

Abstract
The brain is the central organ of perceiving and adapting to social and physical stressors via multiple interacting mediators from the cell surface to the cytoskeleton to epigenetic regulation and non-genomic mechanisms. A key result of stress is structural remodeling of neural architecture that may be a sign of successful adaptation, while persistence of these changes when stress ends indicates failed resilience. Excitatory amino acids and glucocorticoids play a key role, along with a growing list of extra- and intracellular mediators, including endocannabinoids and brain derived neurotrophic factor (BDNF). The result is a continually changing pattern of gene expression via epigenetic mechanisms involving histone modifications and CpG methylation/hydroxy-methylation as well as activity of retrotransponsons that may alter genomic stability. Elucidation of the underlying mechanisms of plasticity and vulnerability of the brain provides a basis for understanding the efficacy of interventions for anxiety and depressive disorders as well as age-related cognitive decline.
Keywords: hippocampus, amygdala, prefrontal cortex, structural remodeling, neurogenesis, epigenetics, retrotransposons, cytoskeleton, nuclear pore complex proteins, cell adhesion, resilience, glucocorticoids, excitatory amino acids, brain derived neurotrophic factor (BDNF), corticotrophin releasing factor (CRF), endocannabinoids, circadian rhythms
I. Introduction
The brain is the central organ of stress and adaptation to social and physical stressors because it determines what is threatening, stores memories and regulates the physiological as well as behavioral responses that may be damaging or protective1. The physiological responses that produce adaptation via “allostasis” include not only the hypothalamic-pituitary-adrenal (HPA) axis and the autonomic nervous system, but also their non-linear interactions with the metabolic system and the pro- and anti-inflammatory components of the immune defense system1,2. Exposure to multiple stressors and the dysregulation of the non-linear interactions (e.g., not turning on or off responses efficiently) lead to wear and tear on the body and brain that is termed allostatic load and overload1,3.
Allostasis is the active process of adapting to stressors via mediators such as cortisol and the autonomic, metabolic and immune system that act together in a non-linear fashion to maintain homeostasis2. Allostatic load refers to the cumulative effect of multiple stressors as well as the dysregulation of the non-linear network of allostasis (eg. too much or too little cortisol, or adrenalin or inflammation in response to a challenge). Allostatic overload refers to the cumulative pathophysiology that can result from this dysregulation and too much stress. Allostasis and allostatic load/overload are more precise biological concepts than the word “stress” to describe adaptation and maladaptation to “stressors” and they include the physiological effects of health-promoting and health-damaging behaviors as well as stressful experiences1,2 Health behaviors (i.e.smoking, alcohol, poor diet, or lack of sleep), resulting from the experience of stress, also play a role and contribute to allostatic load/overload1,3.
“Stress” can be divided into “good stress”, “tolerable stress” and “toxic stress”4. Early life stress can alter neural architecture to increase adverse reactions to stressors, leading to toxic stress4. “Biological embedding”4,5 of these effects during critical or sensitive periods of early development has lasting effects through the lifecourse6,7. Among the most important early life experiences are those that involve abuse and neglect, on the one hand, versus the establishment of strong, positive attachment of child to caregiver; these alter the ability of the individual to engage in cooperative social experiences or feel excluded and hostile to the social environment later in life8.
The brain is a target of stressful experiences, and glucocorticoids along with excitatory amino acid neurotransmitters alter neuronal architecture by causing dendritic retraction or expansion and decreased or increased synapse density, depending on the brain region, along with inhibition of dentate gyrus neurogenesis9–11. There are many intra- and intercellular mediators and processes that are involved in changing the brain during stress and recovery from stressful experiences12,13. (Box 1)
BOX 1. Examples of molecules that are necessary/permissive for remodeling.
BDNF - brain derived neurotrophic factor91,92
-
-
facilitator of plasticity or growth
-
-
BDNF over-expression – occludes effects of chronic stress.
-
-
BDNF haploinsufficiency prevents stress-induced plasticity
tPA - tissue plasminogen activator73,74
-
-
Secreted signaling molecule and protease
-
-
required for stress-induced spine loss in hippocampus and medial amygdala
-
-
required for acute stress-induced increase in anxiety; CRF activates tPA secretion.
-
-
CRF in amygdala regulates tPA release.
CRF – corticotrophin releasing factor74,133
-
-
Secreted in hippocampus by interneurons
-
-
Down-regulates thin spines via RhoA signalling
Lipocalin-2 - secreted protein; previously unknown function77,78
-
-
Acute stress induces Lipocalin-2
-
-
Lipocalin-2 down-regulates mushroom spines
-
-
Lipocalin-2 ko increases neuronal excitability and anxiety
-
-
Induced via glucocorticoids
-
-
Regulate emotionality and HPA habituation and shut off\
-
-
CB1 receptor KO increases anxiety and basolateral amygdala dendrite length and causes stress-like retraction of prefrontal cortical dendrites, likely through the regulation of glutamateric transmission.
-
-
Fatty acid aminohydrolase (FAAH) is a key regulator of eCB action.
This review provides an overview of the mechanisms and mediators through which stressors alter brain structure and function. It does so by focusing primarily on three brain regions, the hippocampus, amygdala and prefrontal cortex (PFC), in full recognition of the fact that stress has widespread effects throughout the brain. This review also emphasizes the complex non-linear interactions between different stress mediators that are central to the concept of allostasis and allostatic load/overload3, in which non-linearity applies not only to systemic hormones but also to intra- and extracellular mediators in the brain. Because of this, the many changes caused by stress often result in an inverted U dose response relationship, as represented in Figure 1.
Figure 1. Effects of acute and chronic stress, mediated in part by glutamate and glucocorticoids as well as other molecules described in the text and in BOX 1.
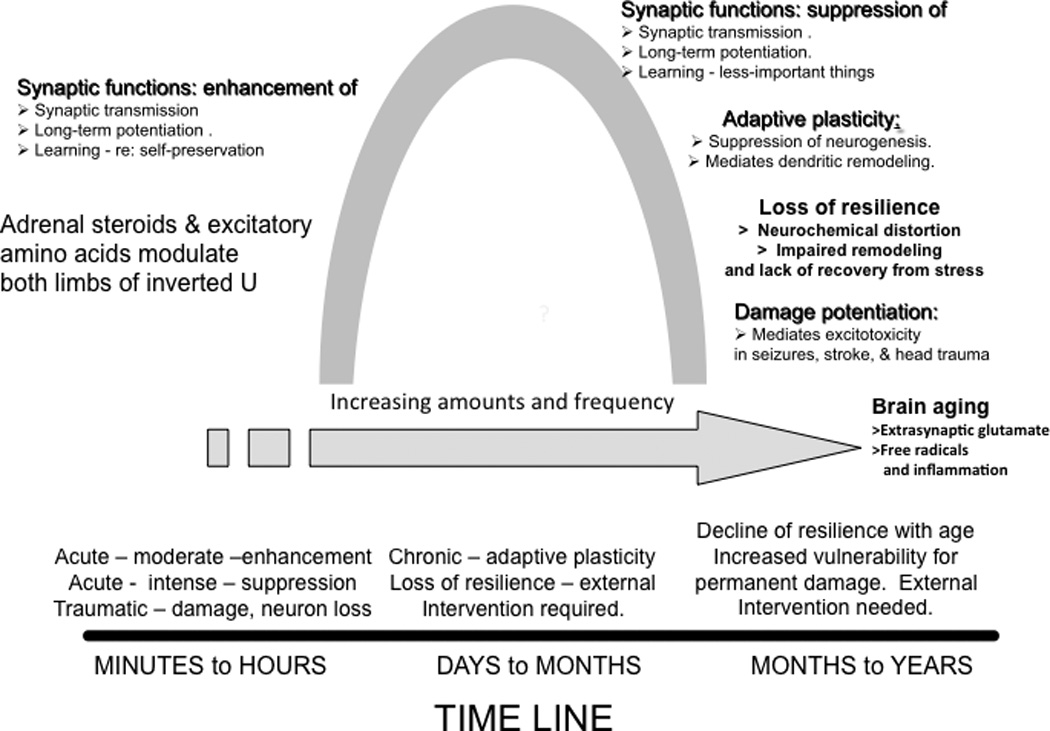
These effects follow an “inverted U shape” curve in dose and time. The time line shows how acute and chronic stress and aging interact with the intensity and duration of stressor.
II. Mechanisms underlying stress effects on the brain
Stressors alter gene expression through multiple mechanisms, including direct effects of glucocorticoids on gene transcription, as well as activation of epigenetic mechanisms in which histone modifications and methylation/hydroxy-methylation of CpG residues in DNA play a role leading to repression and activation of genetic factors, including retrotransposons14,15. Glucocorticoids are not the sole mediators of these effects, with excitatory amino acids and many other cellular mediators playing important roles. Box 1. These mediators span influences from extracellular adhesion molecule, to cytoskeletal elements, and at least one nuclear pore complex protein.
In addition to their critical role in complex behavior and cognition, the hippocampus, amygdala, and PFC have important roles in regulating the autonomic and HPA stress response, and are the main focus of this review. Box 2
Box 2. Overview of stress effects upon the hippocampus, amygdala and prefrontal cortex.
Three regions of the brain shown in the figure have important roles in behavior and cognitive function as well as in regulating the autonomic and HPA stress response and are the main focus of this review.
Glucocorticoid and mineralocorticoid receptors were first recognized in the hippocampal formation16, showing that adrenal steroids affect the brain in more ways than via the hypothalamus, that we now know includes spatial and episodic memory and mood regulation. In the hippocampus, stress and glucocorticoids were first shown to cause dendritic shrinkage and loss of spines. The rediscovery of neurogenesis in the dentate gyrus11 galvanized the widespread interest in the functional role of neuronal replacement in the adult brain. It was in the hippocampus that the role of excitatory amino acids in stress effects was first recognized9.
Effects of acute and chronic stress on the amygdala differ from those in the hippocampus. Acute traumatic stressors were found to cause increased spine density on basolateral amygdala neurons and chronic stress leads to the expansion of basolateral amygdala dendrites137. Yet, the medial amygdala shows a chronic stress-induced loss of spines75. These alterations are implicated in increased anxiety and in PTSD-like behaviors67,137.
Within the prefrontal cortex, chronic stress causes medial PFC neurons to show debranching and shrinkage of dendrites that is associated with cognitive rigidity, while orbitofrontal cortical neurons expand dendrites that may be related to increased vigilance.122,138. The PFC under stress has provided important clues to age-related loss of resilience and impaired memory as well as effects of circadian disruption and extinction of fear memory139.
These 3 brain regions have contributed to our knowledge of cellular and molecular mechanisms and cellular processes that are described in the text and figures, revealing brain regional specializations, as well as common mediators and mechanisms and the complex interactions among the mediators.

A. Stress effects on gene expression in an ever-changing brain
As the first extra-hypothalamic brain structure recognized to have receptors for adrenal steroids16, the hippocampus is an important gateway for understanding the effects of glucocorticoids and stress on gene expression in the brain. Recent technological advances have allowed high-throughput analysis of gene expression changes in response to stress17. For example, microarray analysis of whole hippocampus after acute stress, chronic stress, and stress recovery in mice revealed that acute and chronic stress modulate a core set of genes, but that numerous changes are exclusive to each condition, highlighting how duration and intensity of stress alters reactivity18. Furthermore, corticosterone injections do not yield the same expression profile as acute stress, suggesting that in vivo stressors activate a diverse set of pathways independent of glucocorticoid receptor (GR) activation18. Figure 2. Finally, characterization of expression profiles after extended recovery from 21 days of chronic stress showed that, despite a normalization of anxiety-related behaviors, recovery did not represent a return to the stress-naïve baseline, but rather represents a new state in which reactivity to a novel stressor produces a unique expression profile18. Studies in rats confirm that gene expression profiles can vary significantly from the immediate end of stress to 24 hrs later19, and that chronic stress can alter the transcriptional response to an acute corticosterone injection in dentate gyrus20. Figure 2. Together, these studies demonstrate that a history of stress exposure can have a lasting impact on future stress reactivity and hippocampal function.
Figure 2. Gene expression changes in hippocampus in response to stress and glucocorticoid challenge depend on the prior stress history of the subject.

Hippocampal microarray data reveals stress-induced gene expression changes. (a) Solid bars represent the number of significantly increased genes and hatched bars represent significantly decreased genes identified by pairwise comparisons of each stress group with age-matched controls.
(b) Proportional Venn diagram illustrating the genes significantly altered by both the acute stress, chronic stress, and CORT injection conditions. The numbers of genes unique to each comparison that were increased or decreased are listed next to arrows indicating the direction of change.
(c) Venn diagram of genes altered by each FST condition reveals a core of 95 genes that were always changed by this stressor. The large number of unique gene expression changes in each condition shows that the response to FST is altered by the stress history of the group, with the vast majority of changes occurring when the animal is exposed to a novel stressor immediately after a chronic stress exposure, as also shown in (a).
(d) Scatter plot of normalized expression values for each microarray probe comparing CRS (x axis) with recovery from CRS (y axis). The majority of genes are increased by CRS, but decreased after recovery; however, there are a number of probes that are increased by CRS that remain elevated after recovery or are suppressed by CRS and remain low in recovery. Highlighted probes are those that reached significance when compared with age-matched controls (blue=CRS, red=recovery from CRS, gray=not significant). Several examples of the highlighted genes are listed below the scatter plot by color designation and quadrant. For example, blue points in the lower left quadrant, such as Nrg3 and Scn1b, represent genes that are significantly changed by CRS when compared with unstressed controls and are also decreased after recovery from CRS. Whereas red points in the upper right quadrant, such as Cdk2 and Gria2, are genes that remained significantly different from controls after recovery from CRS, and were also increased immediately following CRS.
(e) Venn diagram illustrating that the number of genes significantly different from controls after recovery from CRS are mostly unique from those significantly altered by CRS. Reprinted from18 by permission.
(f) Pie charts of overrepresented GO-terms among the 576 genes that were differentially expressed upon GC-challenge in naive versus chronically stressed rats. The differentially expressed genes were divided in a group that responded to GCs in both controls and CRS animals (center) or only in controls (left) or in CRS animals (right). The pie charts represent the GO-terms that were overrepresented in the 3 groups of GC-responsive genes and show that after CRS, GC-challenge gives rise to a different gene signature than in control animals. From20 by permission.
Many of the genes altered after glucocorticoid and chronic stress exposure in the hippocampus are known epigenetic regulators21, providing one possible mechanism underlying the persistent alterations in the expression response beyond the end of stress exposure. The continually changing pattern of gene expression is consistent with the finding that, although stress-induced dendritic retraction in PFC neurons appears to be reversible in terms of dendritic length and branching, the recovered neurons are different, in that dendrites that regrow after recovery from stress are more proximal to the cell body than those that retracted22.
B. Epigenetic mediation via post-translational histone modifications
Stress has a clear impact on many types of molecular epigenetic mechanisms, from histone modifications, to DNA methylation/hydroxymethylation and non-coding RNA expression14,23–25. Figure 3. For instance, social defeat stress in rodents causes changes in both histone methylation and acetylation26. Acute and chronic stress promote histone modifications leading to repression or activation of genes related to memory and other processes. Using memory acquired in the forced swim test and Morris water maze, a novel, rapid mechanism was discovered: glucocorticoids, via GR, facilitate signaling of the ERK-MAPK pathway to the downstream nuclear kinases MSK1 and Elk-1 in dentate gyrus granule neurons; and activation of this pathway results in serine10 (S10) phosphorylation and lysine14 (K14) acetylation at histone H3 (H3S10p-K14ac), leading to the induction of the immediate-early genes, c-Fos and Egr-127
Figure 3. Molecular Epigenetic Modifications.

Among the molecular mechanisms that fall under the epigenetic rubric are covalent modifications of the histone proteins which package and control access to the DNA, which include acetylation, methylation, and phosphorylation; as well as a growing number of more exotic modifications. The DNA itself may be methylated or hydroxyl-methylated at cytosine residues. A suite of non-coding RNA species such as microRNA (miRNA), piwi-interacting RNA (piRNA) and long non-coding RNA (lncRNA) also act to convey epigenetic information and to co-ordinate interactions between DNA and the transcriptional and chromatin modification machinery. It is worth noting that many of these mechanisms appear to have evolved in part from, or as a consequence of the presence of transposable elements in eukaryotic genomes. Adapted from24.
Unlike other immediate early genes, FosB/ΔFosB is increased and remains elevated in the nucleus accumbens (NAc) after social defeat stress and is deficient in those animals that show depressive like behavior, as well as in postmortem human depressed patients. Moreover, increased FosB/ΔFosB expression in NAc protects animals from the deleterious effects of chronic stress28. Epigenetically, H3K9me2 is enriched at the FosB promoter in NAc of human depressed patients relative to controls, implicating this repressive epigenetic modification in the repression of FosB. Moreover, in mice ZFP-induced enrichment of H3K9me2 at FosB in NAc was not only sufficient to reduce FosB/ΔFosB expression, but also induced depression- and anxiety-like behaviors after social stress28.
A current practical application of this approach is the investigation of rapidly acting antidepressants28 that act, at least in part, via epigenetic mechanisms, as does electroconvulsive therapy31. An epigenetic mechanism connects excitatory amino acid function with neural remodeling and stress-related behavior in both a genetic and stress-induced rodent model of anxiety- and depressive-like behavior where down-regulation of the presynaptic inhibitor of neuronal glutamate release, mGlu2 receptors, in hippocampus is a key biomarker32. In that connection, drugs that modify glutamate overflow, like ketamine, acetyl-L-carnitine and riluzole, have been show to exert rapid antidepressant-like effects in animal models29 and in humans30.
The novel antidepressant candidate acetyl-L-carnitine (LAC) appears to act inside and outside of the nucleus to exert fast antidepressant responses: LAC corrects mGlu2 deficits by increasing acetylation of the histone H3K27 bound to Grm2 promoter gene as well as the NFkB-p65 sub-unit31. Using the same animal models, 14 days of treatment with the tricyclic antidepressant clomipramine were needed to promote antidepressant responses, which, disappeared when the treatment was stopped, while LAC antidepressant effects were still evident after 2 weeks of drug withdrawal. The persistent effects of LAC highlight the involvement of stable molecular adaptations that are reflected at the level of histone modifications in controlling mGlu2 transcription in hippocampus.
C. Transposons and retrotransposons
Acute restraint stress also has repressive epigenetic effects in the hippocampus and most prominently in the dentate gyrus via trimethylation of lysine 27 and lysine 9 on histone H3. The latter is associated with repression of a number of retrotransposable elements (RTE) and reduction of the coding and non-coding RNA normally produced by the repressed DNA, so far only in hippocampus32,33. This repression is lost with repeated stress, suggesting that those RTEs may impair genomic stability under conditions of chronic stress15.
Retrotransposons constitute a ten fold larger fraction of mammalian genomes than protein coding genes and appear to be unusually active in brain and steroidogenic tissues15. Consequently, they have recently attracted increasing attention from neuroscientists who have shown that they contribute to neural diversity, cell fate and development as well as brain disease15,34–36. In addition to transposons’ mobility, they also seem to contribute the largest fraction of functional elements to what might be referred to as “the RNA genome”37–39. This other genome is composed of genes for non-coding RNA’s that are being found to govern a growing number of cellular processes, including development, cell differentiation, chromosome imprinting and the regulation of the epigenetic machinery40,41; thus RTE derived RNAs represent a substantial store of both genetic and epigenetic information.
Barbara McClintock, who discovered transposons over 60 years ago, noted that they were important contributors to an organism’s ability to deal with environmental stress42,43, and this insight appears to hold true with regard to the neurobiology of stress, though as with many other aspects of stress, transposons likely have both adaptive34 and possibly deleterious effects since dysregulation of these elements has been observed in both human PTSD patients and animal models of stress disorders44–46. Brain transposons therefore appear to represent a significant new frontier for stress research.
D. Role of excitatory amino acids
Excitatory amino acids, particularly glutamate, play a key role in structural as well as functional changes in the brain. Figure 4. Initial studies of restraint stress, in which chronic stress causes shrinkage of apical dendrites of hippocampal CA3 neurons, showed that acute restraint stress elevates extracellular glutamate levels via a process that is absent in adrenalectomized animals, implicating a role for the adrenal cortex47. Indeed, corticosterone acts directly via membrane associated MR and GR to cause glutamate release29,48,49. Importantly, blocking NMDA receptors and interfering with excitatory stimulation of ion channels blocks stress induced dendritic remodeling within the hippocampus, similar to blockade of adrenal steroid synthesis50,51. Similarly, a stress-induced NMDA-dependent dendritic remodeling has been reported in medial PFC neurons52. Excess glutamatergic activity, without adequate reuptake in the aftermath of trauma from seizures, ischemia and head trauma, leads to permanent neuronal loss by a process that is exacerbated by glucocorticoids53. These relationships can be summarized in an inverted U-shaped dose and time response curve (Figure 1).
Figure 4. The tripartite glutamate synapse.

Neuronal glutamate (Glu) is synthesized de novo from glucose (not shown) and from glutamine (Gln) supplied by glial cells. Glutamate is then packaged into synaptic vesicles by vesicular glutamate transporters (vGluTs). SNARE complex proteins mediate the interaction and fusion of vesicles with the presynaptic membrane. Metabotropic glutamate receptors of class II, such as mGlu2 and mGlu3, regulate neuronal release of glutamate into the synaptic space. After release into the synaptic space, glutamate binds to ionotropic glutamate receptors (NMDA receptors (NMDARs) and AMPA receptors (AMPARs)) and metabotropic glutamate receptors (mGluR1 to mGluR8) on the membranes of both postsynaptic and presynaptic neurons and glial cells. Upon binding, the receptors initiate various responses, including membrane depolarization, activation of intracellular messenger cascades, modulation of local protein synthesis and, eventually, gene expression (not shown). Surface expression and function of NMDARs and AMPARs is dynamically regulated by protein synthesis and degradation and receptor trafficking between the postsynaptic membrane and endosomes. The insertion and removal of postsynaptic receptors provide a mechanism for long-term modulation of synaptic strength. Glutamate is cleared from the synapse through excitatory amino acid transporters (EAATs) on neighboring glial cells (EAAT1 and EAAT2) and, to a lesser extent, on neurons (EAAT3 and EAAT4). Within the glial cell, glutamate is converted to glutamine by glutamine synthetase and the glutamine is subsequently released by System N transporters and taken up by neurons through System A sodium-coupled amino acid transporters to complete the glutamate–glutamine cycle. Reprinted from29 by permission.
In that connection, the shrinkage of apical dendrites as a result of stress in CA3 pyramidal neurons can be thought of as a protective mechanism against permanent damage and neuron loss that is caused by the metastable dentate gyrus-CA3 feed forward and feedback circuitry that is the basis of its function54 and yet makes it vulnerable to seizure-induce damage55. This is well illustrated in hibernation, a state of low energy supply to the brain, that is accompanied by rapidly reversible (within hours), shrinkage of CA3 apical dendrites in the hippocampus56,57. This hypothesis is further substantiated by studies in which removal of polysialic acid residues from neural cell adhesion molecule (NCAM) leads to marked increases in dendritic length of CA3 neurons and increased vulnerability to excitotoxic damage, supporting the notion that shorter dendrites reduce vulnerability of CA3 neurons to over-stimulation58.
E. Role of glucocorticoids via multiple intracellular sites and mechanisms
Glucocorticoids produce both genomic and non-genomic effects in the brain via multiple sites and pathways. In addition, glucocorticoids have biphasic effects in which the timing and the level of GR expression are critical29,59. Glucocorticoid actions via genomic mechanisms involve both direct interactions with glucocorticoid response elements (GRE) and indirect actions via tethering to other transcription factors60. Glucocorticoids can directly stimulate release of excitatory amino acids via membrane-associated receptors, and they can indirectly regulate both glutamate and GABA release through induction of local synthesis of endocannabinoids61 (see below),
In addition, glucocorticoids can also translocate GR to mitochondria together with the anti-apoptotic protein B-cell-lymphoma 2 (Bcl-2) where they together promote Ca2+ sequestration, regulate mitochondrial oxidation and free radical formation and membrane potential, three independent measures of mitochondrial function. Bcl-2 is able to inhibit the formation of Bax-containing pores on the mitochondrial outer membrane and reduce the release of calcium and cytochrome C from the mitochondria. Importantly, these 3 Cort effects show and inverted U shape dose response curve and are biphasic, and high glucocorticoid levels cause a failure of this mechanism over 72 hours leading to increased free-radical formation62 n Figure 5.
Figure 5. Biphasic effect of glucocorticoids (Cort) in regulating mitochondrial function.
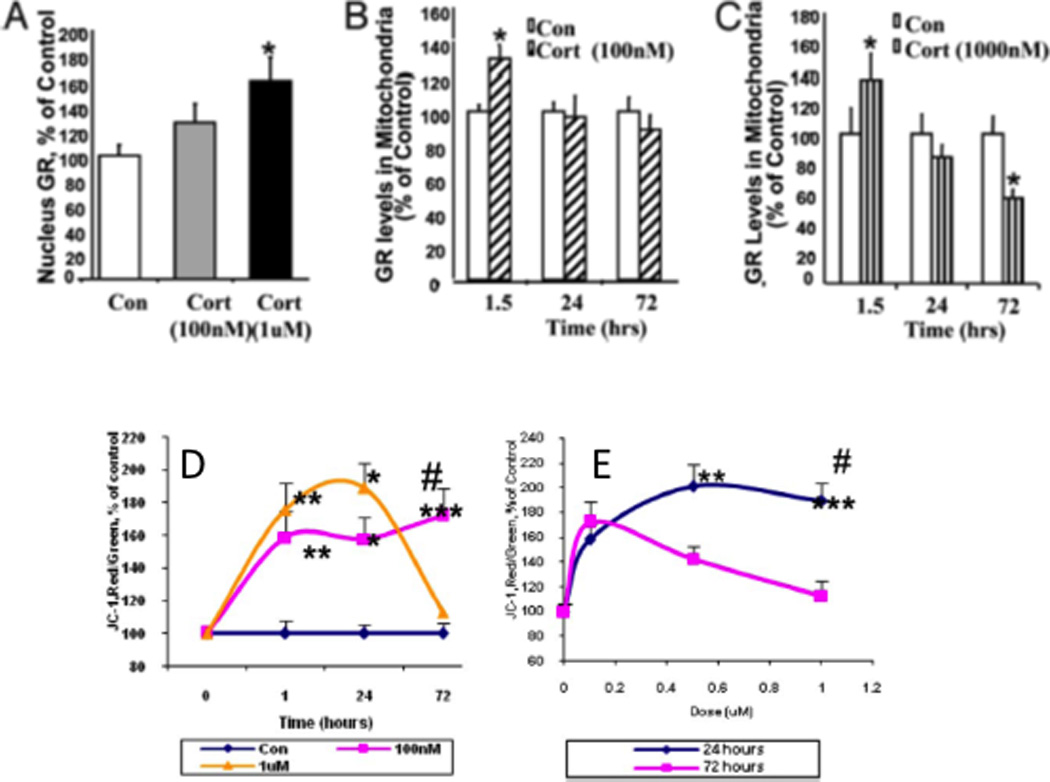
(A) Cort readily penetrate the cell membrane and interact with cytoplasmic glucocorticoid receptors (GRs), showing a dose-dependent increase in GR translocation into cell nuclei; (B), GR translocation into mitochondria as a complex with the anti-apoptotic protein, Bcl-2, where they upregulate mitochondrial calcium levels, membrane potential and oxidation; this is stabilized at 100nM dose (B) and (C) decreases with time at the high 1000nM Cort dose, where, after a 3 day treatment, high Cort leads to decreased GR and Bcl-2 levels in mitochondria (c). CORT modulates membrane potential, measured by Janus-1 (JC-1) staining in a dose- and time-dependent manner: (D) Time course of JC-1 staining after Cort treatment shows sustained potential at 100nM dose and loss of potential at high 1000nM dose; (E) Dose-dependent curve for JC-1 staining after Cort treatment showing that both low and high Cort maintain potential at 24h but high Cort causes failure of membrane potential at 72h. This regulation of mitochondrial function by Cort parallels neuroprotection; that is, treatment with low doses of Cort has a neuroprotective effect, whereas treatment with high Cort enhanced kainic acid (KA)-induced toxicity of cortical neurons62,140, consistent with the “glucocorticoid endangerment” hypothesis141. Modified from62 by permission.
Just as location of GR action is an essential consideration, the level of expression of GR is also very important. Genetically-induced over-expression of GR in forebrain leads to increased lability of mood-related behaviors, yet also confers greater responsiveness to antidepressant drugs63; while genetic knockdown of GR has the opposite effect64. Epigenetic regulation of GR activity also has significant functional implications, as increased CpG methylation within the GR promotor is associated with a sub-optimal HPA stress response and is associated with poor maternal care in rodents and early life abuse in human suicide victims65,66.
Glucocorticoid actions are biphasic, as illustrated above for mitochondria62, and timing is important. For example, in several animal models of traumatic stress induced PTSD-like delayed anxiety and traumatic stress induced spine synapse formation in basolateral amygdala (BLA), a timed elevation of glucocorticoids, prior to the induction of stress, prevents these effects67. Data on human PTSD supports a protective role for adequate glucocorticoid levels at time of traumatic stress68,69. Yet repeated high dose glucocorticoid treatment mimics chronic stress and induces dendritic lengthening in BLA70 emphasizing the differences between acute and chronic elevations in glucocorticoids.
Regarding mineralocorticoid receptors (MR) which have both genomic actions as well as non-genomic actions to stimulate glutamate release48, mice that spontaneously showed increased anxiety have elevated expression of hippocampal MR, which mediated a stress-induced suppression of mGlu2 expression and increased levels of anxiety- and depression-like behavior71. Importantly, blocking MR receptors and interfering with glucocorticoids stimulation of glutamate activity blocks stress induced mood abnormalities. Yet, the nature of the experiences of the animals that develop higher MR is not known but may involve epigenetic influences early in life, such as maternal care and stressors in the neonatal nesting environment72. The epigenetic allostasis model points out to developmental origins of individual differences in the responses to stress and implies that unknown early-life epigenetic influences program each individual to different trajectories of behavioral and physiological responses to later stressful life events. Figure 6.
Figure 6.

Individual differences in naïve C57Bl6 mice in anxiety-related behavior reveal animals more sensitive to stress-induced down-regulation of hippocampal mGlu2 expression, a biomarker of depressive-like behavior and antidepressant response. The use of the light-dark test as a screening method (A) allows identification of clusters of animals with a different baseline anxiety profile (B), along with differences in mineralocorticoid receptor (MR) levels in hippocampus (C). The susceptible mice that are characterized by higher baseline MR levels show reduced hippocampal mGlu2 expression associated with exacerbation of anxious and of depressive-like behaviors after acute (D) and chronic stress, respectively. Conversely, individuals with lower baseline MR levels cope better with stress and show adaptation in mGlu2 receptor expression in hippocampus. The epigenetic allostasis model (E) points to the developmental origins of these individual differences, suggesting that as yet unknown epigenetic influences early in life may lead to alterations in MR hippocampal levels71. In (F) representative mechanisms of action of acetyl-L-carnitine (LAC): ours and other laboratories have shown that the decrease in mGlu2 receptors either following stress exposure or in a genetic animal model of depression is rapidly corrected by 3 days of intraperitoneal administration of the novel antidepressant candidate LAC via acetylation of either the H3K27 bound to Grm2 promoter, which codifies for mGlu2 receptors, or the NFkB/p65 sub-unit32.
F. Involvement of secreted signaling molecules
In addition to glucocorticoids, secreted signaling molecules play an important role in the remodeling of neural tissue during stress. BOX 1. Corticotrophin releasing factor (CRF), which is better known for its role in governing secretion of ACTH and glucocorticoids, plays a key role in stress-induced dendritic remodeling in the CA1 region of the hippocampus73,74. Findings over the past decade have also implicated new players in the regulation of dendritic remodeling. For instance, tissue plasminogen activator (tPA) is a secreted signaling molecule as well as protease that is implicated in stress-induce dendritic remodeling and spine loss in medial amygdala as well as in the CA1 hippocampus. Specifically, tPA-ko mice fail to show chronic stress impairment of memory and spine reduction in the CA173,75. Linking these two factors together, there is evidence that in the amygdala tPA release is stimulated by CRF76. Similarly, Lipocalin-2 is a novel modulator of spine plasticity with different effects in amygdala and hippocampus77,78. Acute stress increases Lipocalin-2 levels, and Lipocalin-2 down-regulates mushroom spines and generally inhibits actin motility in hippocampus. Remarkably, deletion of Lipocalin-2 increases neuronal excitability and anxiety, and, in amygdala, the absence of lipocalin-2 increases the basal number of spines and prevents a stress-induced increase in spine density.
Endocannabinoids are another class of signaling molecules that importantly regulate multiple aspects of the stress response. In addition to contributing to the termination79 of the acute response to stress, as well as habituation to repeated stress80, endocannabinoids also appear to be important for the regulation of structural plasticity under conditions of repeated stress. BOX 1. For example, cannabinoid 1 (CB1) receptor deficient mice exhibit reductions in prefrontal cortical dendritic length and complexity, while having enhanced and more complex dendritic arbors within the BLA, which parallels the effects of chronic stress81,82. More importantly, chronic stress and corticosterone treatment are both known to impair endocannabinoid signaling at multiple levels, through both a down-regulation of the CB1 receptor83 as well as a reduction in the levels of the endocannabinoid, anandamide, mediated by an increase in its hydrolysis by the enzyme fatty acid amide hydrolase (FAAH)93,84,.
Given the parallels between genetic deletion of the CB1 receptor and the ability of chronic stress to impair endocannabinoid signaling, it is interesting to note that elevating anandamide/CB1 receptor signaling, through genetic or pharmacological impairment of FAAH, retards the ability of chronic stress to produce dendritic hypertrophy in the BLA as well as concomitant changes in emotional behavior93,94,85–87. Collectively, these data indicate that endocannabinoid signaling buffers against many of the effects of stress, and appears to be important for limiting the effects of chronic stress on structural plasticity within these identified limbic circuits. At a mechanistic level, this is likely due to the ability of CB1 receptor signaling to gate glutamatergic release as it has been shown that CB1 receptor deficient mice exhibit greater changes in glutamatergic signaling and excitotoxicity within the PFC following chronic stress88. Moreover, similar to the protective effects of CB1 receptor activation identified within the amygdala, administration of a CB1 receptor agonist during repeated stress can reduce the increase in glutamatergic signaling, the induction of pro-inflammatory cytokines and lipid peroxidation within the PFC88. As such, the release of endocannabinoids during stress may temper changes in structural plasticity by limiting the magnitude of glutamate release in response to stress, and under conditions of chronic stress, when this system becomes compromised, the loss of this endogenous buffer facilitates excess glutamate release and the ensuing changes in dendritic morphology. Linking this model with the previously described factors, it is interesting to note that in addition to promoting tPA release, CRF has also been found to induce anandamide hydrolysis by FAAH89 suggesting the possibility that CRF could act as an orchestrator of multiple signaling molecules, all of which converge in structural changes within the brain following chronic stress.
Recent studies have suggested that blood-based biomarkers may be able to predict aspects of brain signaling associated with trauma-related effects in both males and females, specifically with respect to convergence onto GR signaling pathways. After a predator-scent-stress (PSS) exposure, male and female rats were classified into vulnerable (i.e., “PTSD-like”) and resilient (i.e. minimally affected) phenotypes on the basis of their performance on a variety of behavioral measures90. Using genome-wide expression profiling in blood, amygdala, and hippocampus, glucocorticoid signaling was the only convergent pathway associated with individual differences in susceptibility. Moreover, corticosterone treatment 1h after PSS-exposure prevented anxiety and hyperarousal 7 days later in both sexes, consistent with prior findings in the same as well as in another PTSD animal model67,68, confirming GR involvement in sequelae of traumatic stress.
G. Roles of brain-derived neurotrophic factor (BDNF)
Brain derived neurotrophic factor (BDNF) plays an important role in dendritic remodeling in both hippocampus and BLA. BOX 1. BDNF overexpressing mice show increases dendritic length in both CA3 and BLA, which occludes the effects of chronic stress to decrease dendritic branching in CA3 and increase it in BLA91. On the other hand, in WT animals, chronic stress causes a down-regulation of BDNF in CA3 hippocampus and an up-regulation of BDNF in the BLA. While the increase in BLA persists after 21d post stress, the effect in CA3 normalizes92. These intriguing timing issues become more interesting when one considers that after a single acute stress, BDNF expression rises and stays elevated for 10d in the BLA, while that in CA3 only shows a transient increase92. This increase in the BLA is associated with both increased anxiety and increased density of spines in BLA neurons93. The mechanism of these effects on BDNF remain enigmatic, but are not entirely mediated by glucocorticoid actions as corticosterone levels increased after both acute and chronic stress and remained elevated after chronic, but not after acute stress. Thus, it is clear that BDNF-mediated signaling is involved in the structural effects of stress but the direction and nature of signaling is region-specific, stress specific and is influenced by epigenetic modifications94 along with post translational modifications9592. The epigenetic mechanisms controlling BDNF expression are influenced by maternal separation early in life, which, in turn, leads to changes in BDNF expression and epigenetic regulation via histone acetylation and methylation over the life course with consequences for anxiety-like behaviors96.
H. Cellular processes in remodeling of neural architecture
The neuronal surface, cytoskeleton and nuclear envelope are each implicated in the mechanisms of stress-induced retraction and expansion of dendrites and synapse turnover. The polysialated form of neural cell adhesion molecule (PSA-NCAM) is expressed in the CA3 and DG region of the hippocampus and is believed to denote the capacity for adaptive structural plasticity in many parts of the CNS97–99. Repeated stress causes retraction of CA3 hippocampal dendrites accompanied by a modest increase in PSA-NCAM expression, possibly the result of glucocorticoid mediation100. Using EndoN to remove PSA from NCAM, Sandi reported impairment of consolidation of contextual fear conditioning101. Using the same treatment, we reported considerable expansion of the dendritic tree in both CA3 and CA1 and a marked increase in excitotoxicity and damage to CA3 neurons; repeated stress still caused some dendrite retraction after PSA removal58. Thus, while PSA-NCAM is a facilitator of plasticity, the PSA moiety appears to also limit the extent of dendritic growth and yet is not necessary for dendritic retraction under stress.
Two other classes of cell adhesion molecules are reported to change with chronic stress, with behavioral consequences. Neuroligins (NLGNs) are important for proper synaptic formation and functioning, and are critical regulators of the balance between neural excitation/inhibition (E/I), and chronic restraint stress reduced hippocampal NLGN-2 levels, in association with reduced sociability and increased aggression102,103. This occurred along with a reduction of NLGN-2 expression throughout the hippocampus, detectable in different layers of the CA1, CA3, and DG subfields. Intra-hippocampal administration of neurolide-2 that interferes with the interaction between NLGN-2 and neurexin led to reduced sociability and increased aggression, thus mimicking effects of chronic stress102.
Chronic restraint stress also increases activity of matrix metalloproteinase-9 (MMP-9) in the CA1. MMP-9 carries out proteolytic processing of another cell adhesion molecule, nectin-3. Chronic stress reduced nectin-3 in the perisynaptic CA1, but not in the CA3, with consequences for social exploration, social recognition and for a CA1-dependent cognitive task. Implicated in this is a stress-related increase in extracellular glutamate and NMDA receptor mediation of MMP-9104. These findings are reminiscent of the CA1-specific effects of tissue plasminogen activator mediating stress effects on spine density in CA173.
Actin polymerization plays a key role in filopodial extension and spine synapse formation as well as plasticity within the synapse itself105, and cytoskeletal remodeling is an important factor in the effects of stress and other environmental manipulations. Hibernation in European hamsters and ground squirrels results in rapid retraction of dendrites of CA3 pyramidal neurons and equally rapid expansion when hibernation torpor is reversed56,57 The retraction of dendrites is accompanied by increases in a soluble phosphorylated form of tau that may indicate disruption of the cytoskeleton, which permits the dendrite shortening and possible protection from excitotoxicity; at the same time, PSA-NCAM expression is lost during hibernation torpor reducing the capacity for plasticity106. This model highlights the important role that tau plays in normal cytoskeletal function, a fact that should be emphasized when attempting to understand its role in pathology107.
Even though dendrite retraction and regrowth would appear to involve a reversible depolymerization and repolymerization of the cytoskeleton, there are other processes that point to the importance of nuclear factors. A recent example is the unexpected role of a cell nuclear pore complex protein, NUP-62, in the stress-induced dendritic remodeling in the CA3 region of hippocampus108. First identified as a gene that was down regulated in the prefrontal cortex of depressed patients109, NUP-62 was also found to be reduced in response to chronic stress in CA3 neurons of rodents108. Importantly, the levels of other nuclear pore complex genes were unchanged with chronic stress, supporting the specificity of its role in stress remodeling. Subsequent in vitro studies confirmed that the down-regulation of NUP-62 is associated with dendritic retraction and this effect is regulated at the molecular level by NUP-62 phosphorylation at a PYK2 site which results in its retention in the cytoplasm108. A role of NUP-62 in maintaining chromatin structure for transcription is suggested as well as in nucleo-cytoplasmic transport108.
III. Stress - not always what we think it is
Just as stress is not a unitary phenomenon at the cell and circuit level, neither is it at the level of the whole organism. As noted in the Introduction, a key aspect of stress effects on the brain and body is the non-linear interaction of multiple mediators of stress and adaptation that is part of the concept of allostasis1, which refers to the active process of maintaining homeostasis via output of hormones and ANS activity along with immune and metabolic system mediators and the mediators in the brain that are the main focus of this review. When one mediator system changes, the others adjust, and the resulting output can be distorted, as in chronic inflammation or a flat cortisol diurnal rhythm caused by sleep deprivation or depression2. Moreover, the actions of any one mediator may depend on the actions of other mediators. For example, glucocorticoids and excitatory amino acids are both involved in stress-induced suppression of neurogenesis, which was found not only in rodents but also in tree shrews and rhesus monkeys110–112. Yet, glucocorticoid levels alone do not predict the direction of neurogenesis, as shown by studies of male sexual behavior, which results in increased neurogenesis but also high glucocorticoid levels; in this scenario oxytocin appears to play an important role, emphasizing the importance of understanding the interaction of these distinct signaling molecules113,114.
In seeking to understand where and how stress affects neural circuits, it has become evident that “when” these mediators act is also an important consideration. In most vertebrate species, plasma glucocorticoids rise just before the active phase. This rhythm is largely driven by changes in the amplitude and frequency of the ultradian secretion of glucocorticoids115. Indeed, the natural ultradian fluctuations of glucocorticoids mediate turnover of a subset of synapses in cerebral cortex; and inhibiting the fluctuations with a minimal dose of dexamethasone impairs spine turnover116. Moreover, these diurnal changes in spine formation and removal are important for motor learning117.
In addition to ultradian pulses, circadian (or diurnal) rhythms are a crucial factor that impact the stress response. Rhythmic HPA function seems to be necessary for normal initiation and termination of ACTH and other stress mediators118. Epidemiologically, disrupted sleep and circadian rhythms lead to increased risk for development of psychiatric, cardiovascular or other physiological syndromes in shift workers or populations under-going chronic circadian disruption119. Housing mice in a light-dark cycle of 20 hrs (10h light/10h dark) compared to standard 24h cycles to drive circadian disruption (CD), results in metabolic signs of allostatic load120, with increased weight, adiposity and leptin levels, as well as an imbalance between insulin and plasma glucose, suggesting a pre-diabetic state121. The metabolic changes are accompanied by changes in PFC cellular morphology, mirroring those observed in chronic stress122, with CD animals having shrunken and less complex apical dendritic trees of cells in layer II/III of the medial PFC123. Figure 7. In addition, circadian disrupted mice show altered responses to endotoxin challenge with lipopolysaccharide124 highlighting the similarities between chronic circadian disruption and chronic stress. The mechanisms by which these systems interact is not yet fully understood, but do not appear to be driven simply by elevated glucocorticoids.
Figure 7. Mice cannot adjust to a 10:10 light:dark cycle as indicated by body temperature and locomotor activity rhythms.

This circadian disruption, as in shift work, leads to increase body fat and leptin and insulin resistance, along with remodeling of apical dendrites of prefrontal cortical neurons and indications of cognitive rigidity. Data from123.
Intriguingly, glucocorticoids are able to regulate circadian clock gene expression in several brain regions125 as well as in liver126. As such, disruption of normal oscillatory profiles of glucocorticoids could lead to desynchronized activity between different brain regions as well as peripheral organ systems. This dissonance is thought to contribute to several pathologies that are similar to the effects of chronic stress, including obesity and metabolic syndrome119. Thus circadian disruption is both a “stressor” by increasing allostatic load/overload, and it is a risk factor for other stressful experiences, emphasizing the importance of timing in glucocorticoid actions through the brain and body.
IV. Conclusions and future directions
The response of the brain to stressors is a complex process involving multiple interacting mediators that utilizes both genomic and non-genomic mechanisms from the cell surface to the cytoskeleton to epigenetic regulation via the cell nucleus. Resilience in the face of stress is a key aspect of a healthy brain, even though gene expression shows a brain that continually changes with experience127. Therefore recovery of stress-induced changes in neural architecture after stress is not a “reversal” but a form of neuroplastic adaptation that also may be impaired in mood disorders and reduced with aging. Figure 1. Resilience may be thought of as an active process that implies ongoing adaptive plasticity without external intervention128.
On the other hand, resilience is decreased and vulnerability is increased by adverse childhood experiences (ACE) that lead to “biological embedding” of trajectories of response to stressful life events4 throughout the life course6 which contribute disproportionately to allostatic overload in the form of physical and mental health disorders over the life course7. Evidence from CpG methylation of DNA indicates the embedded influence of early adversity66. From the original definition of epigenetics129 as the emergence of characteristics of each individual of each species, not evident from prior stages of development, interventions for ACE cannot “roll back the clock” but rather may be able change the trajectory of brain and body development in a more positive direction.
Can the effects of stress on the brain be treated even though there are no "magic bullets" like penicillin for stress related disorders6? For psychiatric illnesses such as depression and anxiety disorders, including PTSD, it is necessary to complement and even replace existing drugs and adopt strategies that center around the use of targeted behavioral therapies along with treatments, including pharmaceutical agents, that open up “windows of plasticity" in the brain and facilitate the efficacy of the behavioral interventions5,130,131. To that extent, meeting the demands imposed by stressful experiences via various coping resources can lead to growth, adaptation, and learning to promote resilience and improved mental health132,128.
Acknowledgments
The McEwen lab acknowledges research support from NIH R01 MH41456 and the Hope for Depression Research Network and by the American Foundation for Suicide Prevention to Carla Nasca and NRSA Award F32 MH102065 to Jason Gray.
Footnotes
The authors declare that there are no conflicts of interest.
References
- 1.McEwen BS. Protective and Damaging Effects of Stress Mediators. New England J. Med. 1998;338:171–179. doi: 10.1056/NEJM199801153380307. [DOI] [PubMed] [Google Scholar]
- 2.McEwen BS. Protective and damaging effects of stress mediators: central role of the brain. Dial. in Clin. Neurosci.: Stress. 2006;8:367–381. doi: 10.31887/DCNS.2006.8.4/bmcewen. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.McEwen BS, Wingfield JC. The concept of allostasis in biology and biomedicine. Horm. & Behav. 2003;43:2–15. doi: 10.1016/s0018-506x(02)00024-7. [DOI] [PubMed] [Google Scholar]
- 4.Shonkoff JP, Boyce WT, McEwen BS. Neuroscience, molecular biology, and the childhood roots of health disparities. JAMA. 2009;301:2252–2259. doi: 10.1001/jama.2009.754. [DOI] [PubMed] [Google Scholar]
- 5.McEwen BS. Brain on stress: How the social environment gets under the skin. Proc Natl Acad Sci U S A. 2012;109(Suppl 2):17180–17185. doi: 10.1073/pnas.1121254109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Halfon N, Larson K, Lu M, Tullis E, Russ S. Lifecourse health development: past, present and future. Maternal and child health journal. 2014;18:344–365. doi: 10.1007/s10995-013-1346-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Felitti VJ, et al. Relationship of childhood abuse and household dysfunction to many of the leading causes of death in adults. The Adverse Childhood Experiences (ACE) Study. Am J Prev Med. 1998;14:245–258. doi: 10.1016/s0749-3797(98)00017-8. [DOI] [PubMed] [Google Scholar]
- 8.Tost HCD, Meyer-Lindenberg A. Environmental influence in the brain, human welfare and mental health. Nat Neurosci. 2015 doi: 10.1038/nn.4108. [DOI] [PubMed] [Google Scholar]
- 9.McEwen BS. Stress and hippocampal plasticity. Ann. Rev. Neurosci. 1999;22:105–122. doi: 10.1146/annurev.neuro.22.1.105. [DOI] [PubMed] [Google Scholar]
- 10.McEwen BS, Chattarji S. Handbook of Neurochemistry and Molecular Neurobiology. Springer-Verlag; 2007. pp. 572–593. [Google Scholar]
- 11.Cameron HA, Gould E. In: Receptor Dynamics in Neural Development. Shaw C, editor. CRC Press; 1996. pp. 141–157. [Google Scholar]
- 12.McEwen BS. Physiology and neurobiology of stress and adaptation: Central role of the brain. Physiol. Rev. 2007;87:873–904. doi: 10.1152/physrev.00041.2006. [DOI] [PubMed] [Google Scholar]
- 13.McEwen BS. Stress, sex, and neural adaptation to a changing environment: mechanisms of neuronal remodeling. Ann N Y Acad Sci. 2010;1204(Suppl):E38–E59. doi: 10.1111/j.1749-6632.2010.05568.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Meaney MJ, Szyf M. Environmental programming of stress responses through DNA methylation: life at the interface between a dynamic environment and a fixed genome. Dialogues Clin. Neurosci. 2005;7:103–123. doi: 10.31887/DCNS.2005.7.2/mmeaney. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hunter RG, Gagnidze K, McEwen BS, Pfaff DW. Stress and the dynamic genome: Steroids, epigenetics, and the transposome. Proc Natl Acad Sci U S A. 2014 doi: 10.1073/pnas.1411260111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.McEwen BS, Weiss J, Schwartz L. Selective retention of corticosterone by limbic structures in rat brain. Nature. 1968;220:911–912. doi: 10.1038/220911a0. [DOI] [PubMed] [Google Scholar]
- 17.Rubin TG, Gray JD, McEwen BS. Experience and the ever-changing brain: what the transcriptome can reveal. Bioessays. 2014;36:1072–1081. doi: 10.1002/bies.201400095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gray JD, Rubin TG, Hunter RG, McEwen BS. Hippocampal gene expression changes underlying stress sensitization and recovery. Mol Psychiatry. 2014;19:1171–1178. doi: 10.1038/mp.2013.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang K, et al. Transcriptome profiling analysis reveals region-distinctive changes of gene expression in the CNS in response to different moderate restraint stress. J Neurochem. 2010;113:1436–1446. doi: 10.1111/j.1471-4159.2010.06679.x. [DOI] [PubMed] [Google Scholar]
- 20.Datson NA, et al. Previous history of chronic stress changes the transcriptional response to glucocorticoid challenge in the dentate gyrus region of the male rat hippocampus. Endocrinology. 2013;154:3261–3272. doi: 10.1210/en.2012-2233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.de Kloet ER, Fitzsimons CP, Datson NA, Meijer OC, Vreugdenhil E. Glucocorticoid signaling and stress-related limbic susceptibility pathway: about receptors, transcription machinery and microRNA. Brain Res. 2009;1293:129–141. doi: 10.1016/j.brainres.2009.03.039. [DOI] [PubMed] [Google Scholar]
- 22.Goldwater DS, et al. Structural and functional alterations to rat medial prefrontal cortex following chronic restraint stress and recovery. Neuroscience. 2009;164:798–808. doi: 10.1016/j.neuroscience.2009.08.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hunter RG, McEwen BS. Stress and anxiety across the lifespan: structural plasticity and epigenetic regulation. Epigenomics. 2013;5:177–194. doi: 10.2217/epi.13.8. [DOI] [PubMed] [Google Scholar]
- 24.Griffiths BB, Hunter RG. Neuroepigenetics of stress. Neuroscience. 2014;275:420–435. doi: 10.1016/j.neuroscience.2014.06.041. [DOI] [PubMed] [Google Scholar]
- 25.Reul JM. Making memories of stressful events: a journey along epigenetic, gene transcription, and signaling pathways. Frontiers in psychiatry. 2014;5:5. doi: 10.3389/fpsyt.2014.00005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tsankova NM, et al. Sustained hippocampal chromatin regulation in a mouse model of depression and antidepressant action. Nat Neurosci. 2006;9:519–525. doi: 10.1038/nn1659. doi:nn1659 [pii] 10.1038/nn1659. [DOI] [PubMed] [Google Scholar]
- 27.Reul JMHM, Chandramohan Y. Epigenetic mechanisms in stress-related memory formation. Psychoneuroendocrinology. 2007;32:S21–S25. doi: 10.1016/j.psyneuen.2007.03.016. [DOI] [PubMed] [Google Scholar]
- 28.Heller EA, et al. Locus-specific epigenetic remodeling controls addiction- and depression-related behaviors. Nat Neurosci. 2014;17:1720–1727. doi: 10.1038/nn.3871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Popoli M, Yan Z, McEwen BS, Sanacora G. The stressed synapse: the impact of stress and glucocorticoids on glutamate transmission. Nat Rev Neurosci. 2012;13:22–37. doi: 10.1038/nrn3138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Monteggia LM, Zarate C., Jr Antidepressant actions of ketamine: from molecular mechanisms to clinical practice. Current opinion in neurobiology. 2015;30:139–143. doi: 10.1016/j.conb.2014.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nasca C, et al. L-acetylcarnitine causes rapid antidepressant effects through the epigenetic induction of mGlu2 receptors. Proc Natl Acad Sci U S A. 2013;110:4804–4809. doi: 10.1073/pnas.1216100110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hunter RG, McCarthy KJ, Milne TA, Pfaff DW, McEwen BS. Regulation of hippocampal H3 histone methylation by acute and chronic stress. Proc. Natl. Acad. Sci. USA. 2009;106:20912–20917. doi: 10.1073/pnas.0911143106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hunter RG, et al. Acute stress and hippocampal histone H3 lysine 9 trimethylation, a retrotransposon silencing response. Proc Natl Acad Sci U S A. 2012;109:17657–17662. doi: 10.1073/pnas.1215810109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Erwin JA, Marchetto MC, Gage FH. Mobile DNA elements in the generation of diversity and complexity in the brain. Nat Rev Neurosci. 2014;15:497–506. doi: 10.1038/nrn3730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Reilly MT, Faulkner GJ, Dubnau J, Ponomarev I, Gage FH. The role of transposable elements in health and diseases of the central nervous system. J Neurosci. 2013;33:17577–17586. doi: 10.1523/JNEUROSCI.3369-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hunter RG, McEwen BS, Pfaff DW. Environmental stress and transposon transcription in the mammalian brain. Mob Genet Elements. 2013;3:e24555. doi: 10.4161/mge.24555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Khalil AM, et al. Many human large intergenic noncoding RNAs associate with chromatin-modifying complexes and affect gene expression. Proc Natl Acad Sci U S A. 2009;106:11667–11672. doi: 10.1073/pnas.0904715106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Johnson R, Guigo R. The RIDL hypothesis: transposable elements as functional domains of long noncoding RNAs. Rna. 2014;20:959–976. doi: 10.1261/rna.044560.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kelley D, Rinn J. Transposable elements reveal a stem cell-specific class of long noncoding RNAs. Genome biology. 2012;13:R107. doi: 10.1186/gb-2012-13-11-r107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Khalil AM, Rinn JL. RNA-protein interactions in human health and disease. Seminars in cell & developmental biology. 2011;22:359–365. doi: 10.1016/j.semcdb.2011.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pauli A, Rinn JL, Schier AF. Non-coding RNAs as regulators of embryogenesis. Nat Rev Genet. 2011;12:136–149. doi: 10.1038/nrg2904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.McClintock B. Chromosome organization and genic expression. Cold Spring Harbor symposia on quantitative biology. 1951;16:13–47. doi: 10.1101/sqb.1951.016.01.004. [DOI] [PubMed] [Google Scholar]
- 43.McClintock B. The significance of responses of the genome to challenge. Science. 1984;226:792–801. doi: 10.1126/science.15739260. [DOI] [PubMed] [Google Scholar]
- 44.Rusiecki JA, et al. DNA methylation in repetitive elements and post-traumatic stress disorder: a case-control study of US military service members. Epigenomics. 2012;4:29–40. doi: 10.2217/epi.11.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ponomarev I, Rau V, Eger EI, Harris RA, Fanselow MS. Amygdala transcriptome and cellular mechanisms underlying stress-enhanced fear learning in a rat model of posttraumatic stress disorder. Neuropsychopharmacology. 2010;35:1402–1411. doi: 10.1038/npp.2010.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ponomarev I, Wang S, Zhang L, Harris RA, Mayfield RD. Gene coexpression networks in human brain identify epigenetic modifications in alcohol dependence. J Neurosci. 2012;32:1884–1897. doi: 10.1523/JNEUROSCI.3136-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lowy MT, Gault L, Yamamoto BK. Adrenalectomy attenuates stress-induced elevations in extracellular glutamate concentrations in the hippocampus. J Neurochem. 1993;61:1957–1960. doi: 10.1111/j.1471-4159.1993.tb09839.x. [DOI] [PubMed] [Google Scholar]
- 48.Karst H, et al. Mineralocorticoid receptors are indispensable for nongenomic modulation of hippocampal glutamate transmission by corticosterone. Proc. Natl. Acad. Sci. USA. 2005;102:19204–19207. doi: 10.1073/pnas.0507572102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Treccani G, et al. Stress and corticosterone increase the readily releasable pool of glutamate vesicles in synaptic terminals of prefrontal and frontal cortex. Mol Psychiatry. 2014;19:433–443. doi: 10.1038/mp.2014.5. [DOI] [PubMed] [Google Scholar]
- 50.Magarinos AM, McEwen BS. Stress-induced atrophy of apical dendrites of hippocampal CA3c neurons: involvement of glucocorticoid secretion and excitatory amino acid receptors. Neuroscience. 1995;69:89–98. doi: 10.1016/0306-4522(95)00259-l. [DOI] [PubMed] [Google Scholar]
- 51.Watanabe Y, Gould E, McEwen BS. Stress induces atrophy of apical dendrites of hippocampus CA3 pyramidal neurons. Brain. Res. 1992;588:341–344. doi: 10.1016/0006-8993(92)91597-8. [DOI] [PubMed] [Google Scholar]
- 52.Martin KP, Wellman CL. NMDA receptor blockade alters stress-induced dendritic remodeling in medial prefrontal cortex. Cereb Cortex. 2011;21:2366–2373. doi: 10.1093/cercor/bhr021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sapolsky RM, Krey LC, McEwen BS. The Neuroendocrinology of Stress and Aging: The Glucocorticoid Cascade Hypothesis. Endocr. Rev. 1986;7:284–301. doi: 10.1210/edrv-7-3-284. [DOI] [PubMed] [Google Scholar]
- 54.Lisman JE. Relating hippocampal circuitry to function: Recall of memory sequences by reciprocal dentate-CA3 interactions. 1999;22:233–242. doi: 10.1016/s0896-6273(00)81085-5. [DOI] [PubMed] [Google Scholar]
- 55.McEwen BS. Stress and hippocampal plasticity. Annu. Rev. Neurosci. 1999;22:105–122. doi: 10.1146/annurev.neuro.22.1.105. [DOI] [PubMed] [Google Scholar]
- 56.Magarinos AM, McEwen BS, Saboureau M, Pevet P. Rapid and reversible changes in intrahippocampal connectivity during the course of hibernation in European hamsters. Proc. Natl. Acad. Sci. USA. 2006;103:18775–18780. doi: 10.1073/pnas.0608785103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Popov VI, Bocharova LS, Bragin AG. Repeated changes of dendritic morphology in the hippocampus of ground squirrels in the course of hibernation. Neuroscience. 1992;48:45–51. doi: 10.1016/0306-4522(92)90336-z. [DOI] [PubMed] [Google Scholar]
- 58.McCall T, et al. Depletion of polysialic acid from neural cell adhesion molecule (PSA-NCAM) increases CA3 dendritic arborization and increases vulnerability to excitotoxicity. Exp Neurol. 2013;241:5–12. doi: 10.1016/j.expneurol.2012.11.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Joels M. Corticosteroid effects in the brain: U-shape it. Trends Pharmacol. Sci. 2006;27:244–250. doi: 10.1016/j.tips.2006.03.007. [DOI] [PubMed] [Google Scholar]
- 60.Revollo JR, Cidlowski JA. Mechanisms generating diversity in glucocorticoid receptor signaling. Ann N Y Acad Sci. 2009;1179:167–178. doi: 10.1111/j.1749-6632.2009.04986.x. [DOI] [PubMed] [Google Scholar]
- 61.Hill MN, McEwen BS. Involvement of the endocannabinoid system in the neurobehavioural effects of stress and glucocorticoids. Prog. Neuro-Psychopharm. & Biol. Psychiat. 2009 doi: 10.1016/j.pnpbp.2009.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Du J, et al. Dynamic regulation of mitochondrial function by glucocorticoids. Proc. Natl. Acad. Sci. USA. 2009;106:3543–3548. doi: 10.1073/pnas.0812671106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wei Q, et al. Glucocorticoid receptor overexpression in forebrain: A mouse model of increased emotional lability. Proc. Natl. Acad. Sci. USA. 2004;101:11851–11856. doi: 10.1073/pnas.0402208101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Jacobson L. Forebrain glucocorticoid receptor gene deletion attenuates behavioral changes and antidepressant responsiveness during chronic stress. Brain Res. 2014;1583:109–121. doi: 10.1016/j.brainres.2014.07.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Szyf M, Weaver ICG, Champagne FA, Diorio J, Meaney MJ. Maternal programming of steroid receptor expression and phenotype through DNA methylation in the rat. Front. Neuroendocrin. 2005;26:139–162. doi: 10.1016/j.yfrne.2005.10.002. [DOI] [PubMed] [Google Scholar]
- 66.McGowan PO, et al. Epigenetic regulation of the glucocorticoid receptor in human brain associates with childhood abuse. Nature Neurosci. 2009;12:241–243. doi: 10.1038/nn.2270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Rao RP, Anilkumar S, McEwen BS, Chattarji S. Glucocorticoids protect against the delayed behavioral and cellular effects of acute stress on the amygdala. Biol Psychiatry. 2012;72:466–475. doi: 10.1016/j.biopsych.2012.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zohar J, et al. High dose hydrocortisone immediately after trauma may alter the trajectory of PTSD: interplay between clinical and animal studies. Eur Neuropsychopharmacol. 2011;21:796–809. doi: 10.1016/j.euroneuro.2011.06.001. [DOI] [PubMed] [Google Scholar]
- 69.Schelling G, Roozendaal B, De Quervain DJ-F. Can posttraumatic stress disorder be prevented with glucocorticoids? Ann. N. Y. Acad. Sci. 2004;1032:158–166. doi: 10.1196/annals.1314.013. [DOI] [PubMed] [Google Scholar]
- 70.Mitra R, Sapolsky RM. Acute corticosterone treatment is sufficient to induce anxiety and amygdaloid dendritic hypertrophy. Proc. Natl. Acad. Sci. USA. 2008;105:5573–5578. doi: 10.1073/pnas.0705615105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Nasca C, Bigio B, Zelli D, Nicoletti F, McEwen BS. Mind the gap: glucocorticoids modulate hippocampal glutamate tone underlying individual differences in stress susceptibility. Mol Psychiatry. 2014 doi: 10.1038/mp.2014.96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Francis D, Diorio J, Liu D, Meaney MJ. Nongenomic transmission across generations of maternal behavior and stress responses in the rat. Science. 1999;286:1155–1158. doi: 10.1126/science.286.5442.1155. [DOI] [PubMed] [Google Scholar]
- 73.Pawlak R, et al. Tissue plasminogen activator and plasminogen mediate stress-induced decline of neuronal and cognitive functions in the mouse hippocampus. Proc. Natl. Acad. Sci. USA. 2005;102:18201–18206. doi: 10.1073/pnas.0509232102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Chen Y, Fenoglio KA, Dube CM, Grigoriadis DE, Baram TZ. Cellular and molecular mechanisms of hippocampal activation by acute stress are age-dependent. Mol. Psychiat. 2006;11:992–1002. doi: 10.1038/sj.mp.4001863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Bennur S, et al. Stress-induced spine loss in the medial amygdala is mediated by tissue-plasminogen activator. Neuroscience. 2007;144:8–16. doi: 10.1016/j.neuroscience.2006.08.075. [DOI] [PubMed] [Google Scholar]
- 76.Matys T, et al. Tissue plasminogen activator promotes the effects of corticotropin releasing factor on the amygdala and anxiety-like behavior. Proc. Natl. Acad. Sci. USA. 2004;101:16345–16350. doi: 10.1073/pnas.0407355101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Mucha M, et al. Lipocalin-2 controls neuronal excitability and anxiety by regulating dendritic spine formation and maturation. Proc Natl Acad Sci U S A. 2011;108:18436–18441. doi: 10.1073/pnas.1107936108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Skrzypiec AE, et al. Stress-induced lipocalin-2 controls dendritic spine formation and neuronal activity in the amygdala. PLoS One. 2013;8:e61046. doi: 10.1371/journal.pone.0061046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Hill MN, et al. Recruitment of prefrontal cortical endocannabinoid signaling by glucocorticoids contributes to termination of the stress response. J Neurosci. 2011;31:10506–10515. doi: 10.1523/JNEUROSCI.0496-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Hill MN, et al. Endogenous cannabinoid signaling is essential for stress adaptation. Proc Natl Acad Sci U S A. 2010;107:9406–9411. doi: 10.1073/pnas.0914661107. doi:0914661107 [pii] 10.1073/pnas.0914661107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Hill MN, Hillard CJ, McEwen BS. Alterations in corticolimbic dendritic morphology and emotional behavior in cannabinoid CB1 receptor-deficient mice parallel the effects of chronic stress. Cereb Cortex. 2011;21:2056–2064. doi: 10.1093/cercor/bhq280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Lee TT, Filipski SB, Hill MN, McEwen BS. Morphological and behavioral evidence for impaired prefrontal cortical function in female CB1 receptor deficient mice. Behav Brain Res. 2014;271:106–110. doi: 10.1016/j.bbr.2014.05.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Hill MN, et al. Downregulation of endocannabinoid signaling in the hippocampus following chronic unpredictable stress. Neuropsychopharmacology. 2005;30:508–515. doi: 10.1038/sj.npp.1300601. [DOI] [PubMed] [Google Scholar]
- 84.Bowles NP, et al. Chronic, noninvasive glucocorticoid administration suppresses limbic endocannabinoid signaling in mice. Neuroscience. 2012;204:83–89. doi: 10.1016/j.neuroscience.2011.08.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Lomazzo E, et al. Therapeutic potential of inhibitors of endocannabinoid degradation for the treatment of stress-related hyperalgesia in an animal model of chronic pain. Neuropsychopharmacology. 2015;40:488–501. doi: 10.1038/npp.2014.198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Bortolato M, et al. Antidepressant-like activity of the fatty acid amide hydrolase inhibitor URB597 in a rat model of chronic mild stress. Biol Psychiatry. 2007;62:1103–1110. doi: 10.1016/j.biopsych.2006.12.001. [DOI] [PubMed] [Google Scholar]
- 87.Rossi S, et al. Preservation of striatal cannabinoid CB1 receptor function correlates with the antianxiety effects of fatty acid amide hydrolase inhibition. Mol Pharmacol. 2010;78:260–268. doi: 10.1124/mol.110.064196. [DOI] [PubMed] [Google Scholar]
- 88.Zoppi S, et al. Regulatory role of cannabinoid receptor 1 in stress-induced excitotoxicity and neuroinflammation. Neuropsychopharmacology. 2011;36:805–818. doi: 10.1038/npp.2010.214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Gray JM, et al. Corticotropin-releasing hormone drives anandamide hydrolysis in the amygdala to promote anxiety. J Neurosci. 2015;35:3879–3892. doi: 10.1523/JNEUROSCI.2737-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Daskalakis NP, Cohen H, Cai G, Buxbaum JD, Yehuda R. Expression profiling associates blood and brain glucocorticoid receptor signaling with trauma-related individual differences in both sexes. Proc Natl Acad Sci U S A. 2014;111:13529–13534. doi: 10.1073/pnas.1401660111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Govindarajan A, et al. Transgenic brain-derived neurotrophic factor expression causes both anxiogenic and antidepressant effects. Proc. Natl. Acad. Sci. USA. 2006;103:13208–13213. doi: 10.1073/pnas.0605180103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Lakshminarasimhan H, Chattarji S. Stress leads to contrasting effects on the levels of brain derived neurotrophic factor in the hippocampus and amygdala. PLoS One. 2012;7:e30481. doi: 10.1371/journal.pone.0030481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Mitra R, Jadhav S, McEwen BS, Vyas A, Chattarji S. Stress duration modulates the spatiotemporal patterns of spine formation in the basolateral amygdala. Proc. Nat. Acad. Sci USA. 2005;102:9371–9376. doi: 10.1073/pnas.0504011102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Tsankova N, Renthal W, Kumar A, Nestler EJ. Epigenetic regulation in psychiatric disorders. Nature. Rev. Neurosci. 2007;8:355–367. doi: 10.1038/nrn2132. [DOI] [PubMed] [Google Scholar]
- 95.Gray JD, Milner TA, McEwen BS. Dynamic plasticity: the role of glucocorticoids, brain-derived neurotrophic factor and other trophic factors. Neuroscience. 2013;239:214–227. doi: 10.1016/j.neuroscience.2012.08.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Suri D, et al. Early stress evokes age-dependent biphasic changes in hippocampal neurogenesis, BDNF expression, and cognition. Biol Psychiatry. 2013;73:658–666. doi: 10.1016/j.biopsych.2012.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Seki T, Arai Y. Different polysialic acid-neural cell adhesion molecule expression patterns in distinct types of mossy fiber boutons in the adult hippocampus. J Comp Neurol. 1999;410:115–125. doi: 10.1002/(sici)1096-9861(19990719)410:1<115::aid-cne10>3.0.co;2-c. [DOI] [PubMed] [Google Scholar]
- 98.Rutishauser U. Polysialic acid in the plasticity of the developing and adult vertebrate nervous system. Nature. Rev. Neuroscience. 2008;9:26–35. doi: 10.1038/nrn2285. [DOI] [PubMed] [Google Scholar]
- 99.Theodosis DT, Bonhomme R, Vitiello S, Rougon G, Poulain DA. Cell surface expression of polysialic acid on NCAM is a prerequisite for activity-dependent morphological neuronal and glial plasticity. J. Neurosci. 1999;19:10228–10236. doi: 10.1523/JNEUROSCI.19-23-10228.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Pham K, Nacher J, Hof PR, McEwen BS. Repeated restraint stress suppresses neurogenesis and induces biphasic PSA-NCAM expression in the adult rat dentate gyrus. Eur J Neurosci. 2003;17:879–886. doi: 10.1046/j.1460-9568.2003.02513.x. [DOI] [PubMed] [Google Scholar]
- 101.Sandi C. Stress, cognitive impairment and cell adhesion molecules. Nature. Rev. /Neurosci. 2004;5:917–930. doi: 10.1038/nrn1555. [DOI] [PubMed] [Google Scholar]
- 102.van der Kooij MA, et al. Impaired hippocampal neuroligin-2 function by chronic stress or synthetic peptide treatment is linked to social deficits and increased aggression. Neuropsychopharmacology. 2014;39:1148–1158. doi: 10.1038/npp.2013.315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Wood GE, Young LT, Reagan LP, McEwen BS. Acute and chronic restraint stress alter the incidence of social conflict in male rats. Horm. & Behav. 2003;43:205–213. doi: 10.1016/s0018-506x(02)00026-0. [DOI] [PubMed] [Google Scholar]
- 104.van der Kooij MA, et al. Role for MMP-9 in stress-induced downregulation of nectin-3 in hippocampal CA1 and associated behavioural alterations. Nature communications. 2014;5:4995. doi: 10.1038/ncomms5995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Matus A, Brinkhaus H, Wagner U. Actin dynamics in dendritic spines: A form of regulated plasticity at excitatory synapses. Hippocampus. 2000;10:555–560. doi: 10.1002/1098-1063(2000)10:5<555::AID-HIPO5>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- 106.Arendt T, et al. Reversible paired helical filament-like phosphorylation of tau is an adaptive process associated with neuronal plasticity in hibernating animals. J. Neurosci. 2003;23:6972–6981. doi: 10.1523/JNEUROSCI.23-18-06972.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Morris M, Maeda S, Vossel K, Mucke L. The many faces of tau. Neuron. 2011;70:410–426. doi: 10.1016/j.neuron.2011.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Kinoshita Y, et al. Role for NUP62 depletion and PYK2 redistribution in dendritic retraction resulting from chronic stress. Proc Natl Acad Sci U S A. 2014;111:16130–16135. doi: 10.1073/pnas.1418896111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Tochigi M, et al. Gene expression profiling of major depression and suicide in the prefrontal cortex of postmortem brains. Neuroscience research. 2008;60:184–191. doi: 10.1016/j.neures.2007.10.010. [DOI] [PubMed] [Google Scholar]
- 110.Gould E, McEwen BS, Tanapat P, Galea LAM, Fuchs E. Neurogenesis in the dentate gyrus of the adult tree shrew is regulated by psychosocial stress and NMDA receptor activation. J. Neurosci. 1997;17:2492–2498. doi: 10.1523/JNEUROSCI.17-07-02492.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Gould E, Tanapat P, McEwen BS, Flugge G, Fuchs E. Proliferation of granule cell precursors in the dentate gyrus of adult monkeys is diminished by stress. Proc. Natl. Acad. Sci. USA. 1998;95:3168–3171. doi: 10.1073/pnas.95.6.3168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Cameron HA, Gould E. Adult neurogenesis is regulated by adrenal steroids in the dentate gyrus. Neuroscience. 1994;61:203–209. doi: 10.1016/0306-4522(94)90224-0. [DOI] [PubMed] [Google Scholar]
- 113.Leuner B, Glasper ER, Gould E. Sexual experience promotes adult neurogenesis in the hippocampus despite an initial elevation in stress hormones. PLoS One. 2010;5:e11597. doi: 10.1371/journal.pone.0011597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Leuner B, Caponiti JM, Gould E. Oxytocin stimulates adult neurogenesis even under conditions of stress and elevated glucocorticoids. Hippocampus. 2012;22:861–868. doi: 10.1002/hipo.20947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Lightman SL, et al. The significance of glucocorticoid pulsatility. European journal of pharmacology. 2008;583:255–262. doi: 10.1016/j.ejphar.2007.11.073. [DOI] [PubMed] [Google Scholar]
- 116.Liston C, Gan WB. Glucocorticoids are critical regulators of dendritic spine development and plasticity in vivo. Proc Natl Acad Sci U S A. 2011;108:16074–16079. doi: 10.1073/pnas.1110444108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Liston C, et al. Circadian glucocorticoid oscillations promote learning-dependent synapse formation and maintenance. Nat Neurosci. 2013;16:698–705. doi: 10.1038/nn.3387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Akana SF, Jacobson L, Cascio CS, Shinsako J, Dallman MF. Constant corticosterone replacement normalizes basal adrenocorticotropin (ACTH) but permits sustained ACTH hypersecretion after stress in adrenalectomized rats. Endocrinology. 1988;122:1337–1342. doi: 10.1210/endo-122-4-1337. [DOI] [PubMed] [Google Scholar]
- 119.Karatsoreos IN, McEwen BS. Timing is everything: a collection on how clocks affect resilience in biological systems. F1000Research. 2014;3:273. doi: 10.12688/f1000research.5756.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Seeman T, Epel E, Gruenewald T, Karlamangla A, McEwen BS. Socio-economic differentials in peripheral biology: cumulative allostatic load. Ann N Y Acad Sci. 2010;1186:223–239. doi: 10.1111/j.1749-6632.2009.05341.x. [DOI] [PubMed] [Google Scholar]
- 121.Karatsoreos IN, McEwen BS. Psychobiological allostasis: resistance, resilience and vulnerability. Trends in cognitive sciences. 2011;15:576–584. doi: 10.1016/j.tics.2011.10.005. [DOI] [PubMed] [Google Scholar]
- 122.Radley JJ, et al. Chronic behavioral stress induces apical dendritic reorganization in pyramidal neurons of the medial prefrontal cortex. Neuroscience. 2004;125:1–6. doi: 10.1016/j.neuroscience.2004.01.006. [DOI] [PubMed] [Google Scholar]
- 123.Karatsoreos IN, Bhagat S, Bloss EB, Morrison JH, McEwen BS. Disruption of circadian clocks has ramifications for metabolism, brain, and behavior. Proc Natl Acad Sci U S A. 2011;108:1657–1662. doi: 10.1073/pnas.1018375108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Phillips DJ, Savenkova MI, Karatsoreos IN. Environmental disruption of the circadian clock leads to altered sleep and immune responses in mouse. Brain Behav Immun. 2014 doi: 10.1016/j.bbi.2014.12.008. [DOI] [PubMed] [Google Scholar]
- 125.Lamont EW, Robinson B, Stewart J, Amir S. The central and basolateral nuclei of the amygdala exhibit opposite diurnal rhythms of expression of the clock protein Period2. Proc Natl Acad Sci U S A. 2005;102:4180–4184. doi: 10.1073/pnas.0500901102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Reddy AB, et al. Glucocorticoid signaling synchronizes the liver circadian transcriptome. Hepatology. 2007;45:1478–1488. doi: 10.1002/hep.21571. [DOI] [PubMed] [Google Scholar]
- 127.McEwen BS, Gray J, Nasca C. Recognizing Resilience: Learning from the Effects of Stress on the Brain. Neurobiology of stress. 2015;1:1–11. doi: 10.1016/j.ynstr.2014.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Russo SJ, Murrough JW, Han MH, Charney DS, Nestler EJ. Neurobiology of resilience. Nat Neurosci. 2012;15:1475–1484. doi: 10.1038/nn.3234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Waddington CH. The epigenotype. Endeavoour. 1942;1:18–20. [Google Scholar]
- 130.Bavelier D, Levi DM, Li RW, Dan Y, Hensch TK. Removing brakes on adult brain plasticity: from molecular to behavioral interventions. J Neurosci. 2010;30:14964–14971. doi: 10.1523/JNEUROSCI.4812-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Castren E, Rantamaki T. The role of BDNF and its receptors in depression and antidepressant drug action: Reactivation of developmental plasticity. Dev Neurobiol. 2010;70:289–297. doi: 10.1002/dneu.20758. [DOI] [PubMed] [Google Scholar]
- 132.McEwen BS, Gianaros PJ. Stress- and allostasis-induced brain plasticity. Annu Rev Med. 2011;62:431–445. doi: 10.1146/annurev-med-052209-100430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Yan X-X, Toth Z, Schultz L, Ribak CE, Baram TZ. Corticotropin-releasing hormone (CRH)-containing neurons in the immature rat hippocampal formation: Light and electron microscopic features and colocalization with glutamate decarboxylase and parvalbumin. Hippocampus. 1998;8:231–243. doi: 10.1002/(SICI)1098-1063(1998)8:3<231::AID-HIPO6>3.0.CO;2-M. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Hill MN, McEwen BS. Involvement of the endocannabinoid system in the neurobehavioural effects of stress and glucocorticoids. Prog Neuropsychopharmacol Biol Psychiatry. 2010;34:791–797. doi: 10.1016/j.pnpbp.2009.11.001. doi:S0278-5846(09)00387-X [pii] 10.1016/j.pnpbp.2009.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Gunduz-Cinar O, Hill MN, McEwen BS, Holmes A. Amygdala FAAH and anandamide: mediating protection and recovery from stress. Trends in pharmacological sciences. 2013;34:637–644. doi: 10.1016/j.tips.2013.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Hill MN, et al. Disruption of fatty acid amide hydrolase activity prevents the effects of chronic stress on anxiety and amygdalar microstructure. Mol Psychiatry. 2013;18:1125–1135. doi: 10.1038/mp.2012.90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Vyas A, Mitra R, Rao BSS, Chattarji S. Chronic stress induces contrasting patterns of dendritic remodeling in hippocampal and amygdaloid neurons. J. Neurosci. 2002;22:6810–6818. doi: 10.1523/JNEUROSCI.22-15-06810.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Liston C, et al. Stress-induced alterations in prefrontal cortical dendritic morphology predict selective impairments in perceptual attentional set-shifting. J. Neurosci. 2006;26:7870–7874. doi: 10.1523/JNEUROSCI.1184-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.McEwen BS, Morrison JH. The Brain on Stress: Vulnerability and Plasticity of the Prefrontal Cortex over the Life Course. Neuron. 2013;79:16–29. doi: 10.1016/j.neuron.2013.06.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Du J, McEwen BS, Manji HK. Glucocorticoid receptors modulate mitochondrial function. Commun. & Integr. Biol. 2009;2:1–3. doi: 10.4161/cib.2.4.8554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Armanini M, Hutchings C, Stein B, Sapolsky R. Glucocorticoid endangerment of hippocampal neurons is NMDA-receptor dependent. Brain. Res. 1990;532:7–12. doi: 10.1016/0006-8993(90)91734-x. [DOI] [PubMed] [Google Scholar]