

Abstract
Toll-like receptor-3 (TLR3) senses double-stranded RNA intermediates produced during hepatitis C virus (HCV) replication, leading to activation of interferon regulatory factor-3 (IRF3) and NF-κB and subsequent antiviral and proinflammatory responses. Yet, how this TLR3-dependent pathway operates in hepatocytes is unclear. Upon fractionating cultured hepatocytes into various cellular organelles, we observed that TLR3 predominantly resides in endolysosomes of hepatocytes. To determine the critical regulators of TLR3 signaling in response to HCV infection in human hepatocytes, we isolated endolysosome fractions from mock-infected and HCV-infected hepatoma Huh7.5 cells that had been reconstituted for TLR3 expression, separated these fractions on two-dimensional gels, and identified up-regulated/down-regulated proteins by mass spectrometry. Approximately a dozen of cellular proteins were found to be differentially expressed in endolysosome fractions following HCV infection. Of these, expression of several molecular chaperone proteins was elevated. Knockdown of one of these chaperones, glucose-regulated protein 78 kDa (GRP78), compromised TLR3-dependent induction of interferon-stimulated genes and chemokines following HCV infection or poly(I:C) stimulation in cultured hepatocytes. Consistent with this finding, GRP78 depletion impaired TLR3-mediated establishment of an antiviral state. Mechanistically, although TLR3 trafficking to endolysosomes was not affected, phosphorylated IRF3 diminished faster following GRP78 knockdown. Remarkably, GRP78 transcript was significantly up-regulated in liver biopsies of chronic hepatitis C patients as compared with normal liver tissues. Moreover, the GRP78 expression level correlated with that of RANTES (regulated upon activation, normal T-cell expressed and secreted) and CXCL10, two inflammatory chemokines most frequently elevated in HCV-infected liver. Altogether, our data suggest that GRP78 contributes to TLR3-mediated, IRF3-dependent innate immune response to HCV in hepatocytes.
Keywords: chemokine, GRP78, Hepatitis C virus (HCV), hepatocyte, innate immunity, molecular chaperone, RANTES, Toll-like receptor-3, interferon regulatory factor-3, interferon stimulated gene
Introduction
Infection with the hepatitis C virus (HCV)2 is associated with relatively robust transcriptional induction of interferon (IFN)-stimulated genes (ISGs) and inflammatory mediators in the liver (1, 2). This innate immune response provides the first wave of cellular defense against HCV and orchestrates the development of anti-HCV adaptive immunity, which determines infection outcome (3, 4). In support of this concept, genetic polymorphisms around the IFNL3/IFNL4 gene locus and ISG expression status prior to treatment are associated with spontaneous HCV clearance and/or response to IFN-based therapies (5–9).
During viral infections, pattern recognition receptors such as the retinoic acid-inducible gene-I (RIG-I)-like receptors (RIG-I and MDA5) and Toll-like receptors (TLRs) are innate immune sensors that detect viral components as non-self materials and initiate signaling cascades, leading to activation of the latent transcription factors, IRF3 and NF-κB, which culminates in expression of IFNs, ISGs, cytokines, and chemokines (3, 4). As the major target cell for HCV, the hepatocyte can sense viral RNAs through two parallel pathways, i.e. the RIG-I-like receptors (RIG-I and MDA5) and TLR3 (10). HCV replicates its genome in cytoplasm as a membrane-bound replication complex, producing RNAs bearing 5′-triphosphates and dsRNA intermediates that activate these pattern recognition receptors (3, 4). Although the details of RIG-I signaling in response to 5′-triphosphate-bearing HCV RNAs are relatively well characterized (4), how the TLR3 pathway operates in hepatocytes to detect HCV dsRNAs and activate antiviral responses is poorly understood. In this study, we employed biochemical, proteomics, and loss-of-function approaches to characterize the subcellular compartment of TLR3 and cellular proteins enriched in it after HCV infection. These efforts led to the identification of a molecular chaperone, heat shock 70-kDa protein-5 (glucose-regulated protein, 78 kDa) (GRP78, also known as HSPA5/BiP) as an essential factor controlling hepatocellular IRF3-dependent innate immune responses downstream of TLR3. We found that GRP78 co-fractionated with TLR3 in endolysosomes, and GRP78 expression was up-regulated in HCV-infected hepatoma cells and in liver biopsies of chronic hepatitis C (CHC) patients. Remarkably, hepatic GRP78 expression correlated with expression of RANTES and CXCL10, chemokines that are frequently elevated in HCV-infected livers and whose induction by HCV is largely dependent on TLR3 signaling (11).
Experimental Procedures
Cells, Viruses, and Poly(I:C)
Huh7.5, Huh7.5, and Huh7 cells stably expressing TLR3 (referred to as 7.5-TLR3 and Huh7-TLR3) and PH5CH8 cells were cultured as described (10–12). Primary mouse hepatocytes were isolated and cultured according to a published procedure (13). The JFH1 strain cell culture-derived HCV (14) was propagated in Huh7.5 cells, and infectious titers were determined by a TCID50 assay following immunostaining of NS5A, as described previously (15). A recombinant VSV encoding firefly luciferase (16) (VSV-Luc, provided by Sean Whelan) was propagated in Vero cells. Poly(I:C) and Sendai virus (SeV, Cantell strain) were purchased from Sigma and Charles River Laboratories, respectively.
Plasmids
DNA vectors encoding FLAG-IKKϵ, FLAG-TRIFSA (a super-active TRIF mutant that does not induce apoptosis), and GFP-IRF35D (a constitutive, phospho-mimetic IRF3 mutant) were provided by Kate Fitzgerald, Margaret Offermann, and Nancy Reich, respectively. pDR2.1-FLAG-MAVS, which expresses human MAVS attached to an N-terminal FLAG tag under control of the CMV promoter in the pDream2.1/LIC backbone, was synthesized at GenScript. pEF6-GRP78-V5 containing full-length human GRP78 coding sequence fused to C-terminal V5-His6 epitope tags was constructed by conventional PCR cloning techniques.
Sucrose Gradient Separation
After three PBS washes, cells were scraped from culture vessels and incubated in 5 times of packed cell volume of hypotonic buffer (10 mm Tris-HCl, 10 mm KCl, 5 mm MgCl2, pH 7.5) on ice for 15 min. After centrifugation at 2000 × g for 5 min at 4 °C, cell pellets were resuspended in 2 ml of hypotonic buffer supplemented with protease inhibitors and homogenized in a Dounce homogenizer with tight pestle (∼100 strokes). Following centrifugation at 1100 × g for 8 min at 4 °C, the postnuclear supernatant was collected and incubated on ice for 30 min after addition of Triton X-100 to 1% of final volume. Two milliliters of each sample were layered on top of 8 ml of 10–40% sucrose gradient in TNE buffer (25 mm Tris-HCl, pH 7.5, 150 mm NaCl, 1 mm EDTA) and centrifuged at 40,000 × g for 16 h at 4 °C. One-milliliter fractions were collected from top to bottom of the tube, precipitated with TCA, and subjected to further analyses.
Two-dimensional Gel Electrophoresis Analysis and Protein Identification
The first-dimensional electrophoresis of each protein sample (∼300 μg) was conducted on immobilized pH gradient gel (IPG) strip (pH 3–10, 13-cm) on IPGphor-3 (GE Healthcare). The IPG strip, rehydrated at 50 V for 12 h at 20 °C, was focused using a three-step program (500 V for 1 h with rapid ramping, 1000 V for 1 h with rapid ramping, and 8000 V for 3 h with rapid ramping until 16,000–20,000 V were reached). Subsequently, the IPG strip was incubated in equilibration buffer (6 m urea, 50 mm Tris-HCl, 2% SDS, 30% glycerol, 0.002% bromphenol blue, pH 8.8) containing 1% DTT for 15 min before being washed for an additional 15 min in equilibration buffer containing 2.5% iodoacetamide (Sigma). The second-dimensional separation was carried out on a 12% SDS-PAGE at 100 V for 30 min and then at 200 V until the dye front reached the bottom of the gel. After electrophoresis, gels were stained by Coomassie Blue R-250 and scanned using ImageScanner III (GE Healthcare). Spot detection, spot matching, and quantification analysis were performed. Two-dimensional gel analysis was repeated three times for independent biological replicates. The gel images were normalized according to the total quantity in the analysis set. Student's t test was performed to define the significantly differential spots between HCV-infected and mock-infected groups. Protein spots of interest were excised from gels and subjected to in-gel tryptic digestion, followed by protein identification by mass spectrometry.
RNA Interference
For stable knockdown of GRP78, cells were transduced with a pLKO.1-based lentiviral vector carrying a GRP78-specific shRNA (target sequence, CTTGTTGGTGGCTCGACTCGA), followed by selection in puromycin-containing medium. For transient knockdown of GRP78, we transfected cells with a synthetic GRP78 siRNA (17) (target sequence, GGAGCGCAUUGAUACUAGA) using Lipofectamine 2000 (Invitrogen). siRNAs targeting ALAD and NIF3L1 were obtained from Santa Cruz Biotechnology, and those targeting IRE1, ATF6, and PERK were purchased from Invitrogen. For comparison, a non-targeting negative control siRNA (Ambion) was used in parallel.
RNA Analyses
The expression of RANTES, CXCL10, GRP78, GRP94, HSP60, ISG56, and HCV RNA was analyzed by quantitative reverse transcription-PCR conducted on an iCycler IQ5 real time PCR system (Bio-Rad) using total RNA samples and gene-specific primers (Table 1) and GoTaq qPCR Master Mix (Promega). Post RT-PCR, the relative abundance of each cellular target was normalized to that of 28S rRNA or β-actin by comparing the threshold crossing (Ct) values between each sample and an indicated control. Copy numbers for HCV RNA were calculated by comparing the Ct values of samples with those of a standard curve generated using JFH1 cDNA standards and expressed as copies per μg of total RNA.
TABLE 1.
qPCR primers used in this study
Note: qPCR primers for quantifying transcripts for RANTES, CXCL10, ISG56, 28S, and HCV RNA have been described previously (11, 49, 50).
| Target | Sequence (5′ to 3′) | |
|---|---|---|
| GRP78 | Forward | GGCCGCACGTGGAATG |
| Reverse | AACCACCTTGAACGGCAAGA | |
| GRP94 | Forward | TTCTTTTTGGGAGAGACTTGTTTTG |
| Reverse | TGACCCATAATCCCACATTTTACA | |
| HSP60 | Forward | TTCAGGTTGTGGCAGTCAAG |
| Reverse | CTTCTCCAAACACTGCACCA | |
| IRE1 | Forward | TCAACGCTGGATGGAAGTTTGC |
| Reverse | GGCTGCCATCATTAGGATCTGG | |
| ATF6 | Forward | TCAACTTTTGTGAGCGGGGA |
| Reverse | AATCTAAGGGGAGTGCTGCG | |
| PERK | Forward | AGTCCCCCTGTGTGGTAACT |
| Reverse | TTTACCCGCCAGGGACAAAA | |
| ACTB | Forward | CATGTACGTTGCTATCCAGGC |
| Reverse | CTCCTTAATGTCACGCACGAT |
Protein Analyses
Protein samples were fractionated on SDS-PAGE via conventional procedures and transferred to nitrocellulose membranes. After incubation in 3% nonfat milk in PBS to block nonspecific binding sites, membranes were blotted with the following monoclonal (mAb) and polyclonal (pAb) antibodies: rabbit anti-GRP78 mAb (Cell Signaling Technology) and pAb (Santa Cruz Biotechnology); HSP/chaperone sampler kit (Cell Signaling Technology); rabbit anti-EEA1 pAb (Cell Signaling Technology); rabbit anti-calreticulin pAb (Sigma); mouse anti-LAMP2 mAb (Developmental Studies Hybridoma Bank); mouse anti-FLAG M2 and anti-actin mAbs (Sigma); mouse anti-HCV NS3 mAb (Vector Laboratories); rabbit anti-IRF3 pAb (a gift from Michael David), anti-phospho-IRF3-Ser-396 mAb (Cell Signaling Technology), and anti-phospho-IRF3-Ser-398 mAb (Millipore); mouse anti-OxPhos complex I 39-kDa subunit (CI-39) mAb (Invitrogen); rabbit anti-ISG56 and TLR3 pAbs (12); rabbit anti-ISG15 pAb (a gift from Arthur Haas); and peroxidase-conjugated anti-rabbit and anti-mouse secondary pAbs (Southern Biotech). Protein targets were visualized by enhanced chemiluminescence (EMD Millipore), followed by exposure to x-ray films. Immunofluorescence staining and confocal fluorescence microscopy were performed as described (11, 12).
Patient Samples
Liver biopsies were obtained from 26 patients with CHC prior to initiation of the PEG-IFN/RBV combination therapy at the Department of Infectious Diseases, Henan Provincial People's Hospital in Zhengzhou, China. Inclusion was restricted to patients with CHC (duration of liver disease >6 months) and absence of acute flares as defined by acute alanine aminotransferase elevation >5 times the upper limit of the patient's baseline level. Patients with the following criteria were excluded: retreatment; co-infection with other viruses such as hepatitis B virus or HIV; decompensated liver disease; poorly controlled diabetes; autoimmune or immunologically mediated disease; angiocardiopathy; chronic nephrosis, and organ transplantation. Of the 20 CHC patients whose HCV genotype information was available, 15 were infected with genotype 1b virus, and five with genotype 2a virus. For the control group, liver tissues were collected from 22 patients who underwent surgical resection for benign hepatic tumors or hepatic hemangioma at the Department of Hepatobiliary Surgery, Henan Provincial People's Hospital. Patients with the following criteria were excluded: infection with hepatitis viruses, such as HBV and HCV; decompensated liver disease; liver cirrhosis; steatohepatitis; autoimmune or immunologically mediated disease; and organ transplantation. Details of the patient characteristics are shown in Table 2.
TABLE 2.
Baseline characteristics of patients enrolled in this study
For categorical data, the number of patients in each category is shown. For continuous data, the means ± S.D. are displayed. ALT, alanine aminotransferase; AST, aspartate aminotransferase; ALB, albumin; TBiL, total bilirubin; ALP, alkaline phosphatase; GGT, γ-glutamyl transpeptidase; WBC, white blood cells; HGB, hemoglobin; PLT, platelets; AFP, α-fetal protein.
| Characters | CHC (n = 26) | Control (n = 22) | t/χ2 | p value |
|---|---|---|---|---|
| Gender (male/female)a | 14/12 | 13/9 | 0.133 | 0.776 |
| Age (years)b | 43.23 ± 13.94 | 50.36 ± 14.34 | 1.745 | 0.088 |
| ALT (units/ml)b | 93.88 ± 88.85 | 87.55 ± 106.22 | −0.225 | 0.823 |
| AST (units/ml)b | 54.12 ± 39.17 | 61.23 ± 80.62 | 0.398 | 0.692 |
| ALB (g/liter)b | 42.43 ± 7.18 | 39.79 ± 4.03 | −1.531 | 0.133 |
| TBiL (μmol/liter)b | 13.95 ± 5.08 | 22 ± 25.46 | 1.458 | 0.159 |
| ALP (units/liter) | 86.8 ± 33.95 | 141.73 ± 118.21 | 2.104 | 0.046 |
| GGT (units/liter) | 53.4 ± 49.76 | 120.58 ± 183.87 | 1.661 | 0.110 |
| AFP | 3.85 ± 1.98 | 3.87 ± 5.04 | 0.018 | 0.985 |
| WBC (g/liter)b | 6.36 ± 2.00 | 7.63 ± 3.87 | 1.401 | 0.171 |
| HGB (g/liter)b | 143.88 ± 15.36 | 138.82 ± 15.09 | −1.148 | 0.257 |
| PLT (g/liter)b | 190.08 ± 70.88 | 214.7 ± 76.72 | 1.155 | 0.254 |
a χ2 test.
b Two-sample t test.
This study was performed in accordance with the ethical standards of the Declaration of Helsinki and approved by the Ethical Committee of Henan Provincial People's Hospital. All subjects gave written, informed consent.
Statistical Analyses
Where indicated, normalized expression levels of cellular genes or HCV RNAs were compared between groups using two-sided Student's t tests. Correlation between biomarkers was examined by calculating Pearson's correlation coefficients. A p value of <0.05 was considered statistically significant.
Results
TLR3 Localizes to Endolysosomes of Cultured Human Hepatocytes
We have previously shown that extracellular poly(I:C)-induced antiviral gene expression via the TLR3 pathway in 7.5-TLR3 and PH5CH8 cells was abrogated by bafilomycin A1, an inhibitor of endosomal acidification (11), indicating that TLR3 signaling in hepatocytes is initiated intracellularly in conjunction with maturation of endosomal compartments. Previous studies in non-hepatic cells suggested that TLR3 localizes in the endoplasmic reticulum (ER) of quiescent cells and relocates to dsRNA-containing endosomes upon stimulation (18). To better understand how this pathway operates in hepatocytes, it was necessary to elucidate the subcellular distribution of TLR3 in this cell type. Sucrose density gradient centrifugation was used to separate the cytoplasm of 7.5-TLR3 cells into 10 fractions (Fig. 1A, middle panels, fractions 2–10 are shown). Although the top two fractions comprised almost exclusively lipids that were easily visible after centrifugation, the remaining eight fractions were enriched for different cellular organelles as detected by organelle-specific markers (calreticulin for ER, EEA1 for early endosomes, LAMP2 for late endosomes/lysosomes, and CI-39 for mitochondria, respectively). Immunoblotting of the cellular fractions revealed that TLR3 co-localized with LAMP2 in fractions 5–8, although it co-localized with EEA1 to a limited extent in fraction 5. Confocal fluorescence microscopy of immunostained 7.5-TLR3 cells corroborated the co-localization of TLR3 and LAMP2 (Fig. 1B, upper panels). In comparison, TLR3 only partially co-localized with calreticulin (Fig. 1C), an ER marker. The TLR3 immunostaining was specific, because no such immunolabeling was observed in Huh7.5 cells (Fig. 1D). Endogenous TLR3 also co-localized with LAMP2 in primary mouse hepatocytes (Fig. 1E). We did not observe an obvious change in TLR3 expression or its localization in 7.5-TLR3 cells following infection with HCV (data not shown). These data suggest that TLR3 resides in endolysosomes, particularly in the late endosomes/lysosomes of human and mouse hepatocytes.
FIGURE 1.

Endolysosomal localization of TLR3 in cultured hepatocytes. A, immunoblotting of indicated organelle markers, TLR3 and GRP78, in unfractionated cytoplasm of Huh7.5 cells, 7.5-TLR3 cells, and 7.5-TLR3-derived shG78-3 cells with stable knockdown of GRP78 (left), and in sucrose density gradient centrifugation-separated fractions of 7.5-TLR3 cells (middle), and shG78-3 cells (right). B, confocal fluorescence microscopy of immunostained 7.5-TLR3 cells (upper panels) and shG78-13 cells with stable GRP78 knockdown (lower panels) demonstrating co-localization of TLR3 (green fluorescence) and LAMP2 (red fluorescence). Nuclei were counterstained with DAPI (blue fluorescence). C, confocal fluorescence microscopy illustrating partial co-localization of TLR3 (red fluorescence) and calreticulin (green fluorescence) in immunostained 7.5-TLR3 cells. D, no TLR3 immunolabeling was detected in Huh7.5 cells. E, confocal fluorescence microscopy showing co-localization of TLR3 (green fluorescence) and LAMP2 (red fluorescence) in primary mouse hepatocytes. Nuclei were counterstained with DAPI (blue fluorescence).
Identification of Differentially Expressed Cellular Proteins in Endolysosome Fractions of Huh7.5-TLR3 Cells Following HCV Infection
We hypothesized that upon engagement, TLR3 assembles signaling complexes in association with endolysosomal membranes, and therefore, endolysosome fractions are enriched for proteins participating in TLR3 signaling, which could then be identified by proteomics approaches. Therefore, specifically examining the endolysosome fractions as opposed to whole cell lysates would increase the likelihood of isolating TLR3 signaling proteins or their regulators, which are selectively concentrated in these fractions thus allowing higher sensitivity of detection. The endolysosome fractions were isolated from mock-infected 7.5-TLR3 cells and cells infected with HCV, concentrated by TCA precipitation, and then subjected to two-dimensional gel electrophoresis analysis (Fig. 2A, compare left and right panels). A total of 16 protein spots (13 up-regulated and 3 down-regulated) were found to be differentially expressed by ≥2-fold in three independent experiments. These spots were excised from the gels, and their identities were determined by mass spectrometry (Fig. 2A, see also Table 3 for detailed information). Of the 15 spots (except spot 10) for which high probability hits have been found in the human protein database, nine spots comprised proteins encoding various enzymatic activities (up-regulated, spots 5–9, 12, and 13; down-regulated, spots 14 and 15), three spots were molecular chaperones that were up-regulated by HCV, i.e. GRP94 (spot 1), GRP78 (spot 3), and HSP60 (spot 4), and two spots were proteins involved in translational control (spot 2, EF2, up-regulated; and spot 16, EIF3G, down-regulated). Interestingly, several spots (especially spots 11 and 12) were strongly expressed in endolysosome fractions of infected cells, but their expression was undetectable in mock-infected cells.
FIGURE 2.

Identification of cellular proteins differentially expressed in endolysosomal fractions of 7.5-TLR3 cells following HCV infection. A, two-dimensional gel analysis of differentially expressed proteins in endolysosome fractions (fractions 5–7) between mock-infected and JFH1-infected (m.o.i. = 0.5) 7.5-TLR3 cells at 48 hours post-infection. Spots in red and blue circles were up-regulated and down-regulated proteins, respectively. Their identities, as determined by mass spectrometry, are listed on the right. Notably, for spots 11 and 12, two to three high-probability hits were identified. Proteins marked by asterisks in spots 11 and 12 were likely degradation/cleavage products from their full-length counterparts based on molecular weight. B, immunoblotting of GRP78, HSP60, HSP40, and LAMP2 proteins in various sucrose gradient centrifugation-separated fractions of 7.5-TLR3 cells mock-infected or infected with HCV (JFH1 virus at m.o.i. = 0.25, 48 h.p.i.). A representative of three independent experiments is shown.
TABLE 3.
List of proteins differentially expressed in endolysosome fractions of 7.5-TLR3 cells following HCV infection
Note: Proteins marked by asterisks in spots 11 and 12 were likely degradation/cleavage products from their full-length counterparts based on their known molecular mass.
| Spot no. | Accession no. | Protein name (gene name) |
|---|---|---|
| Up-regulated | ||
| 1 | P14625 | 94-kDa glucose-regulated protein (GRP94/HSP90B1/gp96) |
| 2 | P13639 | Elongation factor2 (EEF2/EF2) |
| 3 | P11021 | 78-kDa glucose-regulated protein (GRP78/HSPA5/BiP) |
| 4 | P10809 | 60-kDa heat shock protein, mitochondrial(HSP60/HSPD1) |
| 5 | P04040 | Catalase (CAT) |
| 6 | P00367 | Glutamate dehydrogenase 1, mitochondrial (GDH1/GLUD1) |
| 7 | P06733 | α-Enolase (ENO1) |
| 8 | P07954 | Fumarate hydratase, mitochondrial (FH) |
| 9 | P22234 | Multifunctional protein ADE2 (ADE2/PAICS) |
| 10 | unknown | Unknown |
| 11 | Q8IZQ1 | WD repeat and FYVE domain-containing protein3(WDFY3/ALFY)* |
| Q9GZT8 | NIF3-like protein1 (NIF3L1/ALS2CR1) | |
| Q15417 | Calponin-3 (CNN3) | |
| 12 | O00754 | Lysosomal α-mannosidase (LAMAN/MAN2B1)* |
| P13716 | δ-Aminolevulinic acid dehydratase (ALAD/ALADH) | |
| 13 | P00338 | l-Lactate dehydrogenase A chain (LDHA/LDH1) |
| Down-regulated | ||
| 14 | Q9Y265 | RuvB-like1 (RUVBL1/TIP49) |
| 15 | P15104 | Glutamine synthetase (GLUL/GLNS) |
| 16 | O75821 | Eukaryotic translation initiation factor 3 subunitG(EIF3G/EIF3S4) |
To confirm the results from two-dimensional gel analysis, fractionated mock-infected and HCV-infected 7.5-TLR3 cells were separated on SDS-PAGE and examined for the abundance of GRP78 and HSP60 by immunoblotting (Fig. 2B). Both chaperones had increased abundance in the four LAMP2-containing fractions (i.e. endolysosomes) of HCV-infected cells as compared with uninfected cells. In contrast, the distribution pattern of another chaperone, HSP40, was unchanged by HCV infection.
Expression of GRP78 Was Up-regulated in HCV-infected Hepatoma Cells
Several molecular chaperones, as exemplified by UNC93B1 and GRP94, have been reported to regulate TLR trafficking or folding (19, 20). We thus turned our attention to the chaperone proteins and determined how their expression was modulated during HCV infection. Using a HSP/chaperone antibody panel, we immunoblotted whole cell lysates of Huh7.5 cells and 7.5-TLR3 cells for various chaperones at different times post-HCV infection (Fig. 3A). The expression levels of HSP40, HSP60, HSP90, and calnexin were unaffected by HCV infection. Thus, the up-regulation of HSP60 protein level in endolysosomes fractions of HCV-infected 7.5-TLR3 cells (Fig. 2) resulted from heightened transport to this compartment. In contrast, total GRP78 protein abundance increased after HCV infection in both Huh7.5 and 7.5-TLR3 cells, although the up-regulation was moderate in the former and significantly more pronounced in the latter (Fig. 3A, GRP78 panel). qPCR analyses (Fig. 3B) showed that expression of GRP78 mRNA in Huh7.5 cells did not change during the first 2 days post-HCV infection but significantly decreased at day 3. In contrast, GRP78 transcript was up-regulated by ∼2-fold in 7.5-TLR3 cells at 24 h post-infection and then subsided at later time points, but it still remained higher than that of Huh7.5 cells at 72 h post-infection. Time course analysis of uninfected cells placed in culture did not show an increase in GRP78 mRNA expression throughout a 5-day period (Fig. 3C). The expression pattern of GRP94 mRNA mirrored that of GRP78 (Fig. 3B), which was not unexpected given that the two genes share a common transcriptional regulation mechanism. Together with the immunoblotting data (Fig. 3A), we infer that the moderate increase in GRP78 protein level in HCV-infected Huh7.5 cells is a result of post-transcriptional modulation, perhaps through increased mRNA stability and/or enhanced translation. As indirect evidence, HSP60 protein abundance remained unchanged after HCV infection (Fig. 3A) despite a decline in its mRNA levels (Fig. 3B). However, in HCV-infected 7.5-TLR3 cells, HCV up-regulates GRP78 expression through a combined effect of post-transcriptional up-regulation and increased transcription. This notion is supported by the observations that the increase in GRP78 protein expression far exceeded its mRNA up-regulation, and that GRP78 protein abundance continued to increase at 48–72 h when its mRNA levels already decreased to levels lower than that of uninfected cells (Fig. 3, A and B). Along this line, it has been reported that the HCV replicon up-regulates the GRP78 promoter by activating the ATF6 branch of unfolded protein response (UPR) and promotes internal ribosome entry site activity, including the one residing in and dictating the translation of GRP78 mRNA (21, 22).
FIGURE 3.

Expression of molecular chaperones in hepatoma cells during HCV infection. A, immunoblotting of indicated chaperones, HCV NS3 and actin in Huh7.5 and 7.5-TLR3 cells at indicated times post-infection with JFH1 virus (m.o.i. = 0.25). Values below the GRP78 panel indicate fold changes relative to the GRP78 protein level at 0 h.p.i. after normalization of actin levels as determined by densitometry analysis using ImageJ software. B, qPCR analysis of GRP78, GRP94, and HSP60 mRNA levels in Huh7.5 and 7.5-TLR3 cells at indicated times post-infection with JFH1 virus (m.o.i. = 1). * and ** indicate statistical differences exist as compared with samples at 0 h.p.i., with a p value of <0.05 and <0.01, respectively. C, qPCR analysis of GRP78 and HSP60 mRNA levels in Huh7.5 and 7.5-TLR3 cells cultured for the indicated days. A representative of two independent experiments is shown. Error bars represent standard deviations of replicate samples.
Knockdown of GRP78 Decreases Induction of ISGs and Chemokines by HCV or Extracellular dsRNA in Huh7.5-TLR3 Cells
Recent evidence has suggested that chaperones may regulate innate immunity. Although not impacting TLR3, GRP94 facilitates the folding and function of multiple TLRs (20). HSP90 and HSP60 were recently reported to regulate IRF3 activation downstream of RIG-I (23, 24). Of the three chaperones of interest (GRP94, GRP78, and HSP60) from our two-dimensional gel analysis, we investigated whether GRP78 has any role in TLR3 signaling in response to HCV infection by siRNA-mediated loss-of-function analysis. We also explored the impact of NIF3L1 and ALAD, two proteins identified for spots 11 and 12, respectively. The results showed that knockdown of GRP78 (see below) or ALAD (data not shown) impaired HCV-induced antiviral gene expression, whereas NIF3L1 silencing had no demonstrable effect (data not shown). In this study, we focused on GRP78. To determine how GRP78 impacts HCV-induced innate immune responses, we infected Huh7.5 and Huh7.5-TLR3 cells with HCV in parallel, with or without transient transfection of GRP78 siRNA/control siRNA, followed by examining ISG15 protein expression by immunoblotting (Fig. 4A) or induction of RANTES transcript by qPCR (Fig. 4B) at 3 d.p.i. In agreement with our previous findings (11, 12), HCV infection up-regulated ISG15 and RANTES expression in a TLR3-dependent manner. GRP78 knockdown, but not control siRNA, reduced HCV-induced RANTES expression by 70% (Fig. 4B) and blunted the moderate up-regulation of ISG15 (Fig. 4A) in 7.5-TLR3 cells. GRP78 knockdown also substantially decreased expression of ISG56 and RANTES mRNAs following stimulation by poly(I:C) (pIC, see below in Figs. 5 and 7), demonstrating that the impact of GRP78 depletion on innate immunity is general to TLR3 engagement and not specific for HCV. Notably, GRP78 silencing reproducibly led to up-regulation of GRP94, regardless of cell type (Figs. 5A and 7, see below), suggesting that it is a cellular mechanism to compensate for the loss of GRP78. In accordance, GRP94 knockdown was found to be accompanied by increased GRP78 expression in Huh7.5 cells (25). Nonetheless, GRP94 up-regulation did not reverse the impairment of TLR3 responses resulting from GRP78 loss. To exclude that our finding reflected off-target effects of one particular siRNA, we stably transduced 7.5-TLR3 cells with a lentiviral shRNA targeting GRP78 mRNA at a region distinct from the GRP78 siRNA and selected two cell clones, shG78-3 and shG78-13, in which GRP78 protein was reduced by 70–80% compared with 7.5-TLR3 cells (Fig. 4C), for analysis. In both clones, GRP78 depletion substantially decreased the induction of ISG56 (Fig. 4C), RANTES (Fig. 4E), and CXCL10 (Fig. 4F) by HCV or poly(I:C). Importantly, neither GRP78 siRNA (Fig. 4A) nor GRP78 shRNA (Fig. 4C) compromised HCV protein expression. Of note, intracellular HCV RNA levels were significantly higher in shG78-3 and shG78-13 cells than in control 7.5-TLR3 cells at 3 d.p.i. (Fig. 4D), which was consistent with decreased ISG expression following GRP78 silencing.
FIGURE 4.

Knockdown of GRP78 impairs TLR3-dependent induction of innate immune responses in 7.5-TLR3 cells. A, immunoblotting of ISG15, GRP78, NS3, and actin in Huh7.5 and 7.5-TLR3 cells, mock/JFH1-infected for 24 h (m.o.i. = 0.1), followed by transfection of control siRNA or GRP78 siRNA for an additional 48 h. Values below the ISG15 panel indicate fold changes relative to the ISG15 protein level in mock-infected cells after normalization of actin levels. B, RANTES expression, as determined by qPCR, under conditions of A. A representative of three independent experiments is shown. Error bars represent standard deviations of replicate samples. ** indicates statistical difference exists between control and GRP78-siRNA-transfected cells, with a p value of <0.01. C, immunoblotting of ISG56, NS3, GRP78, and actin in control 7.5-TLR3 cells and cells stably expressing a GRP78 shRNA (shG78-3 and shG78-13 cells) that were mock-treated and stimulated by poly(I:C) (20 μg/ml for 16 h) or infected with JFH1 virus for 72 h. D–F, qPCR analysis of HCV RNA, RANTES, and CXCL10 levels in 7.5-TLR3 cells, shG78-3, and shG78-13 cells that were mock-treated, stimulated by poly(I:C), or infected with HCV for the indicated days. A representative of two independent experiments is shown. * and ** indicate statistical differences exist as compared with 7.5-TLR3 samples, with a p value of <0.05 and <0.01, respectively.
FIGURE 5.

GRP78 knockdown compromises TLR3-dependent expression of ISG/chemokine in non-neoplastic hepatocytes. A, PH5CH8 cells tranfected with control or GRP78 siRNA were mock-treated, infected with SeV (160 hemagglutination units/ml), or stimulated by poly(I:C) for 7 h. Expression of GRP78, GRP94, ISG56, and RANTES were determined by qPCR. B, qPCR analysis of expression of GRP78, ISG56, and RANTES in control PH5CH8 cells and PH5CH8 pools stably expressing GRP78 shRNA (shG78 cells) that were mock-treated, stimulated by poly(I:C), or infected by SeV. A representative of three independent experiments is shown. Error bars represent standard deviations of replicate samples. * and ** indicate statistical differences exist between control and GRP78-siRNA-transfected cells (or between control and shG78 cells), with a p value of <0.05 and <0.01, respectively.
FIGURE 7.
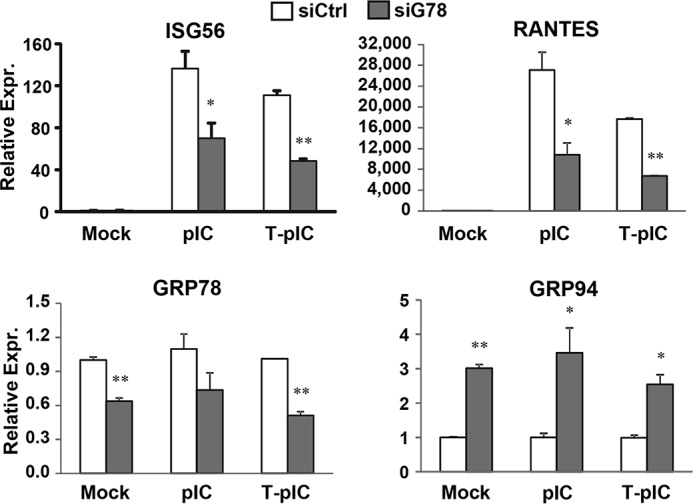
Effects of transient GRP78 knockdown on gene induction by extracellular pIC or poly(I:C) delivered via transfection (T-pIC). 7.5-TLR3 cells were transfected with negative control siRNA or GRP78 siRNA for 48 h, then mock-treated, stimulated by poly(I:C) added to culture medium (pIC, 20 μg/ml), or transfected with poly(I:C) by Lipofectamine (T-pIC, 2 μg/well) for 7 h. ISG56, RANTES, GRP78, and GRP94 mRNA levels were quantified by qPCR. A representative of three independent experiments is shown. Error bars represent standard deviations of replicate samples.* and ** indicate statistical differences exist between control and GRP78-siRNA-transfected cells, with a p value of <0.05 and <0.01, respectively.
To investigate whether overexpression of GRP78 promotes antiviral signaling, we ectopically expressed GRP78 or a control vector in 7.5-TLR3 cells, and we determined by immunoblotting the expression status of ISG15 and ISG56 proteins with and without TLR3 activation by poly(I:C). Overexpression of GRP78 per se did not induce either ISG, nor did it have a demonstrable effect on ISG15 and ISG56 induction following poly(I:C) stimulation (data not shown). Similar results were obtained in HEK293-TLR3 cells when we examined activation of the IFN-β promoter by reporter gene assay (data not shown). Thus, GRP78 overexpression alone is not sufficient to activate antiviral signaling. Given that GRP78 is already abundantly expressed in resting cells, it was not surprising that ectopic overexpression of this protein did not further enhance TLR3 signaling activated by dsRNA.
GRP78 Depletion in Non-neoplastic Human Hepatocytes Impairs Expression of Antiviral Genes via the TLR3 Pathway
Because we found that GRP78 is indispensable for the induction of TLR3-dependent innate immune genes in hepatoma cells, we determined whether this was also the case in PH5CH8 cells, non-neoplastic hepatocytes that mount a robust antiviral response to dsRNA through endogenously expressed TLR3 (10). Transfection of GRP78 siRNA reproducibly curtailed poly(I:C)-induced ISG56 and RANTES mRNAs by 60–70%, which was proportional to the reduction in GRP78 mRNA levels (Fig. 5A). Again, this was observed despite that GRP94 expression was up-regulated by 5–6-fold following GRP78 knockdown. In contrast, SeV-induced expression of antiviral genes was unaffected, implying that GRP78 is dispensable for innate immune responses downstream of RIG-I (Fig. 5A). We obtained similar results when we compared pools of PH5CH8 cells stably transduced with GRP78 shRNA (referred to as PH5shG78 cells) and control PH5CH8 cells (Fig. 5B and data not shown).
GRP78 Knockdown Undermines TLR3-mediated Establishment of an Antiviral State
TLR3 senses viral dsRNAs produced during HCV RNA replication (11), inducing a weak interferon response that inhibits HCV replication in cultured hepatoma cells (12). Our earlier data showed that HCV RNA replicated to significantly higher levels at 3 d.p.i. in shG78-3 and shG78-13 cells than in control 7.5-TLR3 cells (Fig. 4D), suggesting GRP78 depletion compromised TLR3-mediated establishment of an antiviral state against HCV. To corroborate this finding, we transiently transfected Huh7-TLR3 cells with control or GRP78 siRNA, respectively, and monitored the effect on viral RNA replication following HCV infection. It was observed that GRP78 knockdown moderately, yet significantly, increased intracellular HCV RNA levels (Fig. 6A). Next, we assessed whether GRP78 contributes to TLR3-mediated antiviral defense in non-neoplastic hepatocytes by comparing PH5shG78 cells and control PH5CH8 cells. Because PH5CH8 cells are not permissive for HCV infection, we stimulated the cells with extracellular poly(I:C) to engage the TLR3 pathway, and determined the antiviral effects on replication of VSV-Luc, a recombinant VSV encoding firefly luciferase whose replication can be monitored conveniently by luciferase activity assay in lysed cells (Fig. 6B). Despite profound loss of GRP78 protein in PH5shG78 cells compared with control PH5CH8 cells (Fig. 8B, GRP78 panel, compare lanes 2 versus 1), VSV-Luc replication was comparable, regardless of the m.o.i. (Fig. 6B, compare hatched bars with empty bars). Thus, GRP78 per se is dispensable for VSV replication. Prior stimulation by extracellular poly(I:C) established a potent antiviral state, reducing VSV-Luc-encoded luciferase activities by >2–4 logs in control PH5CH8 cells (compare solid bars versus empty bars), with cultures infected at lower multiplicities of infection exhibiting greater antiviral effects. Although poly(I:C) also conferred antiviral protection in PH5shG78 cells (Fig. 6B, compare dotted bars versus hatched bars), it was much less effective than in control PH5CH8 cells. Specifically, the antiviral effect of poly(I:C) was between 11- and 22-fold lower in PH5shG78 cells at all multiplicities of infection tested. Collectively, a pivotal role for GRP78 in TLR3-mediated antiviral defense was established not only in hepatoma cells reconstituted for TLR3 expression but also in non-neoplastic human hepatocytes that harbor the TLR3 pathway physiologically.
FIGURE 6.
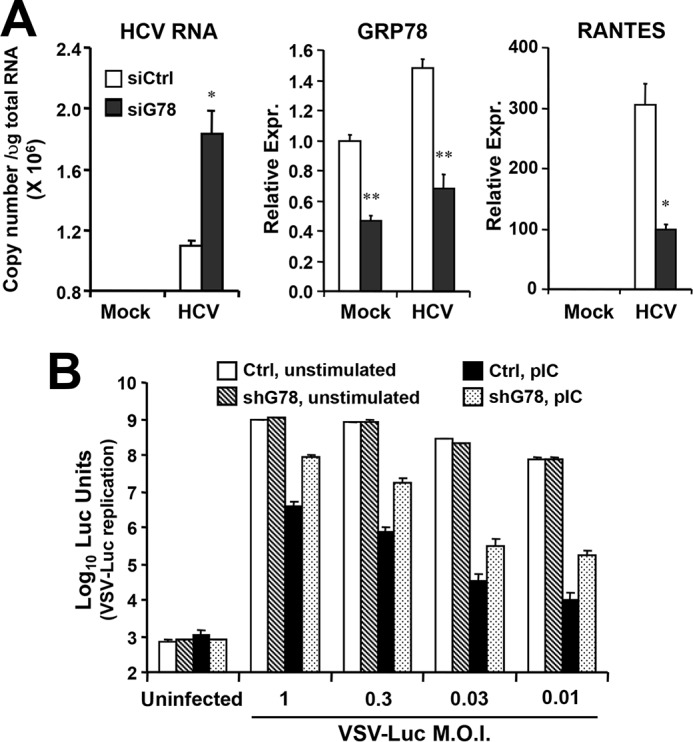
GRP78 knockdown impairs TLR3-mediated establishment of an antiviral state in hepatocytes. A, Huh7-TLR3 cells transiently transfected with control or GRP78 siRNA were mock-infected or infected by JFH1 virus for 72 h (m.o.i. = 0.02), followed by qPCR analysis of intracellular HCV RNA, GRP78, and RANTES levels. * and ** indicate statistical differences exist between control and GRP78-siRNA-transfected cells, with a p value of <0.05 and <0.01, respectively. B, control PH5CH8 and PH5shG78 cells were mock-stimulated or stimulated by poly(I:C) for 10 h, then infected with VSV-Luc at indicated multiplicities of infection for 14 h, followed by cell lysis and luciferase assay. A representative of two independent experiments is shown. Error bars represent standard deviations of replicate samples.
FIGURE 8.

GRP78 is required for sustained IRF3 phosphorylation downstream of TLR3 signaling. A, immunoblotting of ISG15 expression following ectopic expression of indicated signaling proteins in control 7.5-TLR3, shG78-3, and shG78-13 cells. Values below the ISG15 panels indicate fold changes relative to the ISG15 level in 7.5TLR3 samples after normalization of actin abundance. B, immunoblotting of p-IRF3 Ser-396, total IRF3, ISG56, GRP78, and actin in control PH5CH8 and PH5shG78 cells at indicated times post poly(I:C) stimulation. Values below the p-IRF3 panel at each time point indicate fold changes relative to the p-IRF3 abundance in control PH5CH8 cells. A representative of three independent experiments is shown.
The Contribution of GRP78 to TLR3-mediated Antiviral Response Is Not through Facilitation of TLR3 Trafficking to Endolysosomes or Uptake of Extracellular dsRNA
The endosomal TLRs, including TLR3, are required to traffic from ER to endolysosomes for signaling, a process aided by certain chaperones, one such being UNC93B1 (19). We wondered whether GRP78 adopted a similar mechanism to facilitate TLR3 transport. However, subcellular fractionation experiments revealed that the distribution of TLR3 to endolysosomes was similar between 7.5-TLR3 cells and shG78-3 cells with stable GRP78 knockdown (Fig. 1A, compare fractions 5–8 between middle and right panels). In addition, confocal fluorescence microscopy demonstrated that the co-localization of TLR3 and LAMP2 was not different between 7.5-TLR3 cells and shG78-13 cells (Fig. 1B). Apart from residing in the ER and endosomal compartments, GRP78 can traffic to the cell surface and function as a receptor (26). Thus, it is possible that a fraction of GRP78 acts on the plasma membrane to facilitate endocytic uptake of dsRNA. However, confocal microscopy showed that the time course uptake of FITC-labeled poly(I:C) into 7.5-TLR3 and shG78-13 cells was comparable (data not shown). Moreover, when poly(I:C) was complexed with Lipofectamine and transfected into 7.5-TLR3 cells to activate TLR3, the induction of ISG56 and RANTES was still compromised by GRP78 knockdown (Fig. 7, T-pIC). In a previous study we have shown that in these cells TLR3 mediates the induction of these innate immune genes following short term T-pIC stimulation (11). Taken together, we conclude that GRP78 functions downstream of the TLR3-dsRNA recognition step.
TLR3-dependent IRF3 Activation Is Not Sustained in GRP78 Knockdown Cells
To determine where GRP78 acts to regulate TLR3 responses, we ectopically expressed individual components of this signaling pathway and determined how induction of antiviral genes was altered by knockdown of GRP78. Compared with control 7.5-TLR3 cells, cells with stable GRP78 knockdown (shG78-3 and shG78-13) were substantially impaired for ISG15 expression following transfection of TRIF (Fig. 8A, compare lanes 8 and 9 versus 7 and lanes 20 and 21 versus 19) or the IRF3 kinase, IKKϵ (compare lanes 5 and 6 versus 4). Similar results were obtained when we examined ISG56 expression (data not shown). In contrast, ISG15 expression driven by overexpression of the constitutive phospho-mimetic IRF3–5D was not affected (compare lanes 17 and 18 versus 16), suggesting that GRP78 acts downstream of the IRF3 kinase and proximal to IRF3. Notably, ISG15 induction by overexpression of the RIG-I adaptor MAVS was not affected (Fig. 8A, lanes 13–15), consistent with the absence of an effect of GRP78 silencing on SeV-induced ISG/chemokine expression (Fig. 5). Apart from signaling through IRF3, MAVS also induces antiviral gene expression via IRF1 (27). Immunoblotting experiments in poly(I:C)-stimulated PH5CH8 cells and PH5shG78 cells revealed that, although early IRF3 phosphorylation following TLR3 engagement was intact (Fig. 8B, 3 h post-stimulation, compare lanes 4 versus 3), phosphorylated IRF3 was rapidly diminished in PH5shG78 cells as compared with control PH5CH8 cells (9 h post-stimulation, compare lanes 6 versus 5). In aggregate, despite being dispensable for initial IRF3 activation, GRP78 is required for sustained IRF3 phosphorylation, which controls the magnitude of downstream antiviral gene expression.
GRP78 Expression Is Up-regulated in the Liver of CHC Patients and Correlates with Intrahepatic Chemokine Expression
Next, we determined the expression of GRP78 and two chemokines, RANTES and CXCL10, in liver biopsies of CHC patients and in normal liver tissues from patients undergoing surgical resection for benign liver tumors. RANTES and CXCL10 were chosen because they are among the most up-regulated intrahepatic inflammatory mediators in CHC, and their expression is largely dependent on TLR3 signaling (11). qPCR analysis showed that RANTES and CXCL10 expression was significantly higher in liver tissues from the CHC group (Fig. 9A, p < 0.0001 for both targets), and there was significant correlation between expression of the two chemokines (Fig. 9D, r = 0.85, p < 0.001). GRP78 expression was also significantly higher in liver tissues from the CHC group, a 1.9-fold increase from the control group (Fig. 9A, p = 0.0025). The extent of GRP78 mRNA up-regulation was similar to that seen in HCV-infected 7.5-TLR3 cells (Fig. 3B). Of note, because GRP78 is an abundantly expressed chaperone protein required for maintaining cellular homeostasis, an ∼2-fold up-regulation is a sizeable increase in its expression. Remarkably, GRP78 abundance was significantly correlated with that of RANTES (Fig. 9B, r = 0.48, p < 0.001) and CXCL10 (Fig. 9C, r = 0.45, p = 0.001), although the correlations between GRP78 and the chemokines were weak compared with the correlation between the two chemokines. We did not find a correlation between hepatic GRP78 expression and HCV viremia (data not shown). Together, these data demonstrate that GRP78 is up-regulated in HCV-infected patient liver tissues, and its expression is correlated with that of HCV-induced inflammatory chemokines in vivo.
FIGURE 9.

Expression of GRP78, RANTES, and CXCL10 mRNAs was significantly elevated in CHC liver biopsies compared with normal liver tissues (A) and their pairwise correlation (B–D). qPCR was performed to determine the relative expression levels of GRP78, RANTES, and CXCL10 (normalized to β-actin abundance) in liver tissues from CHC patients (n = 26) and control patients (n = 22). Following logarithmic transformation, the expression of each gene was compared between the two patient groups using two-sided Student's t tests (A). B–D, pairwise correlation between the expression of RANTES and GRP78 (B), CXCL10 and GRP78 (C), and CXCL10 and RANTES (D), as determined by Pearson's correlation coefficients (r) and p values. Data from CHC and control patient samples were shown as red diamonds and green dots, respectively.
PERK and ATF6 Contribute to TLR3 Signaling in Hepatocytes
Because GRP78 is a master regulator of the UPR pathway, we further asked the question of whether the three well characterized branches/components of this pathway, i.e. IRE1, ATF6, and PERK, regulate TLR3-mediated host response to HCV. HCV-infected 7.5-TLR3 cells were transiently transfected with control siRNA or siRNA targeting IRE1, ATF6, or PERK, followed by qPCR analysis of RANTES expression. The siRNAs specifically and efficiently repressed the expression of their cognate target mRNAs, without affecting the expression of GRP78 (Fig. 10A). We found that PERK knockdown and to a lesser extent ATF6 depletion diminished RANTES induction by HCV as compared with transfection of control siRNA. In contrast, IRE1 knockdown had no inhibitory effect (Fig. 10B). Similar results were obtained when poly(I:C) was used to engage TLR3 signaling (Fig. 11A). The same phenomenon was observed in poly(I:C)-stimulated PH5CH8 cells (Fig. 11B). Altogether, we conclude that the PERK and, to a lesser extent, ATF6 contribute to TLR3 signaling in hepatocytes.
FIGURE 10.

Impact of knockdown of UPR genes on HCV-induced, TLR3-dependent RANTES expression. A, qPCR analysis of IRE1, ATF6, PERK, and GRP78 mRNA levels in 7.5-TLR3 cells transfected with control siRNA or siRNA specifically targeting ATF6, PERK, or IRE1, for 48 h. B, 7.5-TLR3 cells were mock-infected or infected with JFH1 virus (m.o.i. = 0.1). 8 h later, cells were transfected with negative control siRNA or siRNA specifically targeting IRE1, ATF6, or PERK for additional 48 h. The expression levels of IRE1, ATF6, PERK, GRP78, and RANTES (normalized to 28S rRNA) were determined by qPCR. A representative of two independent experiments is shown. Error bars represent standard deviations of replicate samples. * and ** indicate statistical difference exists as compared with control siRNA transfected cells with a p value of <0.05 and <0.01, respectively.
FIGURE 11.

Impact of knockdown of UPR genes on poly(I:C)-induced, TLR3-dependent RANTES expression. 7.5-TLR3 (A) and PH5CH8 cells (B) were transfected with negative control siRNA or siRNA specifically targeting IRE1, ATF6, PERK, or GRP78 for 48 h, followed by mock treatment or stimulation by pIC (20 μg/ml, added directly to culture medium) for 8 h. The expression levels of IRE1, ATF6, PERK, and RANTES were determined by qPCR. In the RANTES panels, a representative of three independent experiments is shown. Error bars represent standard deviations of replicate samples. * and ** indicate statistical differences exist as compared with control siRNA transfected cells (with poly(I:C) stimulation) with a p value of <0.05 and <0.01, respectively.
Discussion
TLR3 plays an important role in hepatocellular innate immune responses to HCV by recognizing dsRNA intermediates produced during HCV RNA replication, which leads to expression of antiviral ISGs and inflammatory cytokines/chemokines (11, 12). Yet factors regulating this pathway in hepatocytes remain obscure. In this study, we provide evidence that GRP78, an abundantly expressed molecular chaperone involved in protein folding, maturation, and quality control in the ER, is up-regulated by HCV infection in vitro and in vivo and that GRP78 contributes to TLR3-dependent innate immune responses in hepatocytes. Although GRP78 is best known as an ER luminal protein, we found a fraction of GRP78 is enriched in endolysosome fractions of human hepatoma Huh7.5-derived cells, where TLR3 also resides. It is worth noting that GRP78 and several other ER luminal proteins were previously found on autolysosomal and lysosomal membranes isolated from dextran-loaded rat liver (28). Cellular fractionation assay did not show an appreciable change in GRP78 distribution to endolysosome fractions of 7.5-TLR3 cells before and after TLR3 activation by poly(I:C) (data not shown), suggesting that the increased abundance of GRP78 in the endolysosome fractions during HCV infection is more a consequence of increased protein expression. Our data demonstrate that depletion of GRP78 impaired TLR3-mediated expression of IRF3-dependent antiviral ISGs and inflammatory chemokines and the establishment of an antiviral state in hepatocytes. This effect was not stimulation-specific, as observed in poly(I:C)-stimulated and in HCV-infected cells. Moreover, these observations were made in hepatoma cells reconstituted for the TLR3 pathway as well as in non-neoplastic PH5CH8 cells that naturally harbor intact TLR3 signaling resembling primary human hepatocytes. Notably, GRP78 depletion in PH5CH8 cells severely impaired poly(I:C)-induced, TLR3-mediated antiviral state as examined by the impact on VSV replication. In comparison, GRP78 knockdown by siRNA or shRNA in 7.5-TLR3 and Huh7-TLR3 cells led to a moderate yet significant increase in HCV RNA levels. Two reasons may underlie the seemingly different extent of GRP78's contribution to TLR3-mediated immunity between the two hepatocyte culture systems. First, although typically eliciting a robust ISG response in the liver (1, 2), HCV is not a strong inducer of ISG expression in Huh7-derived hepatoma cells, and infection of Huh7/Huh7.5-TLR3 cells led to a weak ISG response that significantly, albeit moderately, inhibits HCV replication (12). Second, compared with PHHs and PH5CH8 cells, Huh7-derived hepatoma cells are severely impaired for innate antiviral responses, although the precise underlying mechanism remains unclear (10, 29).
Significantly, we demonstrated that elevated GRP78 expression correlated with expression of HCV-induced chemokines, RANTES and CXCL10, in CHC liver biopsies, which is consistent with the data obtained from HCV-infected 7.5-TLR3 cells. It should be noted that the correlation between hepatic GRP78 levels and chemokine expression in vivo is relatively weak. The intrahepatic innate immune responses to HCV are the net result of complex cross-talks and interactions between the virus, hepatocytes, other liver-resident parenchymal and non-parenchymal cells, and immune cells infiltrating the liver (4, 30). Although ISGs, cytokines, and chemokines are highly induced in HCV-infected liver (1, 2), the innate immune responses alone are unable to contain the virus (30). Paradoxically, high basal expression of ISGs as a result of activation of the endogenous IFN system is the strongest factor that predicts poor response to IFN-based therapy in CHC patients (31–33). In light of this, the precise role of GRP78 in innate immune responses to HCV in vivo will require further study.
The elevated GRP78 expression in HCV-infected cells is most likely a result of UPR, which is activated by HCV to cope with virus-induced ER stress and protein folding needs (34–36). GRP78, like GRP94, contains ER stress-responsive elements in its promoter that can be activated by the ATF6 transcription factor matured as a result of UPR. Several HCV proteins, including E2, NS2, and NS4B, etc., induce the UPR (37–39) and thereby increase GRP78 expression. Besides inducing the ATF6-dependent GRP78 promoter transcription, subgenomic HCV replicon activates internal ribosome entry site, including the one that controls translation of GRP78 mRNA (21, 22). However, previous studies, the majority of which were conducted in vitro, have yielded inconsistent results concerning the increase in GRP78 expression (40–43). Differences in HCV protein expression levels and cell types may contribute to the discrepancies. Our data, collected from JFH1-infected hepatoma cells and liver biopsies of CHC patients infected with genotype 1b/2a virus, support that GRP78 expression is up-regulated by HCV in hepatocytes, which is consistent with data obtained from genotype 1a (H77) HCV-infected chimeric mice engrafted with human livers (44). Our results also suggest that HCV-induced GRP78 up-regulation involves the concerted action of transcriptional and post-transcriptional controls. The rather moderate up-regulation of GRP78 protein level without an increase in mRNA expression, as we observed in HCV-infected Huh7.5 cells, may help explain previous inconsistent observations concerning HCV's effect on this protein in hepatoma cells. Precisely why GRP78 mRNA up-regulation was seen in HCV-infected 7.5-TLR3 cells and CHC liver biopsies but not in infected Huh7.5 cells is unclear. Regardless, GRP78 mRNA levels peaked at 24 h post-infection in 7.5-TLR3 cells, prior to transcription of HCV-induced inflammatory cytokines/chemokines (11), suggesting GRP78 up-regulation in HCV-infected hepatocytes is not secondary to autocrine/paracrine actions of TLR3-mediated inflammatory mediators/IFN response. Rather, it fits our model that GRP78 acts upstream of these innate immune responses.
Recent studies have suggested pivotal roles for molecular chaperones in regulating TLR signaling. UNC93B1, a polytopic transmembrane chaperone protein, interacts with multiple nucleic acid-sensing TLRs, including TLR3, TLR7, TLR8, and TLR9, etc., and controls transport of these from the ER to endolysosomes such that they become functional pattern recognition receptors (19). Another chaperone, GRP94, forms a complex with and modulates the function of TLR9, TLR4, and TLR11, but not TLR3 (20). However, the mechanism by which GRP78 facilitates TLR3 signaling revealed in this study is apparently different. Knockdown of GRP78 did not alter the endolysosomal localization of TLR3 nor was TLR3 function affected because early IRF3 phosphorylation following poly(I:C) stimulation was intact. We also did not observe that TLR3 and GRP78 formed a complex, prior to or following poly(I:C) stimulation (data not shown). GRP78's involvement was not through aiding dsRNA uptake either. Rather, our data suggest that GRP78 affects the stability of phosphorylated IRF3, which was short-lived when GRP78 was depleted. The endolysosome is increasingly being recognized as an important platform for innate immune signaling, including that via TLR3. Specifically, not only is the acidification of endolysosomes crucial (11), but also downstream phosphorylation and activation of IRF3 require recruitment of TBK1/IKKϵ-containing complexes to the endolysosome compartments (45, 46). Although the exact mechanism(s) requires further investigation, GRP78 may control the stability of phosphorylated IRF3 resulting from TLR3 engagement by chaperoning with it. Surprisingly, GRP78 knockdown had little impact on induction of antiviral genes downstream of the RIG-I/MAVS pathway, either following SeV infection or MAVS overexpression. The underlying reason for this finding is unclear. However, it is worth noting that the magnitude of IRF3 phosphorylation following RIG-I engagement typically is much higher than that induced via TLR3, perhaps rendering the available phospho-IRF3 less sensitive to the effect of GRP78 knockdown. Not mutually exclusive, MAVS can activate IRF1-dependent transcription of numerous well known IRF3-target ISGs/chemokines, including those examined in this study, using peroxisome as the platform (27). Studies are underway to clarify these possibilities.
Being a target gene downstream of UPR activation, GRP78 is also a master regulator of UPR. This chaperone binds to and represses in resting cells the three known sensors of UPR, IRE1, PERK, and ATF6, thereby controlling activation of these three branches of UPR (26). Although UPR activation alone in the absence of dsRNA stimulation/HCV infection and TLR3 expression is insufficient for inducing IRF3 target genes, certain UPR components may contribute in ways that are unexplored. Our data show that knockdown of PERK inhibited TLR3-dependent RANTES induction, producing an effect similar to GRP78 knockdown, regardless of the stimulus (HCV infection or poly(I:C) stimulation), whereas silencing of ATF6 had a lesser yet demonstrable effect. In contrast, IRE1 depletion had no inhibitory effect. Precisely how the PERK and ATF6 axes contribute to TLR3 signaling will require further investigation. In our experiments knockdown of PERK or ATF6 did not affect GRP78 expression, despite impairing TLR3 signaling. We also did not find that overexpression of GRP78 was able to reverse the effect of ATF6 knockdown (data not shown). Thus, the contribution of ATF6 to TLR3 signaling is uncoupled from the effect of GRP78. It remains to be clarified whether the regulation of TLR3-mediated responses by PERK and ATF6 is separate from their roles in the UPR pathway.
In summary, our data reveal a novel role for GRP78 in TLR3-mediated innate immune response in hepatocytes and link two UPR components, i.e. PERK and ATF6, to this intrinsic antiviral pathway. Given the increasing appreciation of UPR's participation in regulating inflammation and cell death/survival responses (47, 48), future investigations are warranted to determine how much GRP78 and UPR regulation (or UPR components) contributes to TLR3-dependent immune control of HCV infection and to what extent they precipitate hepatic inflammation and liver injuries that set the stage for decompensated chronic liver diseases.
Author Contributions
K. L., D. W., and N. L. L. designed the studies and wrote the paper. D. W. and N. L. L. performed most of the experiments and contributed equally to this work. Y. Z. and J. S. collected the patient samples and generated the data shown in Fig. 9. B. L., K. K., and T. T. W. carried out some of the experiments. D. H. conducted statistical analyses of the data from patient samples. J. F. I. and L. L. prepared primary mouse hepatocyte cultures. All authors reviewed the results and approved the final version of the manuscript.
Acknowledgments
We thank Takaji Wakita for JFH1 cDNA, Charles Rice for Huh7.5 cells and NS5A monoclonal antibody, Nobuyuki Kato for PH5CH8 cells, and Sean Whelan for the VSV-Luc virus. We are grateful to Lawrence Pfeffer for critical reading of the manuscript.
This work was support by National Institutes of Health Grants AI069285 (to K. L.) and DK088787 (to T. T. W.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- HCV
- hepatitis C virus
- ISG
- interferon-stimulated gene
- RIG-I
- retinoic inducible gene-I
- MAVS
- mitochondrial antiviral signaling protein, also known as IPS-1/Cardif/VISA
- TRIF
- Toll-IL1 receptor homology domain containing adaptor inducing interferon-β, also known as TICAM1
- ER
- endoplasmic reticulum
- RANTES
- regulated upon activation, normal T-cell expressed and secreted
- SeV
- Sendai virus
- VSV
- vesicular stomatitis virus
- PERK
- protein kinase R-like endoplasmic reticulum kinase, also known as EIF2AK3
- UPR
- unfolded protein response
- m.o.i.
- multiplicity of infection
- pIC
- poly(I:C)
- qPCR
- quantitative PCR
- CHC
- chronic hepatitis C
- TLR
- Toll-like receptor
- d.p.i.
- days post-infection.
References
- 1. Bigger C. B., Brasky K. M., and Lanford R. E. (2001) DNA microarray analysis of chimpanzee liver during acute resolving hepatitis C virus infection. J. Virol. 75, 7059–7066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Su A. I., Pezacki J. P., Wodicka L., Brideau A. D., Supekova L., Thimme R., Wieland S., Bukh J., Purcell R. H., Schultz P. G., and Chisari F. V. (2002) Genomic analysis of the host response to hepatitis C virus infection. Proc. Natl. Acad. Sci. U.S.A. 99, 15669–15674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Li K., and Lemon S. M. (2013) Innate immune responses in hepatitis C virus infection. Semin. Immunopathol. 35, 53–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Horner S. M., and Gale M. Jr. (2013) Regulation of hepatic innate immunity by hepatitis C virus. Nat. Med. 19, 879–888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ge D., Fellay J., Thompson A. J., Simon J. S., Shianna K. V., Urban T. J., Heinzen E. L., Qiu P., Bertelsen A. H., Muir A. J., Sulkowski M., McHutchison J. G., and Goldstein D. B. (2009) Genetic variation in IL28B predicts hepatitis C treatment-induced viral clearance. Nature 461, 399–401 [DOI] [PubMed] [Google Scholar]
- 6. Suppiah V., Moldovan M., Ahlenstiel G., Berg T., Weltman M., Abate M. L., Bassendine M., Spengler U., Dore G. J., Powell E., Riordan S., Sheridan D., Smedile A., Fragomeli V., Müller T., et al. (2009) IL28B is associated with response to chronic hepatitis C interferon-α and ribavirin therapy. Nat. Genet. 41, 1100–1104 [DOI] [PubMed] [Google Scholar]
- 7. Tanaka Y., Nishida N., Sugiyama M., Kurosaki M., Matsuura K., Sakamoto N., Nakagawa M., Korenaga M., Hino K., Hige S., Ito Y., Mita E., Tanaka E., Mochida S., Murawaki Y., et al. (2009) Genome-wide association of IL28B with response to pegylated interferon-α and ribavirin therapy for chronic hepatitis C. Nat. Genet. 41, 1105–1109 [DOI] [PubMed] [Google Scholar]
- 8. Thomas D. L., Thio C. L., Martin M. P., Qi Y., Ge D., O'Huigin C., Kidd J., Kidd K., Khakoo S. I., Alexander G., Goedert J. J., Kirk G. D., Donfield S. M., Rosen H. R., Tobler L. H., et al. (2009) Genetic variation in IL28B and spontaneous clearance of hepatitis C virus. Nature 461, 798–801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Prokunina-Olsson L., Muchmore B., Tang W., Pfeiffer R. M., Park H., Dickensheets H., Hergott D., Porter-Gill P., Mumy A., Kohaar I., Chen S., Brand N., Tarway M., Liu L., Sheikh F., et al. (2013) A variant upstream of IFNL3 (IL28B) creating a new interferon gene IFNL4 is associated with impaired clearance of hepatitis C virus. Nat. Genet. 45, 164–171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Li K., Chen Z., Kato N., Gale M. Jr., and Lemon S. M. (2005) Distinct poly(I-C) and virus-activated signaling pathways leading to interferon-β production in hepatocytes. J. Biol. Chem. 280, 16739–16747 [DOI] [PubMed] [Google Scholar]
- 11. Li K., Li N. L., Wei D., Pfeffer S. R., Fan M., and Pfeffer L. M. (2012) Activation of chemokine and inflammatory cytokine response in hepatitis C virus-infected hepatocytes depends on Toll-like receptor 3 sensing of hepatitis C virus double-stranded RNA intermediates. Hepatology 55, 666–675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wang N., Liang Y., Devaraj S., Wang J., Lemon S. M., and Li K. (2009) Toll-like receptor 3 mediates establishment of an antiviral state against hepatitis C virus in hepatoma cells. J. Virol. 83, 9824–9834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Li W. C., Ralphs K. L., and Tosh D. (2010) Isolation and culture of adult mouse hepatocytes. Methods Mol. Biol. 633, 185–196 [DOI] [PubMed] [Google Scholar]
- 14. Wakita T., Pietschmann T., Kato T., Date T., Miyamoto M., Zhao Z., Murthy K., Habermann A., Kräusslich H. G., Mizokami M., Bartenschlager R., and Liang T. J. (2005) Production of infectious hepatitis C virus in tissue culture from a cloned viral genome. Nat. Med. 11, 791–796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lindenbach B. D., Evans M. J., Syder A. J., Wölk B., Tellinghuisen T. L., Liu C. C., Maruyama T., Hynes R. O., Burton D. R., McKeating J. A., and Rice C. M. (2005) Complete replication of hepatitis C virus in cell culture. Science 309, 623–626 [DOI] [PubMed] [Google Scholar]
- 16. Cureton D. K., Massol R. H., Saffarian S., Kirchhausen T. L., and Whelan S. P. (2009) Vesicular stomatitis virus enters cells through vesicles incompletely coated with clathrin that depend upon actin for internalization. PLoS Pathog. 5, e1000394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Tsutsumi S., Namba T., Tanaka K. I., Arai Y., Ishihara T., Aburaya M., Mima S., Hoshino T., and Mizushima T. (2006) Celecoxib upregulates endoplasmic reticulum chaperones that inhibit celecoxib-induced apoptosis in human gastric cells. Oncogene 25, 1018–1029 [DOI] [PubMed] [Google Scholar]
- 18. Johnsen I. B., Nguyen T. T., Ringdal M., Tryggestad A. M., Bakke O., Lien E., Espevik T., and Anthonsen M. W. (2006) Toll-like receptor 3 associates with c-Src tyrosine kinase on endosomes to initiate antiviral signaling. EMBO J. 25, 3335–3346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kim Y. M., Brinkmann M. M., Paquet M. E., and Ploegh H. L. (2008) UNC93B1 delivers nucleotide-sensing toll-like receptors to endolysosomes. Nature 452, 234–238 [DOI] [PubMed] [Google Scholar]
- 20. Liu B., Yang Y., Qiu Z., Staron M., Hong F., Li Y., Wu S., Li Y., Hao B., Bona R., Han D., and Li Z. (2010) Folding of Toll-like receptors by the HSP90 paralogue gp96 requires a substrate-specific cochaperone. Nat. Commun. 1, 79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Tardif K. D., Mori K., and Siddiqui A. (2002) Hepatitis C virus subgenomic replicons induce endoplasmic reticulum stress activating an intracellular signaling pathway. J. Virol. 76, 7453–7459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Macejak D. G., and Sarnow P. (1990) Translational regulation of the immunoglobulin heavy-chain binding protein mRNA. Enzyme 44, 310–319 [DOI] [PubMed] [Google Scholar]
- 23. Lin L., Pan S., Zhao J., Liu C., Wang P., Fu L., Xu X., Jin M., and Zhang A. (2014) HSPD1 interacts with IRF3 to facilitate interferon-β induction. PLoS ONE 9, e114874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Yang K., Shi H., Qi R., Sun S., Tang Y., Zhang B., and Wang C. (2006) Hsp90 regulates activation of interferon regulatory factor 3 and TBK-1 stabilization in Sendai virus-infected cells. Mol. Biol. Cell 17, 1461–1471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lee S. H., Song R., Lee M. N., Kim C. S., Lee H., Kong Y. Y., Kim H., and Jang S. K. (2008) A molecular chaperone glucose-regulated protein 94 blocks apoptosis induced by virus infection. Hepatology 47, 854–866 [DOI] [PubMed] [Google Scholar]
- 26. Ni M., Zhang Y., and Lee A. S. (2011) Beyond the endoplasmic reticulum: atypical GRP78 in cell viability, signalling and therapeutic targeting. Biochem. J. 434, 181–188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Dixit E., Boulant S., Zhang Y., Lee A. S., Odendall C., Shum B., Hacohen N., Chen Z. J., Whelan S. P., Fransen M., Nibert M. L., Superti-Furga G., and Kagan J. C. (2010) Peroxisomes are signaling platforms for antiviral innate immunity. Cell 141, 668–681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ueno T., Ishidoh K., Mineki R., Tanida I., Murayama K., Kadowaki M., and Kominami E. (1999) Autolysosomal membrane-associated betaine homocysteine methyltransferase. Limited degradation fragment of a sequestered cytosolic enzyme monitoring autophagy. J. Biol. Chem. 274, 15222–15229 [DOI] [PubMed] [Google Scholar]
- 29. Keskinen P., Nyqvist M., Sareneva T., Pirhonen J., Melén K., and Julkunen I. (1999) Impaired antiviral response in human hepatoma cells. Virology 263, 364–375 [DOI] [PubMed] [Google Scholar]
- 30. Park S. H., and Rehermann B. (2014) Immune responses to HCV and other hepatitis viruses. Immunity 40, 13–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Chen L., Borozan I., Sun J., Guindi M., Fischer S., Feld J., Anand N., Heathcote J., Edwards A. M., and McGilvray I. D. (2010) Cell-type specific gene expression signature in liver underlies response to interferon therapy in chronic hepatitis C infection. Gastroenterology 138, 1123–1133 [DOI] [PubMed] [Google Scholar]
- 32. McGilvray I., Feld J. J., Chen L., Pattullo V., Guindi M., Fischer S., Borozan I., Xie G., Selzner N., Heathcote E. J., and Siminovitch K. (2012) Hepatic cell-type specific gene expression better predicts HCV treatment outcome than IL28B genotype. Gastroenterology 142, 1122–1131 [DOI] [PubMed] [Google Scholar]
- 33. Pfeffer L. M., Li K., Fleckenstein J. F., Marion T. N., Diament J., Yang C. H., Pfeffer S. R., Fan M., Handorf E., and Handorf C. R. (2014) An interferon response gene signature is associated with the therapeutic response of hepatitis C patients. PLoS ONE 9, e104202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Chan S. W. (2014) Unfolded protein response in hepatitis C virus infection. Front. Microbiol. 5, 233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ke P. Y., and Chen S. S. (2011) Activation of the unfolded protein response and autophagy after hepatitis C virus infection suppresses innate antiviral immunity in vitro. J. Clin. Invest. 121, 37–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Sir D., Chen W. L., Choi J., Wakita T., Yen T. S., and Ou J. H. (2008) Induction of incomplete autophagic response by hepatitis C virus via the unfolded protein response. Hepatology 48, 1054–1061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. von dem Bussche A., Machida R., Li K., Loevinsohn G., Khander A., Wang J., Wakita T., Wands J. R., and Li J. (2010) Hepatitis C virus NS2 protein triggers endoplasmic reticulum stress and suppresses its own viral replication. J. Hepatol. 53, 797–804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Chan S. W., and Egan P. A. (2005) Hepatitis C virus envelope proteins regulate CHOP via induction of the unfolded protein response. FASEB J. 19, 1510–1512 [DOI] [PubMed] [Google Scholar]
- 39. Zheng Y., Gao B., Ye L., Kong L., Jing W., Yang X., Wu Z., and Ye L. (2005) Hepatitis C virus non-structural protein NS4B can modulate an unfolded protein response. J. Microbiol. 43, 529–536 [PubMed] [Google Scholar]
- 40. Ciccaglione A. R., Costantino A., Tritarelli E., Marcantonio C., Equestre M., Marziliano N., and Rapicetta M. (2005) Activation of endoplasmic reticulum stress response by hepatitis C virus proteins. Arch. Virol. 150, 1339–1356 [DOI] [PubMed] [Google Scholar]
- 41. Deng L., Adachi T., Kitayama K., Bungyoku Y., Kitazawa S., Ishido S., Shoji I., and Hotta H. (2008) Hepatitis C virus infection induces apoptosis through a Bax-triggered, mitochondrion-mediated, caspase 3-dependent pathway. J. Virol. 82, 10375–10385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Sekine-Osajima Y., Sakamoto N., Mishima K., Nakagawa M., Itsui Y., Tasaka M., Nishimura-Sakurai Y., Chen C. H., Kanai T., Tsuchiya K., Wakita T., Enomoto N., and Watanabe M. (2008) Development of plaque assays for hepatitis C virus-JFH1 strain and isolation of mutants with enhanced cytopathogenicity and replication capacity. Virology 371, 71–85 [DOI] [PubMed] [Google Scholar]
- 43. McPherson S., Powell E. E., Barrie H. D., Clouston A. D., McGuckin M., and Jonsson J. R. (2011) No evidence of the unfolded protein response in patients with chronic hepatitis C virus infection. J. Gastroenterol. Hepatol. 26, 319–327 [DOI] [PubMed] [Google Scholar]
- 44. Joyce M. A., Walters K. A., Lamb S. E., Yeh M. M., Zhu L. F., Kneteman N., Doyle J. S., Katze M. G., and Tyrrell D. L. (2009) HCV induces oxidative and ER stress, and sensitizes infected cells to apoptosis in SCID/Alb-uPA mice. PLoS Pathog. 5, e1000291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Chang C. H., Lai L. C., Cheng H. C., Chen K. R., Syue Y. Z., Lu H. C., Lin W. Y., Chen S. H., Huang H. S., Shiau A. L., Lei H. Y., Qin J., and Ling P. (2011) TBK1-associated protein in endolysosomes (TAPE) is an innate immune regulator modulating the TLR3 and TLR4 signaling pathways. J. Biol. Chem. 286, 7043–7051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Chau T. L., Göktuna S. I., Rammal A., Casanova T., Duong H. Q., Gatot J. S., Close P., Dejardin E., Desmecht D., Shostak K., and Chariot A. (2015) A role for APPL1 in TLR3/4-dependent TBK1 and IKKepsilon activation in macrophages. J. Immunol. 194, 3970–3983 [DOI] [PubMed] [Google Scholar]
- 47. Garg A. D., Kaczmarek A., Krysko O., Vandenabeele P., Krysko D. V., and Agostinis P. (2012) ER stress-induced inflammation: does it aid or impede disease progression? Trends Mol. Med 18, 589–598 [DOI] [PubMed] [Google Scholar]
- 48. Hetz C. (2012) The unfolded protein response: controlling cell fate decisions under ER stress and beyond. Nat. Rev. Mol. Cell Biol. 13, 89–102 [DOI] [PubMed] [Google Scholar]
- 49. Wang N., Dong Q., Li J., Jangra R. K., Fan M., Brasier A. R., Lemon S. M., Pfeffer L. M., and Li K. (2010) Viral induction of the zinc finger antiviral protein is IRF3-dependent but NF-κB-independent. J. Biol. Chem. 285, 6080–6090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Shen Y., Li N. L., Wang J., Liu B., Lester S., and Li K. (2012) TRIM56 is an essential component of the TLR3 antiviral signaling pathway. J. Biol. Chem. 287, 36404–36413 [DOI] [PMC free article] [PubMed] [Google Scholar]