forms of aberrant proteins appearing in rqc1Δ cells also constituted proteasomal substrates. To test this hypothesis, we performed a proteasome inhibition with the inhibitor MG-132 in wild-type and rqc1Δ cells and analyzed the stabilization of the TAP-NonStop reporter protein by Western blotting on polysome fractions. A proteasome inhibition in wild-type cells led to the stabilization of the TAP-NonStop in 80S and light fractions of the sucrose gradient (Fig. 7, upper right panel); however, no aggregated forms were detected in the stacking gel, indicating that the aggregation phenomenon is not due to a general defect in proteolysis but a particular phenotype caused by a dysfunction of specific RQC components such as Rqc1, Ltn1, or Cdc48. In sharp contrast, an MG-132 treatment of rqc1Δ cells increased the amount of aberrant protein aggregates visible in the stacking gel in comparison with the nontreated control (Fig. 7, bottom row), which demonstrates that aberrant protein aggregates that appear in the absence of Rqc1 are, at least in part, degraded by the proteasome.
FIGURE 7.
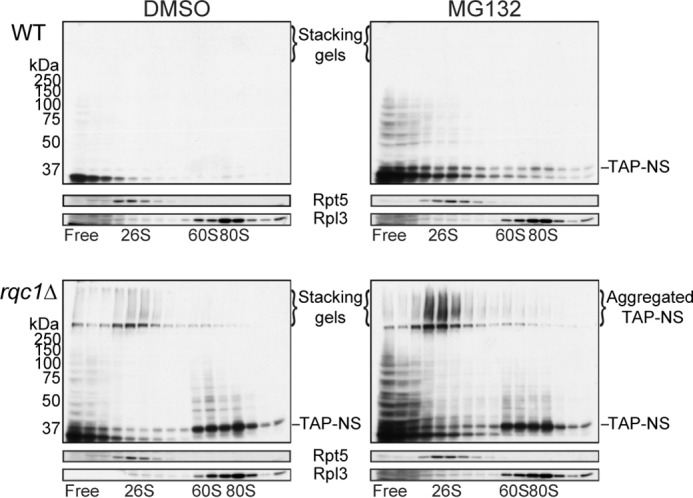
Aberrant protein aggregates are eliminated by the proteasome. Polysome extracts prepared from wild-type and rqc1Δ cells expressing the TAP-NonStop reporter and previously treated with the proteasome inhibitor MG132 or with DMSO as a negative control were separated on a sucrose gradient (10–50%). Both strains are deleted for ERG6. Fractions of the gradient were TCA-precipitated, separated on a 10% acrylamide gel, and analyzed by Western blotting using antibodies against the TAP-NonStop (PAP), mouse polyclonal against Rpl3 (1/5,000), and rabbit polyclonal against Rpt5 from Biomol (1/5,000) as loading controls and indicators of the ribosomal and the proteasomal fractions, respectively. Aggregated forms of the TAP-NonStop accumulating in the stacking gel are annotated. NS, NonStop.
Protein Aggregates Accumulating in rqc1Δ Cells Contain Aggresome-specific Chaperones
Several deposits of cytosolic protein aggregates involving distinct molecular chaperones have been described in yeast (26–28). To investigate toward which type of protein deposit the proteins aggregating in the absence of Rqc1 were addressed, we sought to determine what proteins could be identified in these aberrant protein aggregates. We thus isolated proteins from wild-type and rqc1Δ cells, migrated these samples on a polyacrylamide gel, extracted the proteins that were retained in the stacking gel (Fig. 8A), and analyzed these samples by label-free quantitative mass spectrometry. Protein identifications by LC-MS/MS and their relative label-free quantification revealed significant enrichment of ubiquitin, the co-chaperones Sis1 and Sgt2, and their associated chaperones Ssa1 and Ssa2 (29), as well as Hsp82 in the stacking gel of the rqc1Δ sample compared with the wild type (Fig. 8B and supplemental Data Set S1). Remarkably, all these factors were found in a previous study that characterized the proteins located in the aggresome after expression of Huntingtin exon 1 with an expanded polyglutamine domain in yeast (27), suggesting that aggregates of aberrant proteins that accumulate in the absence of Rqc1 are targeted to cytosolic aggresome-like compartments.
FIGURE 8.
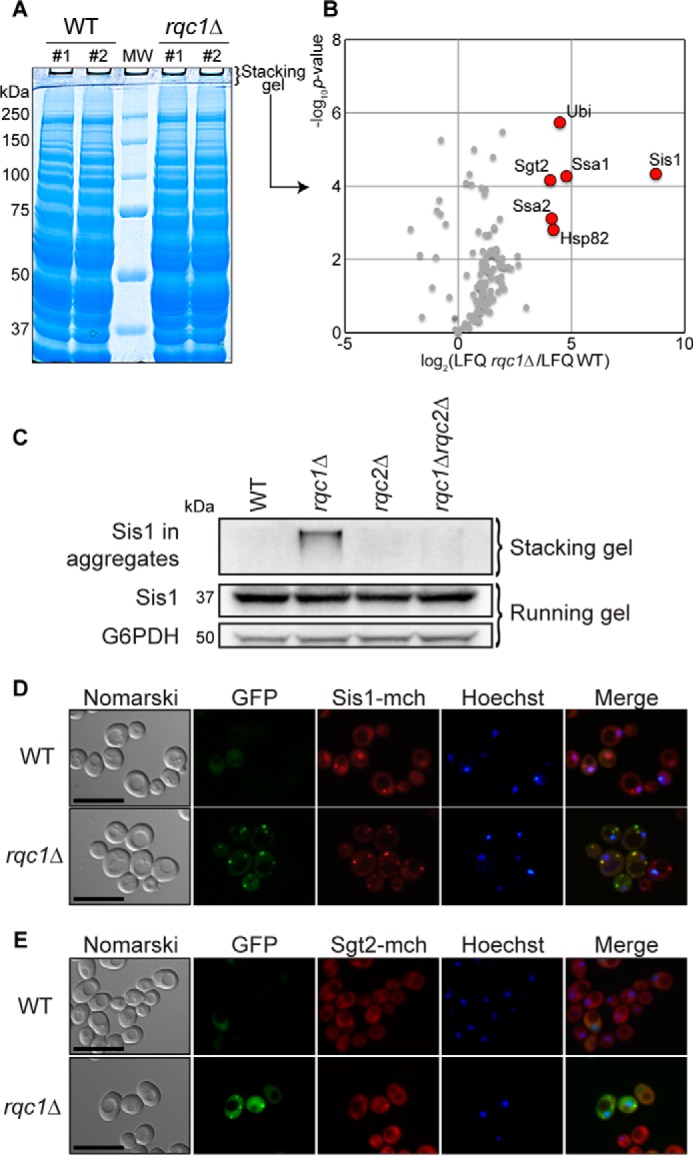
Protein aggregates accumulating in rqc1Δ cells contain aggresome-specific chaperones. A, protein extracts from wild-type and rqc1Δ cells were migrated on a polyacrylamide gel prepared with a 3-mm-thick stacking gel and stained with Coomassie Blue. B, proteins identified by label-free quantitative mass spectrometry in the stacking gel of rqc1Δ cell extracts were compared with the WT strain. The volcano plot shows the fold change (log2 LFQ rqc1Δ/LFQ WT) on the x axis and the p value distribution (−log10 p value) on the y axis for the quantified proteins, calculated using the Student's t test. Each circle indicates an identified protein. The red circles represent the significantly enriched candidates (p value ≥ 0.05). C, total protein extracts from wild-type, rqc1Δ, rqc2Δ, and rqc1Δrqc2Δ cells were separated on 10% polyacrylamide gels. Analyses by Western blotting were performed using rabbit polyclonal antibodies against Sis1 (1/1,000) and G6PDH for loading control. Fractions of Sis1 migrating in the stacking gel or the running gel are specified with curly brackets. D, WT and rqc1Δ Hoechst-stained strains containing a genomic Sis1-mCherry fluorescent fusion and expressing the GFP-NonStop reporter plasmid were analyzed by fluorescence microscopy. Yeast cells are displayed in the left column (Nomarski); GFP, mCherry, and Hoechst fluorescent signals are shown in the middle columns; and a merge is displayed in the right column. Scale bars, 5 μm. E, same as D using a genomic Sgt2-mCherry fluorescent fusion.
To confirm the presence of these aggresome-related chaperones identified by mass spectrometry in aberrant protein aggregates, we performed Western blotting using anti-Sis1 antibodies and analyzed the accumulation of Sis1 in both stacking and running gels from wild-type, rqc1Δ, rqc2Δ, and rqc1Δrqc2Δ strains. Whereas the abundance of Sis1 migrating in the running gel was comparable in all these strains, in the absence of Rqc1 (Fig. 8C), we detected a fraction of Sis1 retained in the stacking gel, which resembles the migration profile observed for the TAP-NonStop aberrant reporter in Fig. 1. These stacking gel-retained forms of Sis1 were not visible for the wild-type, rqc2Δ, and rqc1Δrqc2Δ strains (Fig. 8C), revealing that the presence of Sis1 in stacking gels depends on the activity of Rqc2, as previously observed for TAP-NonStop protein aggregates (Figs. 1B and 3).
Finally, we analyzed the co-localization of Sis1 and Sgt2 with aberrant protein aggregates by fluorescence microscopy by expressing the GFP-NonStop reporter together with fluorescent fusions of these co-chaperones (Sis1-mCherry and Sgt2-mCherry, respectively). In wild-type cells, Sis1-mCherry was more abundant in the nucleus that in the cytosol, whereas Sgt2-mCherry was mainly observed in the cytosol but excluded from the nucleus and the vacuole, according to a merge with nucleus staining (Fig. 8, D and E). As expected, the GFP-NonStop fluorescent signal was weak, and no green foci could be observed in wild-type conditions. In sharp contrast, deletion of RQC1 led to an increase of the GFP-NonStop fluorescent signal and to the presence of green cytosolic foci (as observed in Fig. 2), as well as the appearance of red foci for both Sis1-mCherry and Sgt2-mCherry that co-localized with the GFP-NonStop cytosolic aggregates (Fig. 8, D and E) (note that red foci were also visible for Sis1-mCherry in the wild-type strain, but to a lesser extent than in the rqc1Δ mutant). These results show that Sis1-mCherry and Sgt2-mCherry are specifically recruited to aberrant protein cytosolic aggregates when Rqc1 is impaired. Furthermore, the absence of Rqc1 and the presence of GFP-NonStop aggregates seem to alter the cellular localization of Sis1-mCherry that was not as abundant in the nucleus as in the wild type and relocated in cytosolic protein aggregates (Fig. 8D). Taken together, these results confirm that the co-chaperones Sis1 and Sgt2 are indeed recruited to aberrant protein aggregates that accumulate in the absence of Rqc1 and suggest that aberrant peptides may be targeted to aggresome-like structures when their efficient proteasomal targeting by the RQC complex is impaired.
Discussion
Previous studies identified the Ribosome-bound Quality Control complex as a multiprotein complex that binds stalled 60S ribosomal particles to ubiquitylate aberrant nascent peptides that are subsequently addressed to the proteasome for degradation (13, 14). More recently, a structural study of the RQC complex revealed the Rqc2 function in the addition of CAT-tails to aberrant nascent peptides before their extraction from stalled 60S particles, a process essential to trigger the Hsf1 response to translational stress (17). In this article we showed that Rqc1, Ltn1, and Cdc48 are essential to prevent the aggregation of aberrant nascent peptides (Figs. 1B, 4, and 5). Furthermore, the increased accumulation of aggregated proteins in ribosomal fractions of a polysome gradient observed in the absence of both Rqc1 and Cdc48 suggests that a deficiency in Cdc48 recruitment to stalled 60S particles—and therefore in efficient proteasomal escort of aberrant substrates—is the cause for aberrant protein aggregation (Fig. 4). Interestingly, a deletion of RQC2 could suppress this aggregation phenomenon (Fig. 1B), and complementation with an Rqc2 mutant that is functional for aberrant peptide degradation but nonfunctional for CAT-tail addition revealed that the presence of CAT-tails on aberrant proteins is a sine qua non condition for their aggregation (Fig. 3). Finally, our biochemical analysis of these aggregates revealed that they contain Ltn1-dependent polyubiquitin chains (Fig. 6), are eliminated by the proteasome (Fig. 7), and are recognized by specific co-chaperones (Sis1 and Sgt2) and their associated chaperones (Fig. 8), suggesting that aberrant protein aggregates may be addressed toward cytosolic aggresome-like structures (27) when RQC complex substrates are not efficiently targeted to the proteasome by Cdc48 hexamers (see model in Fig. 9).
FIGURE 9.
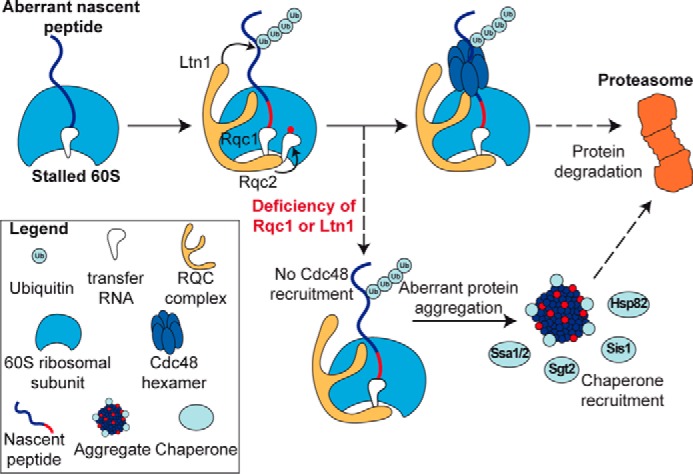
Model for the mechanism of aberrant protein aggregation, recognition, and proteasomal degradation in the absence of Rqc1 or Ltn1. Aberrant nascent peptides are recognized at the 60S level by the RQC complex, which enables its polyubiquitylation by Ltn1 and CAT-tail addition by Rqc2. Rqc1 is then essential for the recruitment of a Cdc48 hexamer to extract the peptide from the 60S exit tunnel and escort it to the proteasome for degradation. The absence of Rqc1 or Ltn1 causes a defect in Cdc48 recruitment, which results in CAT-tail-dependent protein aggregation. These cytosolic aggregates are recognized by specific co-chaperones (Sis1 and Sgt2) and chaperones (Ssa1, Ssa2, and Hsp82). Eventually, aggregated species of aberrant proteins are also eliminated by the proteasome.
Whereas the only previously described role of Rqc1 is to recruit Cdc48 hexamers on stalled 60S particles (14), our study reveals another important function of Rqc1, in preventing the accumulation of aberrant protein aggregates (Figs. 1B and 2). However, because a depletion of Cdc48 also leads to protein aggregate accumulation, it is possible that the aggregation phenotype observed in the rqc1Δ strain is mainly due to a defect in Cdc48 recruitment to stalled 60S subunits in this mutant. In this model, Rqc1 could act as a structural adaptor between the Cdc48 hexamer and the stalled 60S, but one cannot exclude the possibility that the role of Rqc1 in Cdc48 recruitment to stalled 60S particles is indirect, for example, by modifying the structural conformation of the stalled 60S to facilitate Cdc48 binding, or via the recruitment of other as yet unidentified factors. Another hypothesis is that, independently from its function in Cdc48 recruitment, Rqc1 has a direct function in protein aggregate prevention, such as a “chaperone-like” activity or by recruiting chaperones on the stalled 60S, which could explain why the combination of a Cdc48 depletion with an RQC1 deletion drastically increases the amount of aberrant protein aggregates compared with the amounts of aggregates observed in the respective single mutants (Fig. 4). Thus, further research may shed light on the precise role of Rqc1 and Cdc48 in the prevention of aberrant protein aggregation.
Concerning the AAA-ATPase Cdc48, whereas our results clearly demonstrate its importance to prevent the accumulation of aberrant protein aggregates (Fig. 4), one may wonder whether the aggregation phenotype observed upon Cdc48 depletion is due to the chaperone function of this homohexamer or due to a slowdown in the proteasomal escort of aggregation-prone aberrant nascent peptides. Indeed, because Cdc48 has been described as a “ubiquitin-selective chaperone” (30), it is possible that once recruited to stalled 60S particles, the Cdc48 hexamer protects the nascent peptide from aggregating until this substrate is properly delivered to the proteasome to be degraded. This possibility of a ubiquitin-selective chaperone role for Cdc48 in association with the RQC complex would be coherent with our data showing that ubiquitylation of aberrant nascent peptides by Ltn1 promotes their aggregation in the absence of Rqc1 (Figs. 5 and 6). However, because Cdc48 is essential to escort aberrant nascent peptides to the proteasome for degradation (19, 31), the fact that aberrant misfolded proteins accumulate in a polyubiquitylated state upon Cdc48 depletion (14), without being efficiently escorted and degraded by the proteasome, could also explain by itself this aggregation phenotype, regardless of a specific chaperone function of Cdc48 hexamers.
There is a remarkable correlation between the accumulation of aberrant protein aggregates and the triggering of Hsf1 response: indeed, both phenomena arise in the absence of Rqc1 or Ltn1 (Ref. 13 and this study) and require CAT-tail addition by Rqc2 (17). In parallel, proteomic analysis of aggregates enabled us to identify several factors, including Hsp82, whose expression was previously shown to increase upon Hsf1 response (32). Altogether, these results suggest that there could be a functional link between the accumulation of CAT-tails on aberrant peptides, the aggregation of these peptides, and the activation of the Hsf1 response. One early hypothesis is that after CAT-tail addition by Rqc2, the formation of aberrant protein aggregates triggers the Hsf1 response to translational stress to express specific chaperones that may participate in limiting the expansion of potentially toxic aggregates. Conversely, Hsf1 activation by the accumulation of CAT-tails could be an essential step to initiate aggregate formation via the expression of Hsf1-induced chaperones (such as Hsp82) and the relocalization of the co-chaperones Sis1 and Sgt2 to cytosolic foci, as observed in Fig. 8. In this second scenario, the formation of aggresome-like structures in the cytosol could therefore be a cellular stress response that enables the storage of defective proteins that cannot be efficiently degraded by the proteasome, therefore participating in the maintenance of protein homeostasis (3, 21). Finally, although our study underlines the importance of CAT-tail addition to aberrant peptides by Rqc2 for their subsequent aggregation in the absence of Rqc1 (Fig. 3), the biochemical process by which C-terminal alanine-threonine nontemplated sequences cause protein aggregation remains unknown. It is possible that the nonfolded nature of these amino acid stretches triggers the aggregation of the targeted protein, as previously described in the case of polyalanine repeat expansions within specific transcription factors (33). Further characterization of the biochemical properties of CAT-tail-containing proteins could determine how these extensions cause aberrant proteins to aggregate when they are not efficiently degraded.
In conclusion, studies focused on the RQC complex revealed that its function is not limited to polyubiquitylation and proteasomal targeting of aberrant translation products. Indeed, the RQC complex also features important roles in Hsf1 response activation (13), CAT-tail addition to nascent peptides (17), and prevention of cytosolic aggregates (this study) and is therefore a pivotal element of the eukaryotic protein homeostasis maintenance machinery.
Author Contributions
Q. D., E. Z., J. M., and M. F.-R. performed and analyzed the experiments. A. N. generated and analyzed the mass spectrometry data. Q. D., M. F.-R., and A. J. designed the study. Q. D. and M. F.-R. wrote the paper. All authors reviewed the results and approved the final version of the manuscript.
Supplementary Material
Acknowledgments
We thank Onn Brandman for providing RQC2- and RQC2aaa-containing plasmids and Véronique Albanèse and Jonathan Warner for providing the anti-Sis1 and anti-Rpl3 antibodies, respectively. We thank Antonia Doyen for yeast strain construction, Frank Feuerbach for helpful advice in fluorescence microscopy and all the members of the lab for discussions and criticism on the manuscript. We are grateful to the proteomics platform of the Pasteur Institute for the availability of the Orbitrap Velos.
This work was supported by Agence Nationale de la Recherche Grants ANR-2011-BSV6-011-02 and ANR-14-CE-10-0014-01 and funds from the Institut Pasteur and the CNRS. The authors declare that they have no conflicts of interest with the contents of this article.

This article contains supplemental Data Set S1.
- RQC
- ribosome-bound quality control complex
- CAT-tail
- C-terminal alanine-threonine tail
- PAP
- peroxidase-anti-peroxidase.
References
- 1. Richter K., Haslbeck M., and Buchner J. (2010) The heat shock response: life on the verge of death. Mol. Cell 40, 253–266 [DOI] [PubMed] [Google Scholar]
- 2. Johnston J. A., Ward C. L., and Kopito R. R. (1998) Aggresomes: a cellular response to misfolded proteins. J. Cell Biol. 143, 1883–1898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kopito R. R. (2000) Aggresomes, inclusion bodies and protein aggregation. Trends Cell Biol. 10, 524–530 [DOI] [PubMed] [Google Scholar]
- 4. Frischmeyer P. A., van Hoof A., O'Donnell K., Guerrerio A. L., Parker R., and Dietz H. C. (2002) An mRNA surveillance mechanism that eliminates transcripts lacking termination codons. Science 295, 2258–2261 [DOI] [PubMed] [Google Scholar]
- 5. van Hoof A., Frischmeyer P. A., Dietz H. C., and Parker R. (2002) Exosome-mediated recognition and degradation of mRNAs lacking a termination codon. Science 295, 2262–2264 [DOI] [PubMed] [Google Scholar]
- 6. Doma M. K., and Parker R. (2006) Endonucleolytic cleavage of eukaryotic mRNAs with stalls in translation elongation. Nature 440, 561–564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Dimitrova L. N., Kuroha K., Tatematsu T., and Inada T. (2009) Nascent peptide-dependent translation arrest leads to Not4p-mediated protein degradation by the proteasome. J. Biol. Chem. 284, 10343–10352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ito-Harashima S., Kuroha K., Tatematsu T., and Inada T. (2007) Translation of the poly(A) tail plays crucial roles in NonStop mRNA surveillance via translation repression and protein destabilization by proteasome in yeast. Genes Dev. 21, 519–524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. LaRiviere F. J., Cole S. E., Ferullo D. J., and Moore M. J. (2006) A late-acting quality control process for mature eukaryotic rRNAs. Mol. Cell 24, 619–626 [DOI] [PubMed] [Google Scholar]
- 10. Fujii K., Kitabatake M., Sakata T., and Ohno M. (2012) 40S subunit dissociation and proteasome-dependent RNA degradation in nonfunctional 25S rRNA decay. EMBO J. 31, 2579–2589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Shoemaker C. J., and Green R. (2011) Kinetic analysis reveals the ordered coupling of translation termination and ribosome recycling in yeast. Proc. Natl. Acad. Sci. 108, E1392–E1398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Tsuboi T., Kuroha K., Kudo K., Makino S., Inoue E., Kashima I., and Inada T. (2012) Dom34:Hbs1 plays a general role in quality-control systems by dissociation of a stalled ribosome at the 3′ end of aberrant mRNA. Mol. Cell 46, 518–529 [DOI] [PubMed] [Google Scholar]
- 13. Brandman O., Stewart-Ornstein J., Wong D., Larson A., Williams C. C., Li G.-W., Zhou S., King D., Shen P. S., Weibezahn J., Dunn J. G., Rouskin S., Inada T., Frost A., and Weissman J. S. (2012) A ribosome-bound quality control complex triggers degradation of nascent peptides and signals translation stress. Cell 151, 1042–1054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Defenouillère Q., Yao Y., Mouaikel J., Namane A., Galopier A., Decourty L., Doyen A., Malabat C., Saveanu C., Jacquier A., and Fromont-Racine M. (2013) Cdc48-associated complex bound to 60S particles is required for the clearance of aberrant translation products. Proc. Natl. Acad. Sci. U.S.A. 110, 5046–5051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Bengtson M. H., and Joazeiro C. A. (2010) Role of a ribosome-associated E3 ubiquitin ligase in protein quality control. Nature 467, 470–473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lyumkis D., Oliveira dos Passos D., Tahara E. B., Webb K., Bennett E. J., Vinterbo S., Potter C. S., Carragher B., and Joazeiro C. A. (2014) Structural basis for translational surveillance by the large ribosomal subunit-associated protein quality control complex. Proc. Natl. Acad. Sci. U.S.A. 111, 15981–15986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Shen P. S., Park J., Qin Y., Li X., Parsawar K., Larson M. H., Cox J., Cheng Y., Lambowitz A. M., Weissman J. S., Brandman O., and Frost A. (2015) Protein synthesis. Rqc2p and 60S ribosomal subunits mediate mRNA-independent elongation of nascent chains. Science 347, 75–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Shao S., Brown A., Santhanam B., and Hegde R. S. (2015) Structure and assembly pathway of the ribosome quality control complex. Mol. Cell 57, 433–444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Verma R., Oania R. S., Kolawa N. J., and Deshaies R. J. (2013) Cdc48/p97 promotes degradation of aberrant nascent polypeptides bound to the ribosome. eLife 2, e00308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Brandman O., and Hegde R. S. (2016) Ribosome-associated protein quality control. Nat. Struct. Mol. Biol. 23, 7–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Tyedmers J., Mogk A., and Bukau B. (2010) Cellular strategies for controlling protein aggregation. Nat. Rev. Mol. Cell Biol. 11, 777–788 [DOI] [PubMed] [Google Scholar]
- 22. Kushnirov V. V. (2000) Rapid and reliable protein extraction from yeast. Yeast 16, 857–860 [DOI] [PubMed] [Google Scholar]
- 23. Shevchenko A., Tomas H., Havlis J., Olsen J. V., and Mann M. (2006) In-gel digestion for mass spectrometric characterization of proteins and proteomes. Nat. Protoc. 1, 2856–2860 [DOI] [PubMed] [Google Scholar]
- 24. Rigaut G., Shevchenko A., Rutz B., Wilm M., Mann M., and Séraphin B. (1999) A generic protein purification method for protein complex characterization and proteome exploration. Nat. Biotechnol. 17, 1030–1032 [DOI] [PubMed] [Google Scholar]
- 25. Rott R., Szargel R., Haskin J., Shani V., Shainskaya A., Manov I., Liani E., Avraham E., and Engelender S. (2008) Monoubiquitylation of alpha-synuclein by seven in absentia homolog (SIAH) promotes its aggregation in dopaminergic cells. J. Biol. Chem. 283, 3316–3328 [DOI] [PubMed] [Google Scholar]
- 26. Kaganovich D., Kopito R., and Frydman J. (2008) Misfolded proteins partition between two distinct quality control compartments. Nature 454, 1088–1095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Wang Y., Meriin A. B., Zaarur N., Romanova N. V., Chernoff Y. O., Costello C. E., and Sherman M. Y. (2009) Abnormal proteins can form aggresome in yeast: aggresome-targeting signals and components of the machinery. FASEB J. 23, 451–463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Miller S. B., Ho C.-T., Winkler J., Khokhrina M., Neuner A., Mohamed M. Y., Guilbride D. L., Richter K., Lisby M., Schiebel E., Mogk A., and Bukau B. (2015) Compartment-specific aggregases direct distinct nuclear and cytoplasmic aggregate deposition. EMBO J. 34, 778–797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lu Z., and Cyr D. M. (1998) Protein folding activity of Hsp70 is modified differentially by the hsp40 co-chaperones Sis1 and Ydj1. J. Biol. Chem. 273, 27824–27830 [DOI] [PubMed] [Google Scholar]
- 30. Rape M., Hoppe T., Gorr I., Kalocay M., Richly H., and Jentsch S. (2001) Mobilization of processed, membrane-tethered SPT23 transcription factor by CDC48(UFD1/NPL4), a ubiquitin-selective chaperone. Cell 107, 667–677 [DOI] [PubMed] [Google Scholar]
- 31. Richly H., Rape M., Braun S., Rumpf S., Hoege C., and Jentsch S. (2005) A series of ubiquitin binding factors connects CDC48/p97 to substrate multiubiquitylation and proteasomal targeting. Cell 120, 73–84 [DOI] [PubMed] [Google Scholar]
- 32. Morano K. A., Santoro N., Koch K. A., and Thiele D. J. (1999) A trans-activation domain in yeast heat shock transcription factor is essential for cell cycle progression during stress. Mol. Cell. Biol. 19, 402–411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Albrecht A. N., Kornak U., Böddrich A., Süring K., Robinson P. N., Stiege A. C., Lurz R., Stricker S., Wanker E. E., and Mundlos S. (2004) A molecular pathogenesis for transcription factor associated poly-alanine tract expansions. Hum. Mol. Genet. 13, 2351–2359 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.