

Abstract
Autism spectrum disorders (ASD) are clinically heterogeneous and biologically complex. In general it remains unclear, what biological factors lead to changes in the brains of autistic individuals. A considerable number of transcriptome analyses have been performed in attempts to address this question, but their findings lack a clear consensus. As a result, each of these individual studies has not led to any significant advance in understanding the autistic phenotype as a whole. Here we report a meta-analysis of over 1000 microarrays across twelve independent studies on expression changes in ASD compared to unaffected individuals, in both blood and brain tissues. We identified a number of known and novel genes that are consistently differentially expressed across three studies of the brain (71 samples in total). A subset of the highly ranked genes is suggestive of effects on mitochondrial function. In blood, consistent changes were more difficult to identify, despite individual studies tending to exhibit larger effects than the brain studies. Our results are the strongest evidence to date of a common transcriptome signature in the brains of individuals with ASD.
Keywords: gene expression, meta-analysis
Lay Abstract
Research findings reported on the differences between gene expressions of individuals with autism spectrum disorders (ASD) and those without lack a clear consensus. Here we present a meta-analysis across multiple independent studies on expression changes in ASD compared to unaffected individuals, in both blood and brain tissues. We identified some molecular commonalities across brain studies. In blood, consistent changes were more difficult to identify. Our results are the strongest evidence to date of a common expression signature in the brains of individuals with ASD.
Introduction
Autism spectrum disorder (ASD; MIM 209850) encompasses a range of highly heritable but genetically heterogeneous neurodevelopmental diseases (Berg & Geschwind, 2012). ASD is characterized as a set of behavioral phenotypes including social communication deficits, restrictive and repetitive behaviors (Diagnostic and Statistical Manual of Mental Disorders, DSM-5 299.00)(American Psychiatric Association, American Psychiatric Association, & DSM-5 Task Force, 2013). Despite this common (though broadly-defined) behavioural profile, variations within any single gene accounts for only a small fraction of cases (Voineagu, 2012). Thus the mechanistic connection between this genetic diversity and the common phenotypic outcomes are poorly understood. Given this complexity, there appear to be two models for how ASD arises. One is that many different genetic lesions lead to a common set of changes in the brain, which gives rise to a common range of behavioral traits. Alternatively, the behavioral manifestations may be due to widely varying underlying pathologies. The truth may lie between these two extremes, and there has been much effort to identify biomarkers or endophenotypes that unify ASD, or at least provide a scheme for biological stratification intermediate between genotype and behaviour. The search has spanned many modalities, including neuroanatomy, proteomics and transcriptomics. Examples of markers highlighted in imaging studies include facial features (Hammond et al., 2008) and neural responses to facial expressions (Spencer et al., 2011).
In this paper we take up the idea that commonalities among ASD cases might be discerned in the transcriptome, which is an attractive intermediate phenotype for investigation. The hypothesis is that molecular commonalities might be revealed across individuals, helping to explain the autistic phenotype regardless of their genetic background or specific causal variants underlying their autism. In agreement with this, two previous studies reported some convergence in the transcriptomes of independent ASD cohorts (Nishimura et al., 2007; Voineagu et al., 2011). Nishimura et al. (2007) studied ASD individuals with either maternally derived 15q duplications, or fragile–X mutations (FMR1-FM). They reported similarities in the molecular pathways affected. Voineagu et al. (2011) found evidence for convergent molecular abnormalities between gene expressions in post mortem brain samples and an independent cohort from a genome wide association study (GWAS). However, while these reports described some agreements within studies, it is not clear how much agreement there is across studies. For example, Nishimura et al.’s gene list was most enriched for “cell communication”; Voineagu et al. reported enrichment of genes involved in “synaptic function”, “vesicular transport” and “neuronal projection”. Other transcriptome studies have implicated an even more diverse array of biological functions, ranging from circadian rhythms (V. W. Hu, Sarachana, et al., 2009) to metabolism (Ginsberg, Rubin, Falcone, Ting, & Natowicz, 2012). But because no detailed comparison or meta-analysis has been conducted, it remains unclear whether there might be more subtle similarities among these independent studies.
There are many possible reasons why previous studies report different genes and pathways as being affected in ASD, even if there are commonalities present. One is the difference in tissues or cell types analyzed. Another is clinical heterogeneity (Geschwind & Levitt, 2007; McClellan & King, 2010), which might lead to some differences in the research population among studies. Also potentially contributing are methodological differences in the design and implementation of analyses. Finally, small sample sizes of individual studies might not provide sufficient statistical power to uncover subtle perturbations. These issues may mask reproducible aspects of the transcriptome in ASD, which might be revealed by re-examining the original data and performing a meta-analysis. A systematic meta-analysis can overcome sample size limitations and reduce the effects of methodological differences.
To our knowledge, cross-cohort gene expression analyses have only been done in at most two independent ASD cohorts, primarily for cross validation purposes (Kong et al., 2012; Voineagu et al., 2011). Other ASD related meta-analyses are geared towards examining pathogenic variations in whole exomes of individuals (Ben-David & Shifman, 2012; Liu et al., 2013), not transcriptomes. As meta-analysis techniques have been successfully applied in neuropsychiatry (Choi et al., 2008; Mistry, Gillis, & Pavlidis, 2012; Rogic & Pavlidis, 2009) a systematic integration of expression data across multiple independent ASD cohorts will add value to the existing data, and may yield novel insights.
Here we report the meta-analysis of data from twelve ASD transcriptome studies. Together, they comprise over 1000 human samples, 634 of which are from ASD individuals. Despite considerable heterogeneity across cohorts, our analysis reveals genes with consistently altered expression levels in ASD, especially in the brain.
Results
Systematic review shows technical differences and heterogeneity in independent ASD transcriptome studies
We analyzed twelve independent ASD expression-profiling studies and identified differences in microarray preprocessing and data quality control. To ensure comparability among data sets from different laboratories, we corrected for technical variation where possible (Figure 1, Materials and Methods). The resulting data after quality control comprise 634 ASD microarray samples and 457 controls from blood-derived and brain tissues. The studies included are summarized in Table 1.
Figure 1.
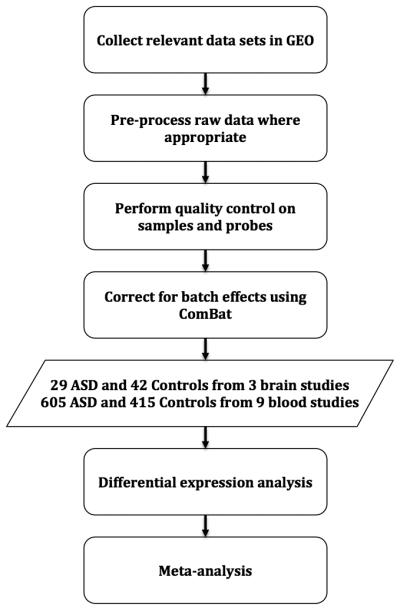
Overview of analysis pipeline.
Table 1.
Data sets used for meta-analysis.
| Data sets | Platform | Reference | Tissue Type | Number of Samples ASD:Control |
|---|---|---|---|---|
| Brain | ||||
| GSE28475 | GPL6883 | (Chow et al., 2012) | Cortex | 13 : 21 |
| GSE28521 | GPL6883 | (Voineagu et al., 2011) | Frontal/ temporal cortex | 12 : 15 |
| GSE38322 | GPL10558 | (Ginsberg et al., 2012) | Occipital cortex | 4 : 6 |
|
| ||||
| 29 : 42 = 71 | ||||
| Blood | ||||
| GSE6575 | GPL570 | (Gregg et al., 2008) | Whole blood | 33 : 11 |
| GSE7329 | GPL1708 | (Nishimura et al., 2007) | Lymphoblastoid cell lines | 7 : 5 |
| GSE15402 | GPL3427 | (V. W. Hu, Sarachana, et al., 2009) | Lymphoblastoid cell lines | 77 : 29 |
| GSE15451 | GPL3427 | (V. W. Hu, Nguyen, et al., 2009) | Lymphoblastoid cell lines | 15 : 12 |
| GSE18123.1 | GPL570 | (Kong et al., 2012) | Whole blood | 64 : 28 |
| GSE18123.2 | GPL6244 | (Kong et al., 2012) | Whole blood | 93 : 63 |
| GSE25507 | GPL570 | (Alter et al., 2011) | Peripheral blood lymphocytes | 80 : 63 |
| GSE32136 | GPL3427 | Unpublished | Lymphoblastoid cell lines | 4 : 4 |
| GSE37772 | GPL6883 | (Luo et al., 2012) | Lymphoblastoid cell lines | 232 : 199 |
|
| ||||
| 605 : 415 = 1020 | ||||
As summarized in Table S2, there were differences among studies in the criteria used to select the pool of ASD individuals. Some individuals were diagnosed based on DSM-IV (American Psychiatric Association, American Psychiatric Association, & Task Force on DSM-IV, 2000); others were determined using the Autism Diagnostic Interview-Revised (ADI-R) (Lord, Rutter, & Le Couteur, 1994) or Autism Diagnostic Observation Schedule (ADOS) (Lord et al., 1989). More importantly, the range of autistic phenotypes included in each cohort differs, particularly among the blood studies.
While some focused on “classical” autism, others included milder forms like Asperger’s syndrome and pervasive developmental disorder not otherwise specified (PDD-NOS). Because ASD is generally more prevalent in males than females (V. W. Hu, Nguyen, et al., 2009; V. W. Hu, Sarachana, et al., 2009), we investigated whether gender imbalance was a factor affecting study designs. Indeed, a few studies showed evidence of gender imbalance, such that the subjects tend to be males with ASD (Table S4). There were no striking differences in the age, race and post-mortem interval (the latter being relevant to brain studies only) between cases and controls of each study (Table S5).
As an initial investigation of similarities across studies, we compared the lists of differentially expressed genes reported in each publication. None of the genes reported overlapped across all brain data sets or blood data sets (there were some overlaps in smaller subsets of studies, Table S13). However, because each publication used different methods for selecting genes, a more careful re-analysis is warranted, as described in the next sections.
Re-analysis for differential expression
The first stage of our meta-analysis was to analyze each data set individually for differential expression. The results are summarized in Table 3. Most data sets had low levels of differential expression, but a few range up to hundreds of significantly differentially expressed genes at a false discovery rate (FDR) of 0.05. We checked if sample size or the fixed FDR threshold could explain the variable amount of differential expression. If one assumes the effect size of ASD on expression is similar across studies, the amount of differential expression (threshold free, estimated from the p-value distribution) should be consistent. A comparison between the estimated proportion of differentially expressed genes (1-π0, see Materials and Methods) and sample size shows that this is clearly not the case for these data (Fig. S3). There are other possible explanations such as phenotype heterogeneity or comorbidities for this phenomenon, but we were unable to identify any explanatory factors from the information available.
Table 3.
Differentially expressed genes in each data set after reanalysis (based on two sided p-values). Only genes with unique mappings and p-values were included in the gene counts. 1 − π0: Estimated proportion of differentially expressed genes.
| Data sets | FDR <0.05 |
Up- regulated |
Down- regulated |
1 - π0 | Total number of genes |
Samples |
|---|---|---|---|---|---|---|
| Brain | ||||||
| GSE28475 | 0 | 0 | 0 | 0.20 | 16598 | 34 |
| GSE28521 | 4 | 1 | 3 | 0.25 | 16598 | 27 |
| GSE38322 | 0 | 0 | 0 | 0.15 | 19558 | 10 |
|
| ||||||
| Blood | ||||||
| GSE6575 | 0 | 0 | 0 | 0.00 | 18305 | 44 |
| GSE7329 | 314 | 160 | 154 | 0.41 | 17159 | 13 |
| GSE15402 | 5 | 1 | 4 | 0.11 | 9821 | 106 |
| GSE15451 | 0 | 0 | 0 | 0.04 | 12066 | 27 |
| GSE18123.1 | 333 | 103 | 230 | 0.27 | 18305 | 92 |
| GSE18123.2 | 57 | 35 | 22 | 0.47 | 18617 | 156 |
| GSE25507 | 2 | 2 | 0 | 0.28 | 18305 | 143 |
| GSE32136 | 2 | 0 | 2 | 0.33 | 9076 | 8 |
| GSE37772 | 0 | 0 | 0 | 0.00 | 16598 | 431 |
We next compared the result of each analysis to that previously published for the same data set, where available. This was done by examining where the differentially expressed genes from the original studies rank in our results (using the area under receiver operating characteristic curve, AU-ROC; equivalent to the Wilcoxon rank-sum test). Despite the extensive additional data cleanup we performed and differences in the statistical analysis methods, our re-analyses were generally concordant with the original reports (Table S6).
Meta-analysis of differential expression
A key observation at this point is that most of the data sets showed clear evidence for differential expression (π0 < 1), but were largely underpowered to separate differentially expressed genes from the background. Thus, it is perhaps not surprising that there was no overlap across any of the studies among the genes (if any) selected at an FDR of 0.05. We hypothesized that there might still be similarities among the studies that would emerge in a combined or meta-analysis. We therefore applied a p-value combination strategy (Materials and Methods), choosing to analyze the blood and brain data sets separately. This approach combines the results for all the data sets without applying any statistical threshold, and thus provides a p-value for all the genes analyzed. The meta-analysis yields four ranked gene lists: one pair each for blood and brain, with separate lists for up- and down-regulation, noting that at this stage they contain all the genes considered without applying a threshold.
We then compared the results of individual study re-analyses to the ranked gene lists. If each data set contributes some signal in the meta-analysis, their results should individually resemble the ranked gene lists. Generally, the trends we observed concur with the amount of differential expression estimated (1-π0) for each data set. Data sets with more differential expression displayed stronger associations with the results of the meta-analyses. As shown in Figure 2A, there is a clear similarity among the three brain data sets in their contributions towards the final gene rankings, as evidenced by the similar trend lines for all three data sets. In contrast, the blood data sets were in lower agreement, with a fraction showing a stronger relationship to the meta-analysis results while others show weak associations (Figure 2B).
Figure 2.

Profiles of meta-analyses gene ranks from the blood and brain: raw p-values for each individual data set are plotted against corrected p-values (FDR) from the meta-analyses. Local Polynomial Regression (LOESS) is used to obtain a smooth fit. The shaded areas represent 95% confidence intervals of the prediction using the t-based approximation (see “stat_smooth” in the ggplot2 R package).
Applying a threshold to these rankings yielded blood and brain “meta-signatures”. At an FDR threshold of 0.05, 30 up-regulated genes and 49 down-regulated genes were found in the brain. The blood meta-analysis yielded 160 up-regulated and 95 down-regulated hits (Tables S7-10). While the studies were balanced for covariates such as age and post-mortem interval (for the brain data), we checked the lists for genes previously reported to be influenced by these factors (Mistry & Pavlidis, 2010). There were minimal overlaps, confirming that our results were not strongly influenced by them. Genes known to be affected by sex differences were removed in the results reported here, though they can be found in the supplement for reference (Materials and Methods). Finally, we investigated whether our results might be influenced by genetic variation within regions assayed by the microarray probes. The presence of a variant in an assayed individual could cause differences in hybridization efficiency, causing apparent changes in gene expression that should instead be interpreted as genetic differences. However, we found no indication that common variants were likely to affect our analysis, especially in the brain data (Table S7, 8) for which there are very few potential variants affecting probes for genes in the meta-signature (see supplement for details).
We further characterized the relative contributions of each data set towards the hits we obtained, to more directly identify any single study that “drives” genes towards significance in the meta-analyses. By assessing the amount of overlap between meta-signatures and differentially expressed genes in each data set, we quantified the contribution of each data set to the meta-analysis (Table 4). Overall, GSE28521 had the strongest impact on the brain meta-analysis; GSE18123.1 and GSE7329 were studies that had a relatively strong influence on the blood meta-analysis. As described in the next section we implemented procedures to find genes robust to the selection of data sets.
Table 4.
Overlap (overlap/total up or down-regulated in data set) between meta-signature (FDR<0.05) and significantly differentially expressed genes per data set (FDR<0.05), as well as enrichment of meta-signatures in the results of individual differential expression analysis. One sided p-values were used to compute FDR here. AU-ROC: area under receiver operating characteristic curve; AP: average precision.
| Up-regulated | AU-ROC | AP(%) | Down-regulated | AU-ROC | AP(%) | |
|---|---|---|---|---|---|---|
| Brain | ||||||
| GSE28475 | 2/3 | 0.92 | 10.77 | 0/0 | 0.90 | 5.60 |
| GSE28521 | 0/0 | 0.96 | 15.33 | 5/5 | 0.94 | 29.47 |
| GSE38322 | 0/0 | 0.90 | 5.70 | 0/0 | 0.84 | 10.80 |
|
| ||||||
| Blood | ||||||
| GSE15402 | 0/1 | 0.78 | 3.31 | 0/29 | 0.71 | 1.35 |
| GSE15451 | 0/0 | 0.54 | 0.80 | 0/0 | 0.55 | 1.22 |
| GSE18123.1 | 16/92 | 0.82 | 8.36 | 28/235 | 0.84 | 15.01 |
| GSE18123.2 | 13/38 | 0.86 | 10.73 | 2/9 | 0.74 | 5.41 |
| GSE25507 | 0/3 | 0.67 | 2.83 | 0/0 | 0.59 | 2.03 |
| GSE32136 | 0/0 | 0.76 | 8.06 | 0/2 | 0.70 | 2.14 |
| GSE37772 | 0/2 | 0.65 | 0.97 | 0/0 | 0.57 | 0.80 |
| GSE6575 | 0/0 | 0.67 | 3.05 | 0/0 | 0.67 | 1.69 |
| GSE7329 | 25/183 | 0.84 | 13.21 | 20/234 | 0.78 | 5.84 |
To see if the meta-signatures in blood and brain are similar, we quantified the reciprocal predictive value of meta-signatures from both tissue types using AU-ROC. There was no indication of a common signature between the blood and brain, supporting our choice in conducting separate analyses.
Robust molecular commonalities in brain data
To focus our attention on the genes that show the strongest concordance across studies, we employed a jackknife procedure (Materials and Methods). Jackknifing yields multiple lists of gene ranks, one for each data set in the meta-analysis, where each list is the result with that data set left out. We initially performed this at the same stringency as the initial meta-analysis, applying an FDR threshold of 0.05 for every jackknife result. With this conservative approach, we identified 10 genes from the blood data for which significant values are not dominated by any single data set, but none from the brain. Because removing data sets reduces power, to establish a less stringent criterion for identifying robust patterns, we define our “core signatures” as the intersection of the top 200 (arbitrary cut off) genes retrieved from each leave-one-out iteration (Mistry et al., 2012). From this analysis, the core blood signature consists of 15 up-regulated genes and 7 down-regulated genes (net estimated corresponding FDRup< 0.12, FDRdown < 0.15). 15 up-regulated genes and 10 down-regulated genes were observed in the core brain signature (net estimated corresponding FDRup < 0.29, FDRdown < 0.24). We visualized these core signature genes using heat maps of the gene expression levels for each sample in the twelve data sets meta-analyzed. The heat maps for the core brain signature showed good concordance across all three brain data sets (Figure 3). In contrast, few genes from the blood analysis exhibited robust concordance when visualized (Fig. S6 and S7).
Figure 3.

Heat map visualizations of core-signatures expression values in each of the brain data sets. Batch corrected expression values were scaled across samples within each data set. Relative expression levels: yellow – high; blue – low.
Two of the brain studies included samples from cerebellum (including some from the same individuals for the neocortex samples), which we treated separately. Since there were only two data sets, a meta-analysis was not feasible. Thus to compare patterns in cerebellum to those in the neocortex, we conducted differential expression analysis on each cerebellum data set (Table S18). An analysis of the rankings of the neocortex core signature genes in the differential expression results from the cerebellum indicated very little if any concordance between cerebellum and neocortex (Fig. S12).
One of the sources of heterogeneity in the blood analysis is the cell types used: five of the studies used Epstein-Barr virus-transformed lymphoblastoid cell lines (LCLs) while four used primary cells. We investigated potential differences between these two groups by conducting the meta-analysis on the LCL subgroup and the non-LCL subgroup, separately (see Supplement and Fig. S13-16 for details). Five genes identified in the full analyses were recovered in the analysis of the LCL data. Several additional signature genes were detected in the LCL data, suggesting some degree of higher homogeneity among these studies, albeit not as strikingly as in brain. In the non-LCL data, the main pattern is driven by two studies from the same laboratory (GSE18123.1 and GSE18123.2) and this was not convincingly observed across other studies (Fig. S13b and S15b).
Functional analyses suggests perturbations in metabolic processes
To explore gene functional themes in our data, we conducted a threshold-free GO term enrichment analysis. None of the functions tested for were significantly enriched in the blood. The brain results were enriched for genes involved in “cellular respiration” (GO:0045333, FDR = 0.11). An analysis using the three jackknifed gene lists from the brain data (that is, meta-analysis of each pair of data sets) showed that this result is robust. Dysregulated genes in this functional group are shown in Table 8. Other top enriched functions were also related to respiration, including GO:0022904 (“respiratory electron transport chain”) and GO:0022900 (“electron transport chain”).
Table 8.
Top genes in the “cellular respiration” GO category at a meta-analysis raw p-value threshold of 0.0001. There is a total of 116 genes in this functional group.
| Gene Symbol | Gene Name | Meta p-value |
|---|---|---|
| ATP5O | ATP synthase, H+ transporting, mitochondrial F1 complex, O subunit |
1.83E-05 |
| UQCRQ | ubiquinol-cytochrome c reductase, complex III subunit VII, 9.5kDa |
5.45E-05 |
| UQCRC1 | ubiquinol-cytochrome c reductase core protein I | 1.84E-04 |
| CYC1 | cytochrome c-1 | 2.90E-04 |
| COX5B | cytochrome c oxidase subunit Vb | 2.98E-04 |
| NDUFA11 | NADH dehydrogenase (ubiquinone) 1 alpha subcomplex, 11, 14.7kDa |
4.38E-04 |
| ATP5L | ATP synthase, H+ transporting, mitochondrial Fo complex, subunit G |
4.53E-04 |
| UQCR10 | ubiquinol-cytochrome c reductase, complex III subunit X |
4.53E-04 |
| UQCRC2 | ubiquinol-cytochrome c reductase core protein II | 5.25E-04 |
| NDUFA13 | NADH dehydrogenase (ubiquinone) 1 alpha subcomplex, 13 |
5.35E-04 |
| SLC25A12 | solute carrier family 25 (aspartate/glutamate carrier), member 12 |
5.37E-04 |
| FH | fumarase hydratase | 7.55E-04 |
| UQCR11 | ubiquinol-cytochrome c reductase, complex III subunit XI |
7.74E04 |
| NDUFS4 | NADH dehydrogenase (ubiquinone) Fe-S protein 4, 18kDa (NADH-coenzyme Q reductase) |
8.29E-04 |
| IDH3A | isocitrate dehydrogenase 3 (NAD+) alpha | 9.06E-04 |
In a complementary approach, we conducted enrichment analysis on our differential expression results of the individual data sets, which we then combined in a meta-analysis at the gene set level, again using a jackknife to test robustness. The results for the brain data were in agreement with our analysis of the gene-level meta-signature, with the top meta-analysis term being “cellular respiration”, though this was not significant after multiple test correction. There was no clear trend in blood, with no statistically significant terms and no significant agreement in the results of individual studies. These results reinforced our conclusion based on the gene-level analysis that the blood data sets are more heterogeneous than the brain studies.
Comparison to known candidate genes in neurodevelopment disorders
A natural question is whether any of the signature genes are known ASD candidates reported in previous genetics or functional studies. We first checked for overall patterns of enrichment based on the ranked gene lists from the blood and brain. We observed enrichment of genes in the Simons Foundation Autism Research Initiative (SFARI) “syndromic” category (FDR = 0.15; see Table 7 for details) in blood. Inspection revealed this was primarily due to the influence of the 15q duplication cohort (GSE7329). We can directly observe the skew in the top two syndromic genes: UBE3A (FDR = 0.004), CDKL5 (FDR = 0.14) (Fig. S9). While UBE3A resides on the 15q11-13 region, CDKL5 (Xp22) does not. The link between 15q duplication and CDKL5 dysregulation is unclear.
Table 7.
All genes and respective p-values in the SFARI syndromic category.
| Gene Symbol | Gene Name | Meta p-value |
|---|---|---|
| UBE3A | ubiquitin protein ligase E3A | 4.72E-06 |
| CDKL5 | cyclin-dependent kinase-like 5 | 1.43E-03 |
| DMD | dystrophin | 2.36E-03 |
| SHANK3 | SH3 and multiple ankyrin repeat domains 3 | 6.58E-03 |
| HOXA1 | homeobox A1 | 1.45E-02 |
| PTEN | phosphatase and tensin homolog | 2.34E-02 |
| TSC1 | tuberous sclerosis 1 | 3.09E-02 |
| DHCR5 | 7-dehydrocholesterol reductase | 3.99E-02 |
| SCN1A | sodium channel, voltage-gated, type I, alpha subunit |
7.29E-02 |
| AHI1 | Abelson helper integration site 1 | 9.63E-02 |
| NF1 | neurofibromin 1 | 9.67E-02 |
| CACNA1C | calcium channel, voltage-dependent, L type, alpha 1C subunit |
0.10 |
| RAI1 | retinoic acid induced 1 | 0.15 |
| ALDH5A1 | aldehyde dehydrogenase 5 family, member A1 | 0.17 |
| MECP2 | methyl CpG binding protein 2 (Rett syndrome) | 0.21 |
| ARX | aristaless related homeobox | 0.22 |
| SLC9A6 | solute carrier family 9, subfamily A (NHE6, cation proton antiporter 6), member 6 |
0.29 |
| ADSL | adenylosuccinate lyase | 0.33 |
| DMPK | dystrophia myotonica-protein kinase | 0.35 |
We repeated this analysis using a list of 798 ASD candidates from Phenocarta (previously known as Neurocarta (Portales-Casamar et al., 2013)), including candidates from several GWAS and other genetics studies, but there was no significant enrichment. This was not unexpected, because we hypothesized any link between gene expression in this diverse cohort and the genetics of ASD is not direct. Among the few Phenocarta ASD candidate genes identified in our meta-signatures are 13 genes in the blood signature (CAMSAP2, UBE3A, CYFIP1, JARID2, PAFAH1B1, FAN1, BRAF, CXCR3, PRDX4, GAP43, GABRA4, CHRM3, BCL2) and one gene in the brain signature (GAS2). We also looked for known candidates in the brain using a relaxed FDR threshold of 0.1. Additional genes found in the brain include ADM, CADM1, STAT3, CD44, CYP19A1, PTCHD1, SLC30A5, SLC25A12, APBA2 and DLX1. None of the existing candidates are common to the meta-signatures of both tissue types.
Meta-signature genes in rare structural variants associated with ASD
The candidate gene lists used in the last section do not, for the most part, include genes covered by rare structural variants associated with ASD, because the precise gene or genes involved are often not known and are thus not documented by SFARI or Phenocarta. To explore the potential links between gene expression and rare structural variations, we assembled ASD-associated copy number variations (CNV) from several sources (Materials and Methods). We first observed that genes in the meta-signatures are distributed widely across the genome. There were no obvious hot spots, and none of the CNVs analyzed were significantly enriched for dysregulated genes (corrected p-value >> 0.05).
Globally, 6.3% of the brain meta-signature genes and 9.8% in blood are located in known CNV regions, which is not a significant enrichment (Table S14). This computation was constrained to genes showing positive associations between expression levels and copy number changes (up-regulated genes within a duplicated region and down-regulated genes within a deleted region). All dysregulated CNV genes are shown in Table 5 and Table 6.
Table 5.
Brain candidate genes within known ASD associated CNVs. CNVs that span the same gene or set of genes are grouped together. Genomic coordinates are from hg18. Overlapped genomic coordinates are lifted over from hg18 to hg19 with UCSC’s LiftOver tool. Lift over failed for coordinates marked with an asterisk.
| Genes | Gain/Loss | Chromosome | CNV Start | CNV End | Reference |
|---|---|---|---|---|---|
| SCIN | Gain | 7 | 12219860 | 17560760 | AGP Consortium (2007) |
|
| |||||
| ABCG2 | Loss | 4 | 86288694 | 101407914 | Jaquemont et al. (2006) |
|
| |||||
| GRK6 | Loss | 5 | 175559839 | 177426530 | Sanders et al (2011) |
|
| |||||
| PANX2 | Loss | 22 | 47898736 47956881 46823508 46765363 |
51162234 51218956 51175739 51119017 |
Sanders et al (2011)
Marshall et al. (2008) Sebat et al. (2007) Sanders et al (2011) |
|
| |||||
| SNRNP25 | Loss | 16 | 60835 | 1313637 | Sanders et al (2011) |
Table 6.
Blood candidate genes within known ASD associated CNVs. CNVs that span the same gene or set of genes are grouped together. Genomic coordinates are lifted over from hg18 to hg19 with UCSC’s LiftOver tool. Lift over failed for coordinates marked with an asterisk.
| Genes | Gain/Loss | Chromosome | CNV Start | CNV End | Reference |
|---|---|---|---|---|---|
| ARL16 | Gain | 17 | 76914079* 76953064* 76953064* |
77771141* 77782267* 77782267* |
Marshall et al (2008)
Pinto et al (2010) Sanders et al (2011) |
|
| |||||
| CSTF2T | Gain | 10 | 53029510 52002204 50892143 |
54738810 61820631 61808505 |
Sanders et al (2011)
Sanders et al (2011) Sebat et al (2007) |
|
| |||||
| CYFIP1 | Gain | 15 | 22684249 22751742 20090262* |
23255910 23249123 21038099* |
Pinto et al (2010)
Pinto et al (2010) Pinto et al (2010) |
|
| |||||
| FAN1 | Gain | 15 | 30936285 30936285 |
32444196 32451488 |
Sanders et al (2011)
Sanders et al (2011) |
|
| |||||
| FUT8-AS1 | Gain | 14 | 62827347 | 66005847 | AGP Consortium (2007) |
|
| |||||
| HCK, C20orf112 |
Gain | 20 | 28251057* | 35143867* | Sanders et al (2011) |
|
| |||||
| IRF2BPL | Gain | 14 | 76938089 | 77854647 | Marshall et al. (2008) |
|
| |||||
| P2RX7, GPR133, KDM2B, MED13L |
Gain | 12 | 115707280 115685617 |
133777650 133779461 |
Marshall et al. (2008)
Sanders et al (2011) |
|
| |||||
| RFC2 | Gain | 7 | 72773570 72662415 72706490 72717647 |
74173250 74144177 74144177 74144177 |
Sanders et al (2011)
Sanders et al (2011) Sanders et al (2011) Sanders et al (2011) |
|
| |||||
| SH2D1B | Gain | 1 | 162169342 | 162867342 | AGP Consortium (2007) |
|
| |||||
| SMARCA2 | Gain | 9 | 185632 | 3383495 | Sanders et al (2011) |
|
| |||||
| TCF7 | Gain | 5 | 132566101 | 134838101 | AGP Consortium (2007) |
|
| |||||
| TXLNA | Gain | 1 | 31125281 | 36307897 | Sanders et al (2011) |
|
| |||||
| UBE3A | Gain | 15 | 25184941 23939207 23639183 23639183 23688944 |
28016015 28024805 28530359 28530359 28422026 |
Jaquemont et al. (2006) AGP Consortium (2007) Pinto et al (2010) Sanders et al (2011) Sanders et al (2011) |
|
| |||||
| UBE3A, CYFIP1 |
Gain | 15 | 22877142 22424462 22646319 22265649 |
28396011 28396011 28396011 28460519 |
Christian et al. (2008) Christian et al. (2008) Christian et al. (2008) Sanders et al (2011) |
|
| |||||
| UBE3A, CYFIP1, FAN1 |
Gain | 15 | 18376200* 18427100* 18376200* 18427100* 18526971* 18526971* |
30298800* 30298847* 30298800* 30298847* 30756771* 30756771* |
Marshall et al. (2008)
Marshall et al. (2008) Sanders et al (2011) Sanders et al (2011) Sebat et al. (2007) Sanders et al (2011) |
|
| |||||
| ZNF611, ZNF702P | Gain | 19 | 53144788 | 53554388 | Marshall et al. (2008) |
|
| |||||
| ZNF721 | Gain | 4 | 338851 | 552862 | Marshall et al. (2008) |
|
| |||||
| ZNF721, SPON2 |
Gain | 4 | 45410 408952 |
3541587 6671958 |
Sanders et al (2011)
AGP Consortium (2007) |
|
| |||||
| CCDC50 | Loss | 3 | 185812357 | 192380293 | Jaquemont et al. (2006) |
|
| |||||
| SLC17A9 | Loss | 20 | 61586179 | 61606318 | Sanders et al (2011) |
|
| |||||
| TSPAN12 | Loss | 7 | 113547764 113741049 |
129034485 12922770 |
Sanders et al (2011)
Marshall et al. (2008) |
Because 15q11-13 duplication is one of the most common CNV aberration in ASD (Miles, 2011), it was unsurprising that we detected dysregulated genes in this region. A closer look at these genes (UBE3A, CYFIP1; Fig. S9) again reveals their sensitivity towards the data set that comprises only autistic subjects with 15q duplications (GSE7329). In other ASD-associated CNVs, we detected genes from the core signatures that are dysregulated in the same direction as the change in copy number: ZNF721 (4p16) in blood; SCIN (7p21.1), SNRNP25 and ABCG2 (4q21) in the brain. However, we conclude that while some of the genes in our signatures are ASD candidate genes or fall in known rare CNV regions, there is no striking overall relationship between the expression patterns and the current state of knowledge of ASD genetics. Again, because the genetic etiologies in our data are presumably diverse overall, transcriptome changes common across cohorts are not necessarily expected to be attributable directly to genes which are mutated in ASD
Discussion
We presented a meta-analysis of autism gene expression profiling studies providing the most comprehensive survey on gene expression in autism available to date. Our main finding is that there are molecular commonalities across multiple independent groups of individuals with ASD. These similarities have, to our knowledge, gone overlooked in individual gene expression studies. Genes we identified as most robustly changed across cohorts were not previously underscored in ASD literature. Here we discuss our findings in the context of other autism research, noting some limitations of the current study and avenues for future work.
The question of whether one should expect some common molecular changes across individuals with ASD is an open one. The studies included in our analysis used a range of criteria to select subjects, but are largely made up of idiopathic cases (the exception being GSE7329). Each study was apparently predicated on the hypothesis that there would be group differences; that is, that there would be a common ASD signature in the data. Thus it is reasonable to hypothesize that there might be similarities across studies, but any lack of similarity could be attributed to cohort or technical differences. Given these challenges, it is striking that we do find some genes showing differences that are relatively consistent across cohorts.
The full biological significance of the genes we identified is currently unclear. However several of the concordant genes (core-signature genes) we found are linked to genetic disorders with neurological implications. Among the genes in the core brain signature are PDYN (prodynorphin) and ABCA1 (ATP-binding cassette, sub-family A). Mutations in PDYN, a gene that codes for an opioid, has causal links to spinocerebellar ataxia (MIM 610245) (Bakalkin et al., 2010). Mutations in ABCA1 are an established cause for Tangier disease (MIM 205400), a disorder which features include neuropathies (Oram, 2000). There were fewer clear hits in the blood data, but several genes stand out (Figure 4). A known ASD candidate BRAF (v-raf murine sarcoma viral oncogene homolog B1) showed consistent dysregulation in at least three cohorts. Other novel candidates in blood are PRKCH (protein kinase C eta, a member of the protein kinase C family) and APBB1 (amyloid beta (A4) precursor protein-binding, family B, member 1 (Fe65)), which have been studied in cellular signaling and Alzheimer’s disease (Q. Hu et al., 1998) respectively.
Figure 4.

Examples of robust genes in the blood core-signature. A) Raw p-values of genes in each individual re-analysis, marked with a triangle if it meets an FDR threshold of 0.05 in that data set; B) log2-transformed expression values of SORL1 for every sample in each data set. Similar plots for other genes are available in the supplement. NS: not significant.
As discussed, results from previously published transcriptome analyses have, at the surface, shown little agreement. We have also described some reasons why this might occur, including differences in clinical properties or technical aspects of the expression analysis. However, we note that some of our candidates were hits reported in the original studies, as well as in other transcriptome studies not included for analysis (Table S13). In fact, two genes were validated with a second independent cohort in the original studies – ZNF322 (zinc finger protein 322) in Kong et al (2013) and PDYN in Voineagu et al (2011), further suggesting bona fide associations with ASD. However these genes were not discussed in these previous publications, perhaps because of their relatively low rankings in the results or the lack of known functional implications. In addition, most existing studies have not dwelled upon the findings of other related studies, either choosing to ignore them or attribute differences to experimental procedures. Our results suggest that in fact many of the molecular or functional differences observed in individual studies are likely to be specific to that study and thus of questionable interpretation when the entire autism spectrum is considered. While inferences made based on our findings are preliminary, the fact that some changes show a tendency to be reproducible opens promising avenues for further research.
Taken as a whole, the expression patterns we observe in brain point to the possibility of effects relating to cellular respiration. Within the cellular respiration group, SLC25A12 (not a hit at an FDR of 0.05 but falls within a relaxed FDR threshold of 0.1), a mitochondrial aspartate/glutamate carrier, was previously reported as a susceptibility gene as it harbors SNPs (single nucleotide polymorphisms) strongly associated with autism (Ramoz et al., 2004). In addition to the genes that were directly annotated with this function, a further examination reveals other highly ranked genes in our data which are known to play regulatory roles in cellular metabolism or mitochondrial related functions. Some of the genes are not directly annotated in the GO functional groups. For instance, P2RX7 (purinergic receptor P2X, ligand-gated ion channel, 7 CNV gene) is involved in purinergic signaling, a pathway that might play a role in mitochondrial dysfunction-associated ASD (Abbracchio, Burnstock, Verkhratsky, & Zimmermann, 2009). Mitochondrial dysfunction (MD) has been a topic of study in some neuropsychiatric disorders (notably bipolar disorder (Andreazza, Shao, Wang, & Young, 2010; Sun, Wang, Tseng, & Young, 2006)). Some have conjectured a 4-5% prevalence of MD in individuals on the autism spectrum (Miles, 2011; Rossignol & Frye, 2012), but there is little direct evidence in the literature. Investigations on mitochondrial DNA mutations in ASD yielded mixed conclusions (Álvarez-Iglesias et al., 2011; Piryaei, Houshmand, Aryani, Dadgar, & Soheili, 2012). In part supported by the enrichment of “cellular respiration” (comprising only nuclear encoded genes), current research seems to indicate a role for nuclear genes in the co-occurrence of MD and ASD (Anitha et al., 2012; Dhillon, Hellings, & Butler, 2011), the genetics of which might not be as simple as other monogenic metabolic disorders with high prevalence of ASD, like Smith-Lemli-Optiz syndrome (MIM 270400). However our analysis of brain transcriptomes showed converging functional consequences of what could be heterogeneous genetic or genomic aberrations underlying the disorders.
Potentially causative rare CNVs are found in up to 20% of ASD cases (Miles, 2011). While several genes we identified are within regions implicated in CNV studies of ASD, there was no overall significant enrichment. It is still possible that the changes in RNA levels we observed are linked indirectly to CNVs or other types of rare genetic variants, which we are not able to determine because the genomic backgrounds for most of the cases in our data set were unknown. Genes suggestive of direct correlations include PANX2, RFC2 and 15q genes, which reside in regions that have recurrent (previously reported in several ASD cases) rare CNVs. RFC2 lies in the 7q11.23 region, deletions of which are associated with Williams-Beuren syndrome (MIM 194050). Duplications of this region, concordant with an up-regulated RFC2 we found, has been strongly linked to autism (Sanders et al., 2011).
An important caveat for our interpretation is the difficulty of attributing any causal role to the changes we observe. They could be sequelae of ASD, or due to comorbid conditions. Most of the studies we used did not provide any details about comorbidities, making this difficult to address in our analysis. Future studies should endeavor to provide such details to allow further dissection of real effects from potential confounds.
In conclusion, our re-analysis reveals subtle but consistent changes in expression in the brains of individuals with ASD. Because the sample size for publicly available brain studies are small, future work could explore whether these changes are replicable in additional cohorts. In blood, the signals were weaker and more heterogeneous than in brain, perhaps in part due to the varying inclusion criteria used among individual studies. Additional work may be needed to clarify the reproducible expression differences in blood. Finally, as more RNA-seq data becomes available, we can also further explore commonalities in ASD transcriptomes at a higher resolution than possible with the technologies used in the present study.
Materials and Methods
Data retrieval, pre-processing and quality control
We retrieved gene expression data sets matching the keywords “autism” or “autistic” from the Gene Expression Omnibus (GEO) (Barrett et al., 2007) on September 10, 2012. There were no additional data sets found in ArrayExpress (Parkinson et al., 2009). Shortlisted data sets include human blood and brain expression profiling studies with case-control experiment designs only. Two studies in the initial pool, GSE4187 and GSE26415 were disqualified for analysis (see supplement for details). The final set of twelve studies (Table 1) consist of data collected on a variety of platforms, including one channel intensity data from Affymetrix and Illumina platforms, and two channel intensity data from Agilent and TIGR platforms (Table 2). To help ensure comparability and consistency in pre-processing methods across studies, we pre-processed the raw expression data using Robust Multi-array Average (RMA) or quantile normalization and log2 transformation implemented in the “affy” (Gautier, Cope, Bolstad, & Irizarry, 2004) or “lumi” (Du, Kibbe, & Lin, 2008) R packages where appropriate.
Table 2.
Summary of platform annotations from Gemma (Zoubarev et al., 2012). The total number of probes and unique genes were obtained from the Gemma platform database.
| Platforms | Platform Name | Gemma Probes | Unique Genes |
|---|---|---|---|
| GPL10558 | Illumina HumanHT-12 V4.0 expression beadchip |
47323 | 21348 |
| GPL1708 | Agilent-012391 Whole Human Genome Oligo Microarray G4112A |
44347 | 19326 |
| GPL3427 | TIGR 40k Human Array | 41472 | 14753 |
| GPL570 | Affymetrix Human Genome U133 Plus 2.0 Array |
54681 | 19763 |
| GPL6244 | Affymetrix Human Gene 1.0 ST Array | 33297 | 20353 |
| GPL6883 | Illumina HumanRef-8 v3.0 expression beadchip |
24526 | 17979 |
The processed data were then subjected to an additional set of quality controls. We first removed samples that are duplicated across datasets (n = 17, see supplement for details). Additionally, we removed eight samples from subjects with syndromic disorders of known genetic etiology, non-ASD cases (e.g., mental retardation - nine samples in GSE6575), samples which were prepared differently than the rest of the samples (e.g., formalin fixed – 36 samples in GSE28475; propanol/PPA treated – 15 samples in GSE32136) and 34 samples in which batch effects were confounded with the case grouping. Two studies included samples from cerebellum (GSE38322 and GSE28521), but some of these samples came from the same individuals as the neocortex samples. This fact together with the dramatic difference in expression pattern (Fig. S1b) led us to consider the cerebellum samples separately.
Two of the studies included technical replicates for some specimens, in which case we computed the mean of the expression values to get a single expression profile for each subject. Outlying samples were identified as those with correlation more than two standard deviations from the mean sample-to-sample expression profile correlation, and removed iteratively until no samples met the threshold for removal. This resulted in the removal of 54 samples, affecting seven studies. Finally, we used ComBat (Johnson, Li, & Rabinovic, 2007) to correct for batch effects (Fig. S2). More details on the quality control and preprocessing procedures are available in the supplement.
Differential expression analysis
We conducted an analysis of variance (ANOVA) for each data set using “limma” in R (Smyth, 2005), using a case-control model. Phenotypic subgroups (savant, mild, etc.) were pooled into one disease group. To consider the direction of expression change in the meta-analyses, we computed one-tailed p-values for probes in each data set. Probes are annotated with platform specific annotations in Gemma (Zoubarev et al., 2012), where gene assignments are made based on current genome annotations obtained via sequence analysis. Each data set is then collapsed to the gene level to allow cross-platform integration. Probes that map to multiple genes or do not map to a gene at all are excluded from the analysis. The proportion of differentially expressed genes (π1 = 1 - π0) was estimated using the qvalue package in R (Storey & Tibshirani, 2003).
Meta-analysis of differentially expressed genes
Fisher’s combined probability test (Fisher, 1948) was applied independently to the blood and brain data sets. Genes were only analyzed if they were represented in at least three data sets in each of the meta-analysis. 19006 and 16591 genes were included in the blood and brain meta-analyses respectively. The resulting p-values were corrected for multiple testing using Benjamini Hochberg’s false discovery rate (FDR) approach (Benjamini & Hochberg, 1995). A second meta-analysis method, ‘Meta-Rank analysis’ gave similar meta-analysis results (see supplement for details of this analysis).
Because of the gender imbalance in some of the data sets, we excluded from downstream analysis genes which were known or strongly suspected to show changes in expression between genders (brain = 202; blood = 116; details in supplement). We note that some of the filtered genes (e.g. USP9Y and KDM5C) have been previously associated with ASD, but we were unconfident we could discriminate gender from disease effects for them in our analysis.
The combined probability method is sensitive to outliers; that is, a single study with a very low p-value can result in statistical significance even when the other studies provide little evidence for rejection of the null. To control for this, we used a jackknife approach to further select for genes that are robust to statistical outliers (a similar approach was used in Mistry et al. (2013)). The jackknife procedure involves repeating the meta-analysis k times, where k is the number of data sets, For each trial k, one data set i is left out, where i ε {1…k}. The agreement among these k jackknife meta-analyses was used as a basis for identifying a “core” signature that excludes genes appearing due to the influence of a single data set (see supplement for details).
Functional enrichment analysis
Gene set enrichment analysis was conducted using ErmineJ 3.0 (http://erminej.chibi.ubc.ca) (Lee, Braynen, Keshav, & Pavlidis, 2005). ErmineJ accounts for the “multifunctionality” bias of gene sets (http://erminej.chibi.ubc.ca/help/tutorials/multifunctionality/,(Gillis & Pavlidis, 2011)). It prioritizes gene sets that are less affected by this bias. The enrichment analysis input for each gene is the better of the two one-tailed test scores (up-regulated and down-regulated p-values). Further specifications of enrichment runs are provided in the supplement. We also tested for enrichment of candidate gene categories from the Simons Foundation Autism Research Initiative (SFARI) database (www.sfari.org, retrieved in December 2012). Only five out of seven SFARI gene categories were included in the analysis. The “High Confidence” category had no genes; the “Not Supported” category is irrelevant because these genes show no association with ASD.
Literature-derived candidate genes
Known ASD candidate genes were downloaded from Phenocarta (phenocarta.chibi.ubc.ca, February, 2013), a knowledge base of gene and phenotype associations aggregated from various sources, such as SFARI Gene (AutDB), OMIM (Online Mendelian Inheritance in Man) and RGD (Rat Genome Database) (Portales-Casamar et al., 2013). We obtained 798 unique genes, including candidate genes from model organisms which were mapped to their human homologs using HomoloGene (ftp://ftp.ncbi.nih.gov/pub/HomoloGene, build 67)(Wheeler et al., 2007). Additional analysis and details are provided in the supplement.
CNV enrichment analysis
We collated copy number variation data from the Autism Chromosomal Rearrangement Database (ACRD) (Marshall et al., 2008), Sanders et al (Sanders et al., 2011) (Table S4 in original study) as well as Pinto et al (Pinto et al., 2010) (Table S8 in original study), obtaining 1023 CNVs. We then merged similar CNVs to obtain a total of 732 (Gain=391, Loss=340, Unknown=1) regions used in our analysis (details provided in the supplement).
Supplementary Material
Acknowledgements
We are grateful to all investigators and institutions who made their data publicly available, as well as Dr. Christian Marshall (The Centre for Applied Genomics, The Hospital for Sick Children, Toronto) who shared data from the Autism Chromosomal Rearrangement Database. We thank Dr. Suzanne Lewis for providing feedback on the manuscript. We also thank all lab members, including Patrick Tan, Dr. Meeta Mistry and Dr. Elodie Portales-Casamar, for providing valuable support and discussion.
Grant Information
This work was supported by NeuroDevNet, the NIH (grant GM076990 to P.P.), and the Canadian Institutes of Health Research Strategic Training Program in Bioinformatics (C.C.).
Footnotes
Conflict of Interest
None.
Literature Cited
- Abbracchio MP, Burnstock G, Verkhratsky A, Zimmermann H. Purinergic signalling in the nervous system: an overview. Trends in Neurosciences. 2009;32(1):19–29. doi: 10.1016/j.tins.2008.10.001. doi:10.1016/j.tins.2008.10.001. [DOI] [PubMed] [Google Scholar]
- Álvarez-Iglesias V, Mosquera-Miguel A, Cuscó I, Carracedo Á, Pérez-Jurado LA, Salas A. Reassessing the role of mitochondrial DNA mutations in autism spectrum disorder. BMC Medical Genetics. 2011;12:50. doi: 10.1186/1471-2350-12-50. doi:10.1186/1471-2350-12-50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- American Psychiatric Association, American Psychiatric Association, & DSM-5 Task Force . Diagnostic and statistical manual of mental disorders DSM-5. American Psychiatric Association; Arlington, VA: 2013. Retrieved from http://dsm.psychiatryonline.org/book.aspx?bookid=556. [Google Scholar]
- American Psychiatric Association, American Psychiatric Association, & Task Force on DSM-IV . Diagnostic and statistical manual of mental disorders DSM-IV-TR. American Psychiatric Association; Washington, DC: 2000. Retrieved from http://dsm.psychiatryonline.org/book.aspx?bookid=22. [Google Scholar]
- Andreazza AC, Shao L, Wang J-F, Young LT. Mitochondrial complex I activity and oxidative damage to mitochondrial proteins in the prefrontal cortex of patients with bipolar disorder. Archives of General Psychiatry. 2010;67(4):360–368. doi: 10.1001/archgenpsychiatry.2010.22. doi:10.1001/archgenpsychiatry.2010.22. [DOI] [PubMed] [Google Scholar]
- Anitha A, Nakamura K, Thanseem I, Yamada K, Iwayama Y, Toyota T, Mori N. Brain region-specific altered expression and association of mitochondria-related genes in autism. Molecular Autism. 2012;3(1):12. doi: 10.1186/2040-2392-3-12. doi:10.1186/2040-2392-3-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakalkin G, Watanabe H, Jezierska J, Depoorter C, Verschuuren-Bemelmans C, Bazov I, Verbeek DS. Prodynorphin mutations cause the neurodegenerative disorder spinocerebellar ataxia type 23. American Journal of Human Genetics. 2010;87(5):593–603. doi: 10.1016/j.ajhg.2010.10.001. doi:10.1016/j.ajhg.2010.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrett T, Troup DB, Wilhite SE, Ledoux P, Rudnev D, Evangelista C, Edgar R. NCBI GEO: mining tens of millions of expression profiles—database and tools update. Nucleic Acids Research. 2007;35(suppl 1):D760–D765. doi: 10.1093/nar/gkl887. doi:10.1093/nar/gkl887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-David E, Shifman S. Combined analysis of exome sequencing points toward a major role for transcription regulation during brain development in autism. Molecular Psychiatry. 2012 doi: 10.1038/mp.2012.148. doi:10.1038/mp.2012.148. [DOI] [PubMed] [Google Scholar]
- Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. Journal of the Royal Statistical Society Series. 1995;(57):289–300. [Google Scholar]
- Berg JM, Geschwind DH. Autism genetics: searching for specificity and convergence. Genome Biology. 2012;13(7):247. doi: 10.1186/gb-2012-13-7-247. doi:10.1186/gb4034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi KH, Elashoff M, Higgs BW, Song J, Kim S, Sabunciyan S, Webster MJ. Putative psychosis genes in the prefrontal cortex: combined analysis of gene expression microarrays. BMC Psychiatry. 2008;8:87. doi: 10.1186/1471-244X-8-87. doi:10.1186/1471-244X-8-87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhillon S, Hellings JA, Butler MG. Genetics and Mitochondrial Abnormalities in Autism Spectrum Disorders: A Review. Current Genomics. 2011;12(5):322–332. doi: 10.2174/138920211796429745. doi:10.2174/138920211796429745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du P, Kibbe WA, Lin SM. lumi: a pipeline for processing Illumina microarray. Bioinformatics. 2008;24(13):1547–1548. doi: 10.1093/bioinformatics/btn224. doi:10.1093/bioinformatics/btn224. [DOI] [PubMed] [Google Scholar]
- Fisher R. Combining independent tests of significance. American Statistician. 1948;2:30. [Google Scholar]
- Gautier L, Cope L, Bolstad BM, Irizarry RA. affy--analysis of Affymetrix GeneChip data at the probe level. Bioinformatics (Oxford, England) 2004;20(3):307–315. doi: 10.1093/bioinformatics/btg405. doi:10.1093/bioinformatics/btg405. [DOI] [PubMed] [Google Scholar]
- Geschwind DH, Levitt P. Autism spectrum disorders: developmental disconnection syndromes. Current Opinion in Neurobiology. 2007;17(1):103–111. doi: 10.1016/j.conb.2007.01.009. doi:10.1016/j.conb.2007.01.009. [DOI] [PubMed] [Google Scholar]
- Gillis J, Pavlidis P. The Impact of Multifunctional Genes on “Guilt by Association” Analysis. PLoS ONE. 2011;6(2):e17258. doi: 10.1371/journal.pone.0017258. doi:10.1371/journal.pone.0017258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ginsberg MR, Rubin RA, Falcone T, Ting AH, Natowicz MR. Brain Transcriptional and Epigenetic Associations with Autism. PLoS ONE. 2012;7(9):e44736. doi: 10.1371/journal.pone.0044736. doi:10.1371/journal.pone.0044736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammond P, Forster-Gibson C, Chudley AE, Allanson JE, Hutton TJ, Farrell SA, Lewis MES. Face–brain asymmetry in autism spectrum disorders. Molecular Psychiatry. 2008;13(6):614–623. doi: 10.1038/mp.2008.18. doi:10.1038/mp.2008.18. [DOI] [PubMed] [Google Scholar]
- Hu Q, Kukull WA, Bressler SL, Gray MD, Cam JA, Larson EB, Deeb SS. The human FE65 gene: genomic structure and an intronic biallelic polymorphism associated with sporadic dementia of the Alzheimer type. Human Genetics. 1998;103(3):295–303. doi: 10.1007/s004390050820. [DOI] [PubMed] [Google Scholar]
- Hu VW, Nguyen A, Kim KS, Steinberg ME, Sarachana T, Scully MA, Lee NH. Gene Expression Profiling of Lymphoblasts from Autistic and Nonaffected Sib Pairs: Altered Pathways in Neuronal Development and Steroid Biosynthesis. PLoS ONE. 2009;4(6):e5775. doi: 10.1371/journal.pone.0005775. doi:10.1371/journal.pone.0005775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu VW, Sarachana T, Kim KS, Nguyen A, Kulkarni S, Steinberg ME, Lee NH. Gene expression profiling differentiates autism case-controls and phenotypic variants of autism spectrum disorders: evidence for circadian rhythm dysfunction in severe autism. Autism Research: Official Journal of the International Society for Autism Research. 2009;2(2):78–97. doi: 10.1002/aur.73. doi:10.1002/aur.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson WE, Li C, Rabinovic A. Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics. 2007;8(1):118–127. doi: 10.1093/biostatistics/kxj037. doi:10.1093/biostatistics/kxj037. [DOI] [PubMed] [Google Scholar]
- Kong SW, Collins CD, Shimizu-Motohashi Y, Holm IA, Campbell MG, Lee I-H, Kohane IS. Characteristics and Predictive Value of Blood Transcriptome Signature in Males with Autism Spectrum Disorders. PLOS ONE. 2012;7(12):e49475. doi: 10.1371/journal.pone.0049475. doi:10.1371/journal.pone.0049475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HK, Braynen W, Keshav K, Pavlidis P. ErmineJ: tool for functional analysis of gene expression data sets. BMC Bioinformatics. 2005;6:269. doi: 10.1186/1471-2105-6-269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, Sabo A, Neale BM, Nagaswamy U, Stevens C, Lim E, Roeder K. Analysis of Rare, Exonic Variation amongst Subjects with Autism Spectrum Disorders and Population Controls. PLoS Genet. 2013;9(4):e1003443. doi: 10.1371/journal.pgen.1003443. doi:10.1371/journal.pgen.1003443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lord C, Rutter M, Goode S, Heemsbergen J, Jordan H, Mawhood L, Schopler E. Autism diagnostic observation schedule: a standardized observation of communicative and social behavior. Journal of Autism and Developmental Disorders. 1989;19(2):185–212. doi: 10.1007/BF02211841. [DOI] [PubMed] [Google Scholar]
- Lord C, Rutter M, Le Couteur A. Autism Diagnostic Interview-Revised: a revised version of a diagnostic interview for caregivers of individuals with possible pervasive developmental disorders. Journal of Autism and Developmental Disorders. 1994;24(5):659–685. doi: 10.1007/BF02172145. [DOI] [PubMed] [Google Scholar]
- Marshall CR, Noor A, Vincent JB, Lionel AC, Feuk L, Skaug J, Scherer SW. Structural Variation of Chromosomes in Autism Spectrum Disorder. American Journal of Human Genetics. 2008;82(2):477–488. doi: 10.1016/j.ajhg.2007.12.009. doi:10.1016/j.ajhg.2007.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClellan J, King M-C. Genetic heterogeneity in human disease. Cell. 2010;141(2):210–217. doi: 10.1016/j.cell.2010.03.032. doi:10.1016/j.cell.2010.03.032. [DOI] [PubMed] [Google Scholar]
- Miles JH. Autism spectrum disorders—A genetics review. Genetics in Medicine. 2011;13(4):278–294. doi: 10.1097/GIM.0b013e3181ff67ba. doi:10.1097/GIM.0b013e3181ff67ba. [DOI] [PubMed] [Google Scholar]
- Mistry M, Gillis J, Pavlidis P. Genome-wide expression profiling of schizophrenia using a large combined cohort. Mol Psychiatry. 2012 doi: 10.1038/mp.2011.172. Retrieved from http://dx.doi.org/10.1038/mp.2011.172. [DOI] [PMC free article] [PubMed]
- Mistry M, Pavlidis P. A cross-laboratory comparison of expression profiling data from normal human postmortem brain. Neuroscience. 2010;167:384–95. doi: 10.1016/j.neuroscience.2010.01.016. doi:S0306-4522(10)00017-5 [pii] 10.1016/j.neuroscience.2010.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishimura Y, Martin CL, Vazquez-Lopez A, Spence SJ, Alvarez-Retuerto AI, Sigman M, Geschwind DH. Genome-Wide Expression Profiling of Lymphoblastoid Cell Lines Distinguishes Different Forms of Autism and Reveals Shared Pathways†. Human Molecular Genetics. 2007;16(14):1682–1698. doi: 10.1093/hmg/ddm116. doi:10.1093/hmg/ddm116. [DOI] [PubMed] [Google Scholar]
- Oram JF. Tangier disease and ABCA1. Biochimica et Biophysica Acta. 2000;1529(1-3):321–330. doi: 10.1016/s1388-1981(00)00157-8. [DOI] [PubMed] [Google Scholar]
- Parkinson H, Kapushesky M, Kolesnikov N, Rustici G, Shojatalab M, Abeygunawardena N, Brazma A. ArrayExpress update—from an archive of functional genomics experiments to the atlas of gene expression. Nucleic Acids Research. 2009;37(suppl 1):D868–D872. doi: 10.1093/nar/gkn889. doi:10.1093/nar/gkn889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinto D, Pagnamenta AT, Klei L, Anney R, Merico D, Regan R, Abrahams BS. Functional impact of global rare copy number variation in autism spectrum disorders. Nature. 2010 doi: 10.1038/nature09146. doi:10.1038/nature09146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piryaei F, Houshmand M, Aryani O, Dadgar S, Soheili Z-S. Investigation of the Mitochondrial ATPase 6/8 and tRNA(Lys) Genes Mutations in Autism. Cell Journal. 2012;14(2):98–101. [PMC free article] [PubMed] [Google Scholar]
- Portales-Casamar E, Ch¿ng C, Lui F, St-Georges N, Zoubarev A, Lai AY, Pavlidis P. Neurocarta: aggregating and sharing disease-gene relations for the neurosciences. BMC Genomics. 2013;14(1):129. doi: 10.1186/1471-2164-14-129. doi:10.1186/1471-2164-14-129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramoz N, Reichert JG, Smith CJ, Silverman JM, Bespalova IN, Davis KL, Buxbaum JD. Linkage and Association of the Mitochondrial Aspartate/Glutamate Carrier SLC25A12 Gene With Autism. American Journal of Psychiatry. 2004;161(4):662–669. doi: 10.1176/appi.ajp.161.4.662. doi:10.1176/appi.ajp.161.4.662. [DOI] [PubMed] [Google Scholar]
- Rogic S, Pavlidis P. Meta-analysis of kindling-induced gene expression changes in the rat hippocampus. Front Neurosci. 2009;3:53. doi: 10.3389/neuro.15.001.2009. doi:10.3389/neuro.15.001.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossignol DA, Frye RE. Mitochondrial dysfunction in autism spectrum disorders: a systematic review and meta-analysis. Molecular Psychiatry. 2012;17(3):290–314. doi: 10.1038/mp.2010.136. doi:10.1038/mp.2010.136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders SJ, Ercan-Sencicek AG, Hus V, Luo R, Murtha MT, Moreno-De-Luca D, Thomson SA. Multiple recurrent de novo CNVs, including duplications of the 7q11.23 Williams syndrome region, are strongly associated with autism. Neuron. 2011;70(5):863–885. doi: 10.1016/j.neuron.2011.05.002. doi:10.1016/j.neuron.2011.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smyth G. Limma: linear models for microarray data. In: Gentleman R, Carey V, Dudoit S, Irizarry R, Huber W, editors. Bioinformatics and Computational Biology Solutions using R and Bioconductor. Springer; New York: 2005. pp. 397–420. [Google Scholar]
- Spencer MD, Holt RJ, Chura LR, Suckling J, Calder AJ, Bullmore ET, Baron-Cohen S. A novel functional brain imaging endophenotype of autism: the neural response to facial expression of emotion. Translational Psychiatry. 2011;1(7):e19. doi: 10.1038/tp.2011.18. doi:10.1038/tp.2011.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storey JD, Tibshirani R. Statistical significance for genomewide studies. Proceedings of the National Academy of Sciences. 2003;100(16):9440–9445. doi: 10.1073/pnas.1530509100. doi:10.1073/pnas.1530509100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun X, Wang J-F, Tseng M, Young LT. Downregulation in components of the mitochondrial electron transport chain in the postmortem frontal cortex of subjects with bipolar disorder. Journal of Psychiatry & Neuroscience: JPN. 2006;31(3):189–196. [PMC free article] [PubMed] [Google Scholar]
- Voineagu I. Gene expression studies in autism: Moving from the genome to the transcriptome and beyond. Neurobiology of Disease. 2012;45(1):69–75. doi: 10.1016/j.nbd.2011.07.017. doi:10.1016/j.nbd.2011.07.017. [DOI] [PubMed] [Google Scholar]
- Voineagu I, Wang X, Johnston P, Lowe JK, Tian Y, Horvath S, Geschwind DH. Transcriptomic analysis of autistic brain reveals convergent molecular pathology. Nature. 2011 doi: 10.1038/nature10110. doi:10.1038/nature10110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheeler DL, Barrett T, Benson DA, Bryant SH, Canese K, Chetvernin V, Yaschenko E. Database resources of the National Center for Biotechnology Information. Nucleic Acids Research. 2007;35:D5–D12. doi: 10.1093/nar/gkl1031. Database. doi:10.1093/nar/gkl1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoubarev A, Hamer KM, Keshav KD, McCarthy EL, Santos JRC, Van Rossum T, Pavlidis P. Gemma: A resource for the re-use, sharing and meta-analysis of expression profiling data. Bioinformatics (Oxford, England) 2012;28(17):2272–3. doi: 10.1093/bioinformatics/bts430. doi:10.1093/bioinformatics/bts430. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.