

Abstract
A missense mutation (T835M) in the uncoordinated-5C (UNC5C) netrin receptor gene increases the risk of late-onset Alzheimer disease (AD) and also the vulnerability of neurons harboring the mutation to various insults. The molecular mechanisms underlying T835M-UNC5C-induced death remain to be elucidated. In this study, we show that overexpression of wild-type UNC5C causes low-grade death, which is intensified by an AD-linked mutation T835M. An AD-linked survival factor, calmodulin-like skin protein (CLSP), and a natural ligand of UNC5C, netrin1, inhibit this death. T835M-UNC5C-induced neuronal cell death is mediated by an intracellular death-signaling cascade, consisting of death-associated protein kinase 1/protein kinase D/apoptosis signal-regulating kinase 1 (ASK1)/JNK/NADPH oxidase/caspases, which merges at ASK1 with a death-signaling cascade, mediated by amyloid β precursor protein (APP). Notably, netrin1 also binds to APP and partially inhibits the death-signaling cascade, induced by APP. These results may provide new insight into the amyloid β-independent pathomechanism of AD.
Keywords: Alzheimer disease, amyloid precursor protein (APP), apoptosis, neurodegenerative disease, signal transduction, UNC5C, amyloid beta-independent, neuronal cell death
Introduction
Alzheimer disease (AD)2 is the most prevalent form of late-onset dementia. The up-regulation of amyloid β (Aβ), which is deposited as an aggregate in senile plaques and forms soluble oligomers in the central nervous system (CNS), is considered to be a hallmark of AD, and numerous studies showed its close relationship with the AD pathogenesis (1–3). Multiple missense mutations in three genes, Aβ precursor protein (APP), presenilin 1 (PS1), and presenilin 2 (PS2), cause autosomal dominant familial AD. A number of studies have shown that constitutive expression of these mutant genes causes an increased production of Aβ and/or an increased ratio of Aβ42, a longer and more toxic isoform of Aβ, to Aβ40 in cells and that transgenic overexpression of these genes causes the formation of senile plaques and dementia in aged mice (2, 4). If used as potential AD drugs in mouse AD models, some Aβ antibodies eliminate Aβ from the CNS and some inhibitors to β- or γ-secretase reduce the production of Aβ in the CNS, thereby mitigating the dementia of these mice (2, 3). Despite these findings, there is still no direct evidence that Aβ causes neuronal toxicity in human AD patients. Although some ongoing clinical trials may be showing promise, the Aβ-reducing therapy has not been proved to be effective in human AD patients until now (5, 6).
Aβ-independent neurotoxicity may contribute to the pathogenesis of AD (7–9). Overexpression of a London-type APP mutant V642I-APP or the binding of TGFβ2 to the extracellular domain of wild-type APP (WT-APP) causes neuronal cell death. These are mediated by a cell death-signaling pathway, consisting of heterotrimeric G protein Go, Rac1, or Cdc42, apoptosis signal-regulating kinase 1 (ASK1), JNK, NADPH oxidase, and caspases, independently of the production of Aβ (10–13).
It was recently reported that a missense mutation of a single-spanning transmembrane protein uncoordinated-5 homologue C (UNC5C; alternative name UNC5H3), a netrin receptor (14–16), increases the risk of late-onset AD (17), although the prevalence of its mutation in AD patients is low (17). Interestingly, T835M-UNC5C did not increase the production of Aβ but increased the vulnerability of neuronal cells to various insults (17). The molecular mechanism underlying the T835M-UNC5C-mediated increase in neuronal toxicity remains uncharacterized.
The netrins and its receptors, UNC5 family proteins, are involved in axonal guidance during embryonal development (14–16). UNC5C and its close relatives UNC5A (UNC5H1), UNC5B (UNC5H2), and UNC5D (UNC5H4) are also known to be “dependence receptors,” which cause cell death in the absence of their ligands (14–16). There have been few studies on the molecular mechanisms underlying UNC5 family protein-induced cell death. It was shown in a previous study that UNC5A induces apoptosis by interacting with the neurotrophin receptor-interacting melanoma-associated antigen homologue (18). Overexpression of UNC5B induces neuronal death by activating death-associated protein kinase (DAPK1) (19) via protein phosphatase 2A-mediated dephosphorylation of DAPK1 (20).
In this study, overexpression of T835M-UNC5C causes overt death in the absence of other neurotoxic insults, whereas overexpression of WT-UNC5C causes low-grade death in F11 neurohybrid cells and SH-SY5Y neuroblastoma cells. A natural ligand for UNC5C, named netrins, and an endogenous AD-linked survival factor calmodulin-like skin protein (CLSP) inhibit this death. Our data also indicate that the T835M-UNC5C-induced neuronal cell death is mediated by an intracellular death-signaling pathway, consisting of DAPK1/protein kinase D (PKD)/ASK1/JNK/NADPH oxidase/caspases.
Experimental Procedures
Protein Analysis
For analysis of cell lysates, cells were washed twice with PBS and suspended in 50 mm HEPES (pH 7.4), 150 mm NaCl, 0.1% Nonidet P-40, and protease inhibitor mixture Complete (Roche Diagnostics). After freezing and thawing twice, the cell lysate was centrifuged at 15,000 rpm for 10 min at 4 °C. The supernatant was submitted to immunoprecipitation with an indicated antibody and/or analysis with standard or Tris-Tricine SDS-PAGE and/or immunoblot analysis. For immunoprecipitation, supernatants were mixed with the antibody, incubated at 4 °C for 2 h, then mixed at 1:1 with slurry of protein G-Sepharose 4B (GE Healthcare, Little Chalfont, UK), and then rotated overnight before washing with the buffer. The whole washed precipitates were boiled in the SDS sample buffer before application to the gels. Ten μg of cell lysates per lane were used for direct immunoblot analysis.
Recombinant Proteins, Antibodies, and Other Reagents
GST-mouse CLSP1 (mCLSP1)-MycHis and GST-human CLSP-MycHis were expressed in Escherichia coli BL-21 at 25 °C for 6 h in 1 mm isopropyl-thio-β-d-galactopyranoside. Purified GST-mCLSP1-MycHis, bound to Glutathione-Sepharose (GE Healthcare), was co-incubated in PBS containing thrombin (1 unit/ml, GE Healthcare) at 25 °C overnight to be released from GST that bound to the glutathione-Sepharose, as shown previously (21). Recombinant GST protein was also purified for the usage as a negative control.
Recombinant mouse netrin1 was purchased from R&D Systems (catalogue no. 1109-N1/CF, Minneapolis, MN). Recombinant human transforming growth factor β2 (TGFβ2) was purchased from PeproTech EC (catalogue no. 100-358, London, UK). A DAPK1 inhibitor (catalogue no. 324788-10MGCN) and a PKD inhibitor (catalogue no. 476495-10MGCN) were purchased from Calbiochem-Novabiochem. An NADPH oxidase inhibitor acetovanillone (or apocynin) (catalogue no. W508454-100G) and a caspase-3 inhibitor Ac-DEVD-CHO (DEVD) (catalogue no. 3172-v) were purchased from Sigma and Peptide Institute (Osaka, Japan), respectively. TALON metal resin was purchased from Clontech (catalogue no. 635502). Antibodies against the indicated peptides and proteins used in this study were purchased from the following companies: horseradish peroxidate-conjugated FLAG epitope (clone M2, catalogue no. 158592-1MG) and DAPK1 (catalogue no. WH0001612M1-100UG), Sigma; APP (22C11, catalogue no. MAB348) and PS1 (catalogue no. MAB 1563), Chemicon (Temecula, CA); phospho-SAPK/JNK (T183/Y185) (clone 81E11, catalogue no. 4668S) and protein kinase D (PKD, catalogue no. 2052S), Cell Signaling Technology (Beverly, MA); JNK (catalogue no. SC-571), Santa Cruz Biotechnology (Santa Cruz, CA); peroxidase-conjugated HA epitope (clone 3F10, catalogue no. 2013819), Roche Diagnostics; and Myc epitope (catalogue no. R950-25), Invitrogen.
Genes and Vectors
The pHA vector, a CMV promoter-driven expression vector harboring a C-terminally HA tag, was generated as follows. The in vitro annealed sense primer, 5′-GCCGGTACCACCATGTACCCATACGATGTTCCAGATTACGCTTGAGGTACCCCG-3′, and the antisense primer, 5′-CGGGGTACCTCAAGCGTAATCTGGAACATCGTATGGGTACATGGTGGTACCGGC-3′, encoding the HA epitope, was inserted into the pFLAG-5a vector (Eastman Kodak) at the KpnI site. A pRK5 expression vector encoding human WT-UNC5C C-terminally tagged with HA was a gift from Dr. Guofa Liu (University of Toledo, Toledo, OH). As a control vector for the pRK5-WT-UNC5C and pRK5-mutant UNC5C constructs, the empty pHA vector was used as it shares the basic components of plasmids. Human DAPK1 and K42A-DAPK1 (dominant-negative DAPK1) cDNAs inserted in the pRK5/Myc vector were gifts from Dr. T. H. Lee (Beth Israel Deaconess Medical Center, Harvard Medical School, Boston). HA-tagged human WT-PKD (ID: 10808), constitutively active (S738E/S742E) PKD (ID: 10810), and kinase-dead (K612W) PKD (ID: 10809) in the pcDNA3 vector (Invitrogen) were purchased from Addgene (Tokyo, Japan). The T835M mutant of UNC5C was constructed using KOD-Plus mutagenesis kit (Toyobo, Tokyo, Japan) with the sense primer, 5′-TGGTCACGGGGCCCAGTGCTTTCAGCATCCCTCTCCCTATCC-3′, and the antisense primer, 5′-TGGTGATGGTGTTCGCAGGATCCAGCAGCGGCAAATCGATGCC-3′. Death domain-defective UNC5C (WT-UNC5CΔDD) and T835M-UNC5CΔDD were also constructed using the same kit with the sense primer, 5′-CAGTATCTCGAGGCCTACCCATACGATGTTCCTGACTATGCG-3′, and the antisense primers, 5′-CGTGACCGTGGTGATGGTGTTCGCAGGATCCAGCAGCGGC-3′ and 5′-CGTGACCATGGTGATGGTGTTCGCAGGATCCAGCAGCGGC-3′, respectively. pRK5-FLAG-HA encoding mouse full-length mouse netrin1 (mNetrin1) was generously donated by Dr. Marko Hyytiäinen (The Haartman Institute, Translational Cancer Biology Research Program and Helsinki University Hospital, University of Helsinki). The mNetrin1 cDNA was inserted into the pEF1/MycHis vector (Invitrogen) at the EcoRI and XbaI sites. Mouse WT-APP and V642I-APP cDNAs inserted in the pcDNA3.1/MycHis were described previously (11–13). The expression vectors for dominant-negative ASK1 and JNK were also described in earlier studies (11–13).
Cells, Cell Death, and Cell Viability
Neuronal cell death assays related to AD were first performed by Yamatsuji et al. (10) and previously described in detail (11–13, 21). Neurohybrid F11 cells were also described earlier (22). F11 cells are the hybrids of rat embryonic day 13 primary cultured neurons and mouse neuroblastoma NTG18 cells. The transient transfection procedure was described previously in detail (10–13, 21). F11 cells, seeded at 7 × 104/well in six-well plates in Ham's F-12 with 18% FBS (HyCloneTM, GE Healthcare) for 12–16 h, were co-transfected with the indicated vectors for 3 h in the absence of serum and were then incubated with Ham's F-12 with 18% FBS for 2 h. Doses of transfected vectors were 0.5 μg unless otherwise mentioned. At 5 h after the onset of the transfection, culture media were replaced by Ham's F-12 with 10% FBS. At 24 h after the transfection, the media were replaced by Ham's F-12 containing N2 supplement (Invitrogen) with or without recombinant Netrin1, CLSP1, or TGFβ2. BSA (Sigma) or GST was used as negative controls. At 72 h after the onset of the transfection, the cells were harvested for the cell viability assays using the WST-8 cell death assay kit (Dojindo, Kumamoto, Japan) or staining with calcein AM (Dojindo), and trypan blue exclusion death assays with their microscopic views taken to show viable cells that were attached to cell plates, as described previously (13, 21, 23).
SH-SY5Y cells were grown in DMEM/Ham's F-12 mixture (DMEM/F-12) containing 10% FBS. SH-SY5Y cells were seeded at 2 × 105/well in six-well plates for 12–16 h, transfected with indicated vectors for 3 h in the absence of serum, and then cultured in DMEM/F-12, 10% FBS with/without a rescue factor. At 24 h after the transfection, the media were replaced with DMEM/F-12 containing N2 supplement with/without a rescue factor. At 48 h after the onset of the transfection, cells were harvested to perform cell viability assays using the staining with calcein AM (Dojindo) and trypan blue exclusion cell mortality assays (23).
Transfection efficiency in F11 cells and SH-SY5Y cells was ∼80%. COS7 cell were grown in DMEM with 10% FBS and used only for the generation of recombinant mouse netrin1 C-terminally tagged with MycHis.
In Vitro Binding Assays by Co-immunoprecipitation
APP, UNC5C, its derivative, or Myc-DAPK1 was overexpressed in F11 cells by transfection. At 24 h after transfection, the cells were harvested for the preparation of cell lysates. Two types of cell lysates were mixed and incubated at 4 °C overnight. The mixed cell lysates was then subjected to immunoprecipitation with a Myc antibody.
In Vitro Binding Assays by Pulldown
APP, UNC5C, or mouse netrin1 was overexpressed in F11 cells or COS7 cells by transfection. At 24 h after transfection of pHA-WT-APP or pRK5-WT-UNC5C to F11 cells, the F11 cells were harvested for the preparation of cell lysates. At 48 h after transfection of pEF1/MycHis-mNetrin1 to COS7 cells, the conditioned media were also harvested for the pulldown with TALON metal beads (Clontech) that bind to the C-terminally His6-tagged mNetrin1. The washed TALON beads were then mixed with one of the cell lysates from the F11 cells and incubated at 4 °C overnight. The mixtures were then subjected to extensive washing. The washed precipitates, the cell lysates, and the beads were then subjected to SDS-PAGE and immunoblot analysis.
In Vitro JNK Kinase Assays
F11 cells, co-transfected with the pcDNA3-FLAG-JNK-1a1 construct with other vectors, were harvested at 48 h after transfection, and lysates were prepared from these cells. The FLAG-JNK-1a1 protein expressed in the lysates was then immunoprecipitated with the FLAG antibody (M2). The immunoprecipitates were used for an in vitro JNK kinase assay using a recombinant c-Jun protein as a substrate, in accordance with the manufacturer's instruction (Kinase STAR JNK activity assay kit, BioVision Research Products), and as described earlier (23). In brief, whole mixtures from the in vitro JNK kinase assay reactions were fractionated with SDS-PAGE, and the levels of phosphorylated FLAG-JNK-1a1, total FLAG-JNK-1a1, and phosphorylated c-Jun (phospho-c-Jun) were analyzed by immunoblot analysis with antibodies against phospho-JNK, FLAG, and phospho-c-Jun.
Statistical Analysis
All cell death experiments were performed in triplicate (n = 3). All values shown in the figures are the means ± S.D. Statistical analysis was performed using one-way analysis of variance, followed by Bonferroni/Dunn post hoc analysis. Data densitometrically obtained using ImageJ software were analyzed with Student's t test. All data were analyzed using StatView (version 5.0.1) software from SAS Institute (Cary, NC).
Results
T835M Mutation Intensifies the UNC5C-induced Neuronal Death That Is Inhibited by Netrin1
The UNC5 family proteins are so-called dependence receptors that induce cell death in the absence of ligand binding (14–16). We found that overexpression of WT-UNC5C caused death in F11 neurohybrid cells (Fig. 1, A–C). Notably, the T835M mutation intensified the WT-UNC5C-induced death (Fig. 1, A and B). As expected, recombinant mouse netrin1 (mNetrin1) inhibited the WT-UNC5C- and the T835M-UNC5C-induced death (Fig. 1, A and B) (14–16). This finding indicates that netrin1 may suppress the UNC5C-induced death by binding to UNC5C on the cell surface and inhibiting the UNC5C-mediated intracellular death signaling. Unexpectedly, however, because of a yet unknown mechanism, the treatment of these cells with netrin1 decreased WT-UNC5C and T835M-UNC5C expression levels (Fig. 1C). This finding indicates that it is also possible that netrin1 may reduce the UNC5C-induced death by decreasing the levels of UNC5C. To confirm that the former mechanism contributes to the netrin-induced inhibition of the T835M-UNC5C-induced death, we performed another experiment in which the levels of UNC5C expression were changed by the alteration of doses of the transfected UNC5C vectors (Fig. 1, D–F). Transfection of 0.25 μg of the T835M-UNC5C-encoding vector, followed by the mock incubation, and transfection of 0.5 μg of the T835M-UNC5C-encoding vector, followed by the incubation with recombinant netrin1, resulted in similar protein expression levels of T835M-UNC5C (Fig. 1F, 5th and 8th lanes). The former transfection caused cell death, whereas the latter transfection did not cause apparent cell death (Fig. 1, D and E, 5th and 8th columns). This result indicates that netrin1 inhibited the UNC5C-induced death by binding to UNC5C on the cell surface and inhibiting the UNC5C-mediated intracellular death signaling.
FIGURE 1.

Overexpression of UNC5C induces neuronal cell death, intensified by the T835M mutation but inhibited by the binding of netrin1. A–C, F11 cells, transfected with the empty pHA vector (vector), pRK5-WT-UNC5C (WT-UNC5C), or pRK5-T835M-UNC5C (T835M-UNC5C), were co-incubated with or without 1.0 μm recombinant mouse netrin1 (mNetrin1) or bovine serum albumin (BSA) for 48 h. This co-incubation was started at 24 h after transfection. At 72 h after transfection, the cells were harvested for trypan blue exclusion assays (A) and WST-8 assays (B). The cell lysates were subjected to SDS-PAGE and immunoblot analysis with the monoclonal antibody to HA (C). D–F, F11 cells, transfected with indicated amounts (μg/well) of the empty pHA vector (vector) or pRK5-T835M-UNC5C, were co-incubated with or without 1.0 μm recombinant mouse Netrin1 (mNetrin1) or BSA for 48 h. The co-incubation was started at 24 h after transfection. At 72 h after transfection, they were harvested for trypan blue exclusion assays (D) and WST-8 assays (E). The cell lysates were subject to SDS-PAGE and immunoblot analysis with a monoclonal antibody against HA (F). ***, p < 0.001; **, p < 0.01; n.s., not significant. WB, Western blot.
CLSP Inhibits the T835M-UNC5C-induced Death in F11 and SH-SY5Y Cells
It was previously shown that neuronal death induced by a London-type familial AD-linked APP mutant V642I-APP or the binding of TGFβ2 to the extracellular domain of WT-APP is mediated by heterotrimeric G protein Go, Rac1, or Cdc42, ASK1, JNK, NADPH oxidase, and caspases in vitro (10–13). Humanin and CLSP are secreted bioactive peptides that inhibit AD-related neuronal death (24) by binding to a heterotrimeric humanin receptor (25, 26) and by inducing expression of SH3BP5, a newly identified endogenous JNK inhibitor (23). In this study, we assessed whether recombinant CLSP is effective against another intracellular signaling pathway underlying the AD-linked neuronal death, induced by overexpression of T835M-UNC5C, and we found that recombinant mouse CLSP1 (mCLSP1) (27) completely inhibited the T835M-UNC5C-induced death (Fig. 2, A–C). This result suggests that the T835M-UNC5C-induced death signaling is also mediated by JNK because JNK is the target of the CLSP-induced survival signaling. Using other neuronal cells, SH-SY5Y cells, we further confirmed that overexpression of T835M-UNC5C induced neuronal cell death and that the T835M-UNC5C-induced neuronal cell death was inhibited by the treatment with recombinant mouse netrin1 and human CLSP (Fig. 2, D–F).
FIGURE 2.
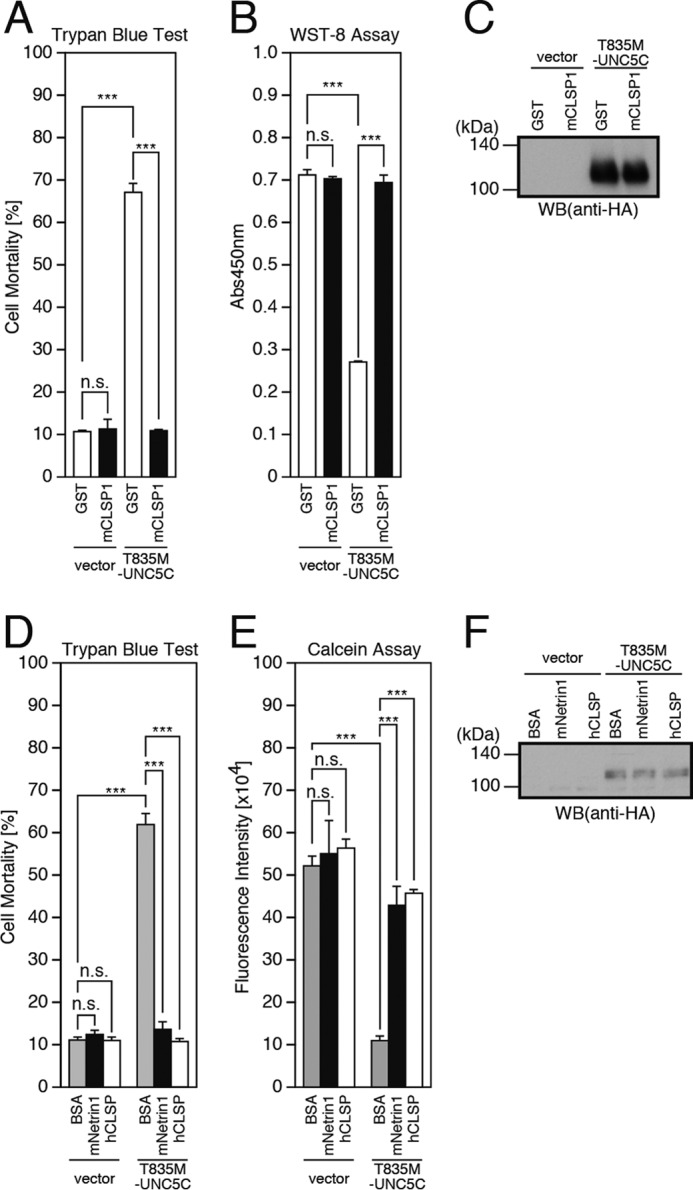
CLSP inhibits T835M-UNC5C-induced death in F11 cells and SH-SY5Y cells. A–C, F11 cells, transfected with the empty pHA vector (vector) or pRK5-T835M-UNC5C, were co-incubated with or without 10 nm recombinant mouse CLSP1 (mCLSP1) or glutathione S-transferase (GST) as a negative control for 48 h. The co-incubation was started 24 h after transfection. At 72 h after transfection, the cells were harvested for trypan blue exclusion assays (A) and WST-8 assays (B). The cell lysates were subjected to SDS-PAGE and immunoblot analysis with a monoclonal antibody against HA (C). D–F, SH-SY5Y cells, transfected with the empty pHA vector (vector) or pRK5-T835M-UNC5C, were co-incubated with or without 1.0 μm recombinant mouse netrin1 (mNetrin1), 10 nm recombinant human CLSP (hCLSP1), or 1.0 μm BSA as a negative control for 48 h. The co-incubation was started at 24 h after transfection. At 24 h after transfection, the cells were harvested for trypan blue exclusion assays (D) and calcein assays (E). The cell lysates were subjected to SDS-PAGE and immunoblot analysis with a monoclonal antibody against HA (F). ***, p < 0.001; n.s., not significant; WB, Western blot.
T835M-UNC5C-induced Death Is Mediated by DAPK1 and PKD
A previous study (19) has reported that overexpression of UNC5B causes cell death by binding to and activating DAPK1 via its death domain. The same study additionally showed that UNC5C also showed weak binding to DAPK1 (19). In this study, the deletion of the C-terminal death domain of UNC5C (UNC5CΔDD) markedly attenuated the T835M-UNC5C-induced death (Fig. 3, A–C). Furthermore, co-immunoprecipitation analysis revealed that DAPK1 bound to UNC5C, whereas DAPK1 did not bind to UNC5C if UNC5C lacked the death domain (Fig. 3D), suggesting that UNC5C is associated with DAPK1 via its death domain and that the interaction of UNC5C with DAPK1 is responsible for the UNC5C-induced death. These findings prompted us to examine whether the UNC5C-induced death is mediated by DAPK1. We treated the cells overexpressing T835M-UNC5C with a DAPK1 inhibitor and found that this treatment almost nullified the T835M-UNC5C-induced death (Fig. 4, A–C). Moreover, co-expression of a dominant-negative DAPK1 (dnDAPK1) inhibited the T835M-UNC5C-induced death (Fig. 4, D and E). These results together indicate that DAPK1 mediates the UNC5C-induced death.
FIGURE 3.
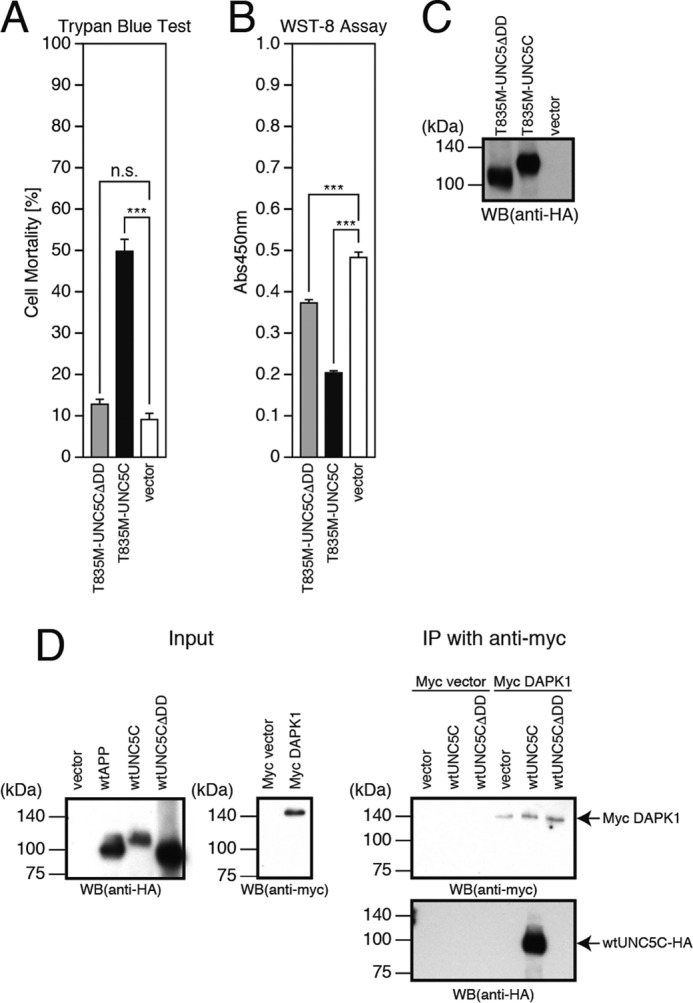
Death domain of UNC5C is essential for UNC5C toxicity. A–C, F11 cells, transfected with the empty pHA vector (vector), pRK5-T835M-UNC5C, or pRK5-T835M-UNC5CΔDD, were harvested at 72 h after transfection for trypan blue exclusion assays (A) and WST-8 assays (B). The cell lysates were then subjected to SDS-PAGE and immunoblot analysis with a monoclonal antibody against HA (C). D, F11 cells were transfected with the pHA vector (vector), pHA-WT-APP (WT-APP) or pRK5-WT-UNC5C (WT-UNC5C), pRK5-WT-UNC5CΔDD (WT-UNC5CΔDD), the empty pRK5/Myc vector (Myc vector), or pRK5/Myc-DAPK1 (Myc DAPK1). At 24 h after transfection, the cells were harvested for the preparation of cell lysates. Two types of cell lysates were mixed and incubated at 4 °C overnight. The mixed cell lysates were then subjected to immunoprecipitation (IP) using a Myc antibody. Washed immunoprecipitates (IP with anti-Myc) and the cell lysates (Input) were then subjected to SDS-PAGE and immunoblot analysis with a Myc antibody or HA antibody. Overexpression of WT-APP was performed as a transfection control. ***, p < 0.001; n.s., not significant; WB, Western blot.
FIGURE 4.

T835M-UNC5C-induced cell death is mediated by DAPK and PKD. A–C, F11 cells, transfected with the empty pHA vector (vector) or pRK5-T835M-UNC5C, were co-incubated with or without 1 μm DAPK1 inhibitor or 1 μm PKD inhibitors (or DMSO as a negative control) for 48 h. This co-incubation was started at 24 h after transfection. At 72 h after transfection, the cells were harvested for trypan blue exclusion assays (A) and WST-8 assays (B). The cell lysates were then subjected to SDS-PAGE and immunoblot analysis with a monoclonal antibody against HA (C). D and E, F11 cells, co-transfected with the empty pHA vector (vector) or pRK5-T835M-UNC5C together with the empty pRK5/Myc vector (vector 1) or pRK5/Myc-dominant-negative DAPK1 (dnDAPK1) or with the empty pcDNA3 vector (vector 2) or pcDNA3-kinase-dead PKD (kdPKD), were harvested at 72 h for trypan blue exclusion assays (D). The cell lysates were then subjected to SDS-PAGE and immunoblot analysis with a monoclonal antibody against HA or a DAPK1 antibody mixed with PKD antibody (E). ***, p < 0.001; n.s., not significant; WB, Western blot.
It was previously reported that under the oxidative stress condition, DAPK1 binds to and phosphorylates PKD, and that PKD is indispensable for the DAPK1-induced JNK phosphorylation (28). Considering these findings (28) and our current findings, shown above (Fig. 2), that CLSP inhibits the T835M-UNC5C-induced cell death, possibly by inactivating JNK (23), we hypothesized the involvement of PKD and JNK in the UNC5C-induced death-signaling pathway. In this study, we first examined the involvement of PKD. The treatment of the cells overexpressing T835M-UNC5C with a PKD inhibitor (Fig. 4, A–C) and the co-overexpression of a kinase-dead PKD almost completely inhibited the T835M-UNC5C-induced death (Fig. 4, D and E). These results indicate that PKD participates in the T835M-UNC5C-induced death as a downstream mediator of DAPK1.
ASK1 and JNK Are Involved in T835M-UNC5C-induced Death-signaling Pathway as Downstream Mediators of PKD
We next examined the involvement of JNK as well as ASK1, an upstream MAPKKK of JNK (11–13), in the T835M-UNC5C-induced death. Co-expression of a dominant-negative ASK1 or JNK almost completely inhibited the T835M-UNC5C-induced death (Fig. 5, A and B). This result suggests that ASK1 and JNK mediate the T835M-UNC5C-induced neuronal death.
FIGURE 5.
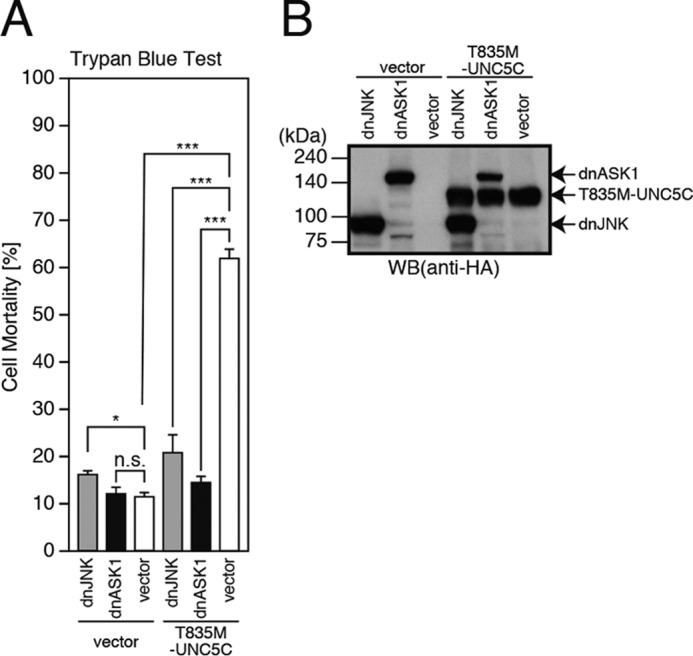
T835M-UNC5C-induced cell death is mediated by ASK1 and JNK. A and B, F11 cells were co-transfected with the empty pHA vector (vector) or pRK5-T835M-UNC5C together with the pcDNA3 vector (vector), pcDNA3-dominant-negative ASK1 (dnASK1), or pcDNA3-dominant-negative JNK (dnJNK). At 72 h, the cells were harvested for trypan blue exclusion assays (A). The cell lysates were subjected to SDS-PAGE and immunoblot analysis with a monoclonal antibody against HA (B). ***, p < 0.001; *, p < 0.05; n.s., not significant; WB, Western blot.
To confirm the involvement of PKD, ASK1, and JNK in the T835M-UNC5C-induced death, we overexpressed constitutively active PKD with or without dominant-negative JNK or ASK1 (dnJNK or dnASK1) in F11 cells. Expression of constitutively active PKD caused death in F11 cells, similarly to that of T835M-UNC5C, and the constitutively active PKD-induced death was largely blocked by co-expression of dnASK1 or dnJNK (Fig. 6, A and B). This result supports the idea that ASK1 and JNK mediate the T835M-UNC5C-induced death-signaling pathway as downstream mediators of PKD.
FIGURE 6.

Constitutively active PKD induces cell death, mediated by ASK1 and JNK. A and B, F11 cells were co-transfected with the empty pHA vector (vector) or pcDNA3-constitutively active PKD (caPKD) together with the pcDNA3 vector (vector2), pcDNA3-dominant-negative ASK1 (dnASK1), or pcDNA3-dominant-negative JNK (dnJNK). At 72 h, the cells were harvested for trypan blue exclusion assays (A). The cell lysates were subjected to SDS-PAGE and immunoblot analysis with a PKD antibody or a monoclonal antibody against HA (B). C, F11 cells were co-transfected with pcDNA3-FLAG-JNK1a1 and the empty pHA vector (vector), pRK5-T835M-UNC5C, or pHA-V642I-APP. At 48 h after transfection, the cells were harvested for the preparation of cell lysates. FLAG-JNK1a1 in the lysates was immunoprecipitated with a FLAG antibody (M2) and used for in vitro kinase assays with c-Jun-derived peptide as a substrate. Whole mixtures were subjected to SDS-PAGE and immunoblot analysis with an HA antibody, phospho-c-Jun antibody, phospho-JNK antibody, or FLAG antibody (M2). Similar results were obtained in three independent experiments. ***, p < 0.001; n.s., not significant; WB, Western blot.
To further confirm the involvement of JNK in the T835M-UNC5C-induced death, we expressed FLAG-JNK1a1 alone or in association with T835M-UNC5C or V642I-APP in F11 cells and immunoprecipitated FLAG-JNK1a1 from the cell lysates. These immunoprecipitates were then used for in vitro JNK kinase assays with c-Jun-derived peptide as a substrate (Fig. 6C) (23). Co-expression of T835M-UNC5C or V642I-APP increased the levels of phosphorylated FLAG-JNK1a1 and phosphorylated c-Jun (Fig. 6C). This result supported our notion that JNK mediates the T835M-UNC5C-induced as well as the V642I-APP-induced death-signaling pathway (10–13).
T835M-UNC5C-induced Death-signaling Pathway Is Mediated by NADPH Oxidase and Caspases
As downstream mediators, NADPH oxidase and caspases were shown previously to be involved in the neuronal death induced by V642I-APP or by the binding of TGFβ2 to the extracellular domain of WT-APP (11–13). In our present analyses, treatment with the NADPH oxidase inhibitor apocynin or the caspase-3 inhibitor DEVD almost completely inhibited the T835M-UNC5C-induced neuronal cell death (Fig. 7, A–C). This result indicates that these mediators are also involved in the pathway, possibly as a downstream mediator of JNK.
FIGURE 7.

NADPH oxidase and caspases are involved in cell death, mediated by T835M-UNC5C. A–C, F11 cells, transfected with the empty pHA vector (vector) or pRK5-T835M-UNC5C were co-incubated with or without 100 μm NADPH oxidase inhibitor acetovanillone (or apocynin; APO) or 100 μm caspase-3 inhibitor Ac-DEVD-CHO (DEVD) for 48 h. This co-incubation was started 24 h after transfection. At 72 h after transfection, the cells were harvested for trypan blue exclusion assays (A) and WST-8 assays (B). The cell lysates were subjected to SDS-PAGE and immunoblot analysis with a monoclonal antibody against HA (C). ***, p < 0.001; n.s., not significant; WB, Western blot.
T835M-UNC5C-induced Death-signaling Pathway Merges with the APP-mediated Death-signaling Pathway at ASK1
Thus, in this study, we have demonstrated that the T835M-UNC5C-induced neuronal death is mediated by DAPK1, PKD, ASK1, JNK, NADPH oxidase, and caspases. In previous studies (11–13), it was shown that the V642I-APP-induced neuronal cell death and the TGFβ2-induced neuronal death via APP are mediated by a heterotrimeric GTP-binding protein Go, Rac1/Cdc42, ASK1, JNK, NADPH oxidase, and caspases. Comparing these results, we could assume that both death signals advance independently during the initial phase, merge at ASK1, and progress via a common death-signaling pathway, mediated by JNK/NADPH oxidase/caspases, during the final phase. Considering that DAPK1 and PKD act as upstream mediators of ASK1 in the former signal transduction pathway, we further examined the possibility that DAPK1 and PKD also participate in the latter signal transduction pathway as an upstream mediator of ASK1. However, the treatment of the cells overexpressing V642I-APP with the DAPK1 or PKD inhibitor did not affect the V642I-APP-induced neuronal death (Fig. 8, A–C). This result indicated that neither DAPK1 nor PKD is involved in the latter signal transduction pathway.
FIGURE 8.

Neither DAPK1 nor PKD is involved in the cell death pathway mediated by APP. A–C, F11 cells, transfected with the empty pcDNA3.1/MycHis vector (vector) or pcDNA3.1/MycHis-V642I-APP (V642I-APP), were co-incubated with or without 1 μm DAPK1 inhibitor or 1 μm PKD inhibitors or DMSO for 48 h. The co-incubation was started at 24 h after transfection. At 72 h after transfection, the cells were harvested for trypan blue exclusion assays (A) and WST-8 assays (B). The cell lysates were subjected to SDS-PAGE and immunoblot analysis with antibody against APP (C). ***, p < 0.001; **, p < 0.01; n.s., not significant; WB, Western blot.
Netrin1 Binds to APP and Partially Attenuates the TGFβ2-induced Death via APP
It was previously shown that netrin1 also binds to a domain of APP containing the Aβ-corresponding region and decreases the production of Aβ (29). In this study, we confirmed that netrin1 binds to APP (Fig. 9A). Considering these findings, we further examined whether netrin1 affects the APP-mediated neuronal cell death, triggered by the binding of TGFβ2 to APP (11, 30). As shown in previous studies (11–13), TGFβ2 treatment induced death in F11 cells overexpressing WT-APP (Fig. 9, B and C). In contrast, the netrin1 treatment did not induce death in F11 cells overexpressing WT-APP (Fig. 9, B and C). Notably, however, the netrin1 treatment decreased the TGFβ2-induced neuronal cell death via APP significantly albeit incompletely (Fig. 9, B and C). These results indicated that netrin1 inhibits the APP-mediated neuronal cell death as well as the UNC5C-induced neuronal cell death.
FIGURE 9.

Netrin1 binds to APP and inhibits TGFβ2-induced neuronal cell death via APP. A, conditioned media, derived from the cells that were transfected with the empty pEF1/MycHis vector (vector) or pEF1/MycHis-mNetrin1 (mouse netrin1 C-terminally tagged with MycHis), were collected for pulldown with TALON beads (Beads) for the preparation of His6-tagged mNetrin1. In parallel, F11 cells were transfected with the empty pHA vector (vector), pRK5-WT-UNC5C (WT-UNC5C-HA), or pHA-WT-APP (WT-APP-HA). 24 h after transfection, the cell lysates were prepared from the cells. The washed TALON beads capturing mNetrin1-MycHis (mNetrin1) or control (vector) were then mixed with one of these cell lysates. The whole mixtures were incubated at 4 °C overnight and then washed. The final beads (Pull-down) as well as the beads and the cell lysates (Input) were used for immunoblot analysis with a monoclonal antibody against HA or Myc. Similar results were obtained in two independent experiments. B and C, F11 cells, transfected with the empty pFLAG vector (vector) or pFLAG-WT-APP (WT-APP), were co-incubated with 1 μm mNetrin1 or BSA together with 20 nm TGFβ2 or PBS. This co-incubation was started 24 h after transfection. 72 h after transfection, they were harvested for trypan blue exclusion assays (B). The cell lysates were subjected to SDS-PAGE and immunoblot analysis with an antibody against APP (C). ***, p < 0.001; n.s., not significant; WB, Western blot.
Discussion
The death-signaling pathway underlying the UNC5C-induced cell death has not been previously characterized. This is possibly because UNC5C induces cell death only weakly in vitro (Fig. 1). However, the AD-linked T835M mutation greatly increases the toxicity by UNC5C (Fig. 1), enabling us to characterize the death-signaling pathway by UNC5C in detail in vitro in our current analyses.
The percentages of dead F11 cells and SH-SY5Y cells that were transfected with the T835M-UNC5C-encoding vector were ∼50% in this study (Figs. 1–3), whereas the percentages of dead HEK293T cells and primary rat hippocampal neurons that exogenously expressed T835M-UNC5C were ∼25 and 50% at the harvest time in the previous study (17). Given that the transfection efficiency in our experiments using F11 cells and SH-SY5Y cells is 80% or more, the death-inducing activity of T835M-UNC5C is estimated to be slightly higher in our neuronal cell lines than in those reported previously (17).
In this study, death-associated protein was first found to interact with and be activated by UNC5C (Figs. 3 and 4). DAPK1 belongs to the DAPK calmodulin-regulated serine/threonine kinase family, whose members include DAPK1, DRP-1, and ZIP kinase. Although DAPK1 is involved in a variety of caspase-dependent and -independent cell deaths as well as autophagy (31), its downstream mediators have not been sufficiently characterized (32). Until now, only one study addressed the DAPK1-mediated cell death-signaling pathway (28), showing that the DAPK1-mediated cell death-signaling pathway during oxidative stress is mediated by PKD and JNK. In this study, we have shown that in the T835M-UNC5C-induced death, PKD works as a downstream mediator of DAPK1 (Fig. 4). A series of experiments, the results of which are shown in Figs. 3–7, led us to conclude that the T835M-UNC5C-induced death-signaling pathway is composed of DAPK1/PKD/ASK1/JNK/NADPH oxidase/caspases.
Other uncharacterized signaling molecules may also be involved in the T835M-UNC5C-induced cell death. For example, a neurotrophin receptor-interacting melanoma-associated antigen (MAGE) homologue named NRAGE was previously identified as an UNC5A-interacting protein that mediates the UNC5A-induced death-signaling pathway in COS cells (18), and possibly further mediated by the degradation of the caspase inhibitor X-chromosome-linked inhibitor of apoptosis protein and/or the activation of the JNK signaling pathway (18). Because the binding affinity of NRAGE to UNC5C appears to be much weaker than that to UNC5A (18), it is less likely that NRAGE is a mediator of T835M-UNC5C-induced death.
It remains undetermined whether PKD directly interacts with ASK1 in the signal transduction pathways. It is possible that some molecules such as Rac1/Cdc42 connect these components as intermediate mediators. If this is true, the merging point between the T835M-UNC5C-induced and the APP-mediated death-signaling pathways may move upstream of ASK1.
It is highly likely that humanin, CLSP, and netrins work as endogenous neuroprotective factors that suppress the AD-related neuronal toxicities. CLSP inhibits both the APP-mediated neuronal cell death (21, 24) and the UNC5C-mediated neuronal cell death (Fig. 2). Netrin1 inhibits the UNC5C-mediated neuronal cell death (Figs. 1 and 2) and partially inhibits the APP-mediated neuronal cell death (Fig. 9B) by binding to APP (Fig. 9A). In an earlier study, it was shown that netrins inhibit the production of Aβ (33). Based on these findings, it could be further assumed that the dysfunction of CLSP, humanin, or netrins may contribute to the AD pathogenesis.
The T835M-UNC5C-induced death-signaling pathway, mediated by DAPK1/PKD/ASK1/JNK/NADPH oxidase/caspases, merges at ASK1 with the APP-mediated death-signaling pathway, which consists of Go/Rac1 or Cdc42/ASK1/JNK/NADPH oxidase/caspases) (Fig. 10) (11–13). This finding suggests that the signal transduction pathway via ASK1/JNK/NADPH oxidase/caspases may be the common late-phase AD-linked cell death-signaling pathway. This notion is consistent with the results of multiple earlier in vivo studies using AD model mice and AD patient-derived brains. JNK is activated in the neurons of AD model mice (33–36) and AD patients (33). NADPH oxidase is activated in the neurons of AD model mice and AD patients (37–39). Multiple studies have also shown that the activation of caspases occurs in the brains of AD patients (40). Interestingly, a study demonstrated that the expression of DAPK increased in the brains of some AD patients (41).
FIGURE 10.

Schematic illustration of the proposed T835M-UNC5C- and V642I-APP-induced neuronal death-signaling pathways. The UNC5C-induced death-signaling pathway is mediated by DAPK1, PKD, ASK1, JNK, NADPH oxidase, and caspases (Figs. 1–8), and the APP-induced death-signaling pathway is mediated by Go, Rac1/Cdc42, ASK1, MAPKK, JNK, NADPH oxidase, and caspases (9–13). The T835M mutation of UNC5C and a London-type mutation of APP, V642I, intensify each death signaling (Fig. 1) (9–13). CLSP binds to the heterotrimeric Humanin receptor and induces expression of SH3BP5, which inhibits the UNC5C- and APP-mediated death signaling by inhibiting JNK (Fig. 2) (21, 23, 24, 26). Netrin1 binds to UNC5C and inhibits the UNC5C-induced death signaling (Fig. 1). Netrin1 binds to APP and partially inhibits the APP-induced death signaling and reduces the production of Aβ (Fig. 9) (29).
The mutant form of UNC5C is present as a risk factor (17) throughout life, although the disease occurs later in the life. The mechanism underlying the effect of aging on the disease onset remains to be investigated. It could be hypothesized that multiple other neuronal toxicities, including Aβ, must accumulate during aging to finally cause neuronal cell death. It is also possible that aging causes the deterioration of some endogenous protective systems that suppress the disease onset in younger ages. Humanin-, CLSP-, and netrin-mediated inhibition of AD-related toxicities may be examples of such protective systems.
There is no direct linkage between the T835M mutation of UNC5C and the Aβ cascade hypothesis. Most importantly, the T835M mutation does not affect the production of Aβ (17). In addition, the T835M mutation not only increases the vulnerability of cells to Aβ but also to other insults in a similar manner (17). These findings indicate that the T835M-UNC5C-induced neuronal cell death occurs independently of Aβ and that this mutation may equally increase the risk for other neurodegenerative diseases. The latter possibility remains to be investigated. Our previous studies also suggest that the cell death-signaling pathway via APP, mediated by a heterotrimeric GTP-binding protein Go, Rac1/Cdc42, ASK1, JNK, NADPH oxidase, and caspases, appears to be independent of Aβ, because artificially mutated APP that does not generate Aβ is still able to induce neuronal cell death (10–13).
Recently, a missense mutation in APP that is protective against AD was reported (13, 42). The AD-protective mutation not only reduces the production of Aβ (42) but also attenuates the APP-mediated intracellular death signal independently of the reduction of Aβ (13). Moreover, a mutation that causes hereditary cerebral hemorrhage with amyloidosis-Dutch type (HCHWA-D) also attenuates the APP-mediated intracellular death signal (13). HCHWA-D, in which amyloidosis occurs in both the vascular walls and the parenchyma of the brain and the rupture of arteries with massive amyloidosis, causes stroke (43). Notably, AD-type neuronal death does not occur in HCHWA-D despite massive parenchymal amyloidosis (44). From the standpoint of neuronal cell death-signaling pathways that are dependent or independent of Aβ, the evidence suggests that the pathogenesis of AD should be reconsidered.
Author Contributions
Y. H., Y. T., S. K., and M. N. performed the experiments. Y. H. and M. M. designed the experiments, analyzed the data, and wrote the manuscript. M. M. directed the study.
Acknowledgments
We are grateful to Takako Hiraki and Tomoko Yamada for essential assistance throughout the study. We thank Dr. Guofa Liu, Dr. Tae Ho Lee, and Dr. Marko Hyytiäinen for expression vectors.
This work was supported in part by Grant-in-aid for Scientific Research (B) 15H04689 (to M. M.) and Grant-in-aid for Scientific Research (C) 25460343 (to Y. H.) from the Japan Society for the Promotion of Science, by a Naito Foundation Natural Science Scholarship (to M. M.), and by a research grant from Akaeda Medical Research Foundation to (to M. M.). The authors declare that they have no conflicts of interest with the contents of this article.
- AD
- Alzheimer disease
- Aβ
- amyloid β
- APP
- amyloid β precursor protein
- CLSP
- calmodulin-like skin protein
- DEVD
- Ac-DEVD-CHO
- UNC5C
- uncoordinated-5C
- Tricine
- N-[2-hydroxy-1,1-bis(hydroxymethyl)ethyl]glycine
- dn
- dominant-negative
- mCLSP
- mouse CLSP.
References
- 1. Hardy J., and Selkoe D. J. (2002) The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science 297, 353–356 [DOI] [PubMed] [Google Scholar]
- 2. Selkoe D. J. (2011) Alzheimer's disease. Cold Spring Harb. Perspect. Biol. 3, a004457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Selkoe D. J. (2013) SnapShot: pathobiology of Alzheimer's disease. Cell 154, 468–468 [DOI] [PubMed] [Google Scholar]
- 4. Chávez-Gutiérrez L., Bammens L., Benilova I., Vandersteen A., Benurwar M., Borgers M., Lismont S., Zhou L., Van Cleynenbreugel S., Esselmann H., Wiltfang J., Serneels L., Karran E., Gijsen H., Schymkowitz J., et al. (2012) The mechanism of γ-secretase dysfunction in familial Alzheimer disease. EMBO J. 31, 2261–2274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Tayeb H. O., Murray E. D., Price B. H., and Tarazi F. I. (2013) Bapineuzumab and solanezumab for Alzheimer's disease: is the “amyloid cascade hypothesis” still alive? Expert Opin. Biol. Ther. 13, 1075–1084 [DOI] [PubMed] [Google Scholar]
- 6. Selkoe D. J. (2013) The therapeutics of Alzheimer's disease: where we stand and where we are heading. Ann. Neurol. 74, 328–336 [DOI] [PubMed] [Google Scholar]
- 7. Cruz J. C., and Tsai L. H. (2004) Cdk5 deregulation in the pathogenesis of Alzheimer's disease. Trends Mol. Med. 10, 452–458 [DOI] [PubMed] [Google Scholar]
- 8. Mattson M. P. (2007) Calcium and neurodegeneration. Aging Cell 6, 337–350 [DOI] [PubMed] [Google Scholar]
- 9. Matsuoka M. (2009) HUMANIN: a defender against Alzheimer's disease. Recent Pat. CNS Drug Discov. 4, 37–42 [DOI] [PubMed] [Google Scholar]
- 10. Yamatsuji T., Matsui T., Okamoto T., Komatsuzaki K., Takeda S., Fukumoto H., Iwatsubo T., Suzuki N., Asami-Odaka A., Ireland S., Kinane T. B., Giambarella U., and Nishimoto I. (1996) G protein-mediated neuronal DNA fragmentation by familial Alzheimer's disease associated V642 mutants of APP. Science 272, 1349–1352 [DOI] [PubMed] [Google Scholar]
- 11. Hashimoto Y., Chiba T., Yamada M., Nawa M., Kanekura K., Suzuki H., Terashita K., Aiso S., Nishimoto I., and Matsuoka M. (2005) Transforming growth factor β2 is a neuronal cell death-inducing ligand for amyloid-β precursor protein. Mol. Cell. Biol. 25, 9304–9317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Tachi N., Hashimoto Y., and Matsuoka M. (2012) MOCA is an integrator of the neuronal death signals that are activated by familial Alzheimer's disease-related mutants of amyloid β precursor protein and presenilins. Biochem. J. 442, 413–422 [DOI] [PubMed] [Google Scholar]
- 13. Hashimoto Y, Matsuoka M. (2014) A mutation protective against Alzheimer's disease renders amyloid β precursor protein incapable of mediating neurotoxicity. J. Neurochem. 130, 291–300 [DOI] [PubMed] [Google Scholar]
- 14. Arakawa H. (2004) Netrin-1 and its receptors in tumorigenesis. Nat. Rev. Cancer 4, 978–987 [DOI] [PubMed] [Google Scholar]
- 15. Moore S. W., Tessier-Lavigne M., and Kennedy T. E. (2007) Netrins and their receptors. Adv. Exp. Med. Biol. 621, 17–31 [DOI] [PubMed] [Google Scholar]
- 16. Larrieu-Lahargue F., Thomas K. R., and Li D. Y. (2012) Netrin Ligands and Receptors: Lessons from neurons to the endothelium. Trends Cardiovasc. Med. 22, 44–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wetzel-Smith M. K., Hunkapiller J., Bhangale T. R., Srinivasan K., Maloney J. A., Atwal J. K., Sa S. M., Yaylaoglu M. B., Foreman O., Ortmann W., Rathore N., Hansen D. V., and Tessier-Lavigne M., Alzheimer's Disease Genetics Consortium, Mayeux R., et al. (2014) A rare mutation in UNC5C predisposes to late-onset Alzheimer's disease and increases neuronal cell death. Nat. Med. 20, 1452–1457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Williams M. E., Strickland P., Watanabe K., and Hinck L. (2003) UNC5H1 induces apoptosis via its juxtamembrane region through an interaction with NARGE. J. Biol. Chem. 278, 17483–17490 [DOI] [PubMed] [Google Scholar]
- 19. Llambi F., Lourenço F. C., Gozuacik D., Guix C., Pays L., Del Rio G., Kimchi A., and Mehlen P. (2005) The dependence receptor UNC5H2 mediates apoptosis through DAP-kinase. EMBO J. 24, 1192–1201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Guenebeaud C., Goldschneider D., Castets M., Guix C., Chazot G., Delloye-Bourgeois C., Eisenberg-Lerner A., Shohat G., Zhang M., Laudet V., Kimchi A., Bernet A., and Mehlen P. (2010) The dependence receptor UNC5H2/B triggers apoptosis via PP2A-mediated dephosphorylation of DAP kinase. Mol. Cell 40, 863–876 [DOI] [PubMed] [Google Scholar]
- 21. Hashimoto Y., Nawa M., Kurita M., Tokizawa M., Iwamatsu A., and Matsuoka M. (2013) Secreted calmodulin-like skin protein inhibits neuronal death in cell-based Alzheimer's disease models via the heterotrimeric Humanin receptor. Cell Death Dis. 4, e555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Platika D., Boulos M. H., Baizer L., and Fishman M. C. (1985) Neuronal traits of clonal cell lines derived by fusion of dorsal root ganglia neurons with neuroblastoma cells. Proc. Natl. Acad. Sci. U.S.A. 82, 3499–3503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Takeshita Y., Hashimoto Y., Nawa M., Uchino H., and Matsuoka M. (2013) SH3-binding protein 5 mediates the neuroprotective effect of the secreted bioactive peptide humanin by inhibiting c-Jun NH2-terminal kinase. J. Biol. Chem. 288, 24691–24704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Matsuoka M. (2015) Protective effects of Humanin and calmodulin-like skin protein in Alzheimer's disease and broad range of abnormalities. Mol. Neurobiol. 51, 1232–1239 [DOI] [PubMed] [Google Scholar]
- 25. Hashimoto Y., Kurita M., Aiso S., Nishimoto I., and Matsuoka M. (2009) Humanin inhibits neuronal cell death by interacting with a cytokine receptor complex or complexes involving CNTF receptor α/WSX-1/gp130. Mol. Biol. Cell 20, 2864–2873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Matsuoka M., and Hashimoto Y. (2010) Humanin and the receptors for humanin. Mol. Neurobiol. 41, 22–28 [DOI] [PubMed] [Google Scholar]
- 27. Hayashi M., Tajima H., Hashimoto Y., and Matsuoka M. (2014) Secreted calmodulin-like skin protein ameliorates scopolamine-induced memory impairment. Neuroreport 25, 725–729 [DOI] [PubMed] [Google Scholar]
- 28. Eisenberg-Lerner A., and Kimchi A. (2007) DAP kinase regulates JNK signaling by binding and activating protein kinase D under oxidative stress. Cell Death Differ. 14, 1908–1915 [DOI] [PubMed] [Google Scholar]
- 29. Lourenço F. C., Galvan V., Fombonne J., Corset V., Llambi F., Müller U., Bredesen D. E., and Mehlen P. (2009) Netrin-1 interacts with amyloid precursor protein and regulates amyloid-β production. Cell Death Differ. 16, 655–663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Bodmer S., Podlisny M. B., Selkoe D. J., Heid I., and Fontana A. (1990) Transforming growth factor-β bound to soluble derivatives of the β amyloid precursor protein of Alzheimer's disease. Biochem. Biophys. Res. Commun. 171, 890–897 [DOI] [PubMed] [Google Scholar]
- 31. Shiloh R., Bialik S., and Kimchi A. (2014) The DAPK family: a structure-function analysis. Apoptosis 19, 286–297 [DOI] [PubMed] [Google Scholar]
- 32. Fujita Y., and Yamashita T. (2014) Role of DAPK in neuronal cell death. Apoptosis 19, 339–345 [DOI] [PubMed] [Google Scholar]
- 33. Mehan S., Meena H., Sharma D., and Sankhla R. (2011) JNK: a stress-activated protein kinase therapeutic strategies and involvement in Alzheimer's and various neurodegenerative abnormalities. J. Mol. Neurosci. 43, 376–390 [DOI] [PubMed] [Google Scholar]
- 34. Zhou Q., Wang M., Du Y., Zhang W., Bai M, Zhang Z., Li Z., and Miao J. (2015) Inhibition of c-Jun N-terminal kinase activation reverses Alzheimer disease phenotypes in APPswe/PS1dE9 mice. Ann. Neurol. 77, 637–654 [DOI] [PubMed] [Google Scholar]
- 35. Barbero-Camps E., Fernández A., Martínez L., Fernández-Checa J. C., and Colell A. (2013) APP/PS1 mice overexpressing SREBP-2 exhibit combined Aβ accumulation and τ pathology underlying Alzheimer's disease. Hum. Mol. Genet. 22, 3460–3476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Savage M. J., Lin Y. G., Ciallella J. R., Flood D. G., and Scott R. W. (2002) Activation of c-Jun N-terminal kinase and p38 in an Alzheimer's disease model is associated with amyloid deposition. J. Neurosci. 22, 3376–3385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Block M. L. (2008) NADPH oxidase as a therapeutic target in Alzheimer's disease. BMC Neurosci. 9, S8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Ansari M. A., and Scheff S. W. (2011) NADPH-oxidase activation and cognition in Alzheimer's disease progression. Free Radic. Biol. Med. 51, 171–178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Hernandes M. S., and Britto L. R. (2012) NADPH oxidase and neurodegeneration. Curr. Neuropharmacol. 10, 321–327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Rohn T. T. (2010) The role of caspases in Alzheimer's disease; potential novel therapeutic opportunities. Apoptosis 15, 1403–1409 [DOI] [PubMed] [Google Scholar]
- 41. Kim B. M., You M.-H., Chen C.-H., Lee S., Hong Y., Kimchi A., Zhou X. Z., and Lee T. H. (2014) Death-associated protein kinase 1 has a critical role in aberrant τ protein regulation and function. Cell Death Dis. 5, e1237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Jonsson T., Atwal J. K., Steinberg S., Snaedal J., Jonsson P. V., Bjornsson S., Stefansson H., Sulem P., Gudbjartsson D., Maloney J., Hoyte K., Gustafson A., Liu Y., Lu Y., Bhangale T., et al. (2012) A mutation in APP protects against Alzheimer's disease and age-related cognitive decline. Nature 488, 96–99 [DOI] [PubMed] [Google Scholar]
- 43. Levy E., Carman M. D., Fernandez-Madrid I. J., Power M. D., Lieberburg I., van Duinen S. G., Bots G. T., Luyendijk W., and Frangione B. (1990) Mutation of the Alzheimer's disease amyloid gene in hereditary cerebral hemorrhage, Dutch type. Science 248, 1124–1126 [DOI] [PubMed] [Google Scholar]
- 44. Wattendorff A. R., Frangione B., Luyendijk W., and Bots G. T. (1995) Hereditary cerebral haemorrhage with amyloidosis, Dutch type (HCHWA-D): clinicopathological studies. J. Neurol. Neurosurg. Psychiatry 58, 699–705 [DOI] [PMC free article] [PubMed] [Google Scholar]