

Abstract
Genome-wide association studies (GWAS) have identified many common single nucleotide polymorphisms (SNPs) associated with colorectal cancer risk. These SNPs may tag correlated variants with biological importance. Fine-mapping around GWAS loci can facilitate detection of functional candidates and additional independent risk variants. We analyzed 11,900 cases and 14,311 controls in the Genetics and Epidemiology of Colorectal Cancer Consortium and the Colon Cancer Family Registry. To fine-map genomic regions containing all known common risk variants, we imputed high-density genetic data from the 1000 Genomes Project. We tested single-variant associations with colorectal tumor risk for all variants spanning genomic regions 250-kb upstream or downstream of 31 GWAS-identified SNPs (index SNPs). We queried the University of California, Santa Cruz Genome Browser to examine evidence for biological function. Index SNPs did not show the strongest association signals with colorectal tumor risk in their respective genomic regions. Bioinformatics analysis of SNPs showing smaller P-values in each region revealed 21 functional candidates in 12 loci (5q31.1, 8q24, 11q13.4, 11q23, 12p13.32, 12q24.21, 14q22.2, 15q13, 18q21, 19q13.1, 20p12.3, and 20q13.33). We did not observe evidence of additional independent association signals in GWAS-identified regions. Our results support the utility of integrating data from comprehensive fine-mapping with expanding publicly available genomic databases to help clarify GWAS associations and identify functional candidates that warrant more onerous laboratory follow-up. Such efforts may aid the eventual discovery of disease-causing variant(s).
Introduction
Genetics play a key role in colorectal cancer (CRC) development [1, 2]; genome-wide association studies (GWAS) have successfully identified many common genetic variants that predict risk [3–19]. Although these variants have modest associations (i.e., per-allele odds ratio less than 1.3), their discovery has reinforced the importance of suspected disease pathways as well as suggested novel ones [20].
An important next step is to characterize the biological importance of these loci. However, single nucleotide polymorphisms (SNPs) identified by GWAS (i.e., index SNPs) are themselves unlikely to be the underlying disease-causing variants; instead, they are expected to tag genomic regions containing correlated SNPs, which may have functional consequences [21, 22]. Laboratory evaluation of all these variants is prohibitively cost- and labor-intensive. Fine-mapping efforts can help inform these experiments by narrowing the size of associated genomic regions likely to contain functional variation [22, 23]. Several recent studies have shown the utility of this approach to refine regions of interest and propose promising functional candidates [14, 17, 24–31].
In addition, some loci may harbor multiple independent risk variants, rather than a single variant. As genomic regions surrounding index SNPs may span more than one linkage disequilibrium block, it is possible these loci harbor additional variants that predict risk independent of the index SNPs. Fine-mapping studies, when conducted within a broader region, can help identify these novel independent risk variants for cancer [14, 31, 32].
In this study of 11,900 colorectal tumor cases and 14,311 controls of European ancestry, we fine-mapped genomic regions harboring 31 known CRC risk variants using both genotyped data and data imputed from the 1000 Genomes Project [33]. This high-density genetic data allowed us to comprehensively examine common (>5%) as well as less common or rare (<5%) genetic variation in these regions. We aimed to narrow the likely region containing the functional variant(s) based on results from association testing, as well as search for novel risk alleles independent of the index SNP. Further, to help inform follow-up laboratory studies, we used a comprehensive bioinformatics-based approach to annotate potential functional candidates.
Materials and Methods
Ethics statement
All participants gave written informed consent and this study has been approved by the Fred Hutchinson Cancer Research Center (FHCRC) Institutional Review Board.
Study population
Details of this study population have been described previously [3, 34] and study-specific descriptions are provided in S1 Text. The study population was derived from studies in the Genetics and Epidemiology of Colorectal Cancer Consortium (GECCO) (13 total: 7 case-control studies nested in prospective cohorts and 6 case-control studies) and the Colon Cancer Family Registry (CCFR) [3, 34]. Study case, control, age, and sex distributions are listed in Table A in S1 Text. We excluded participants of non-European ancestry as determined by principal component analysis [35]. The final study population comprised 11,900 cases (11,074 colorectal cancers, 826 advanced colorectal adenomas) and 14,311 controls.
Colorectal tumor case definition
Detailed information on case and control definitions is provided in S1 Text. Colorectal cancer cases were defined as colorectal adenocarcinoma confirmed by medical records, pathologic reports, or death certificates. Controls for colorectal cancer cases were population-based or selected from cohort participants who provided a blood sample and had no previous diagnosis of colorectal cancer. Advanced colorectal adenoma cases in the Nurses' Health Study and Health Professionals Follow-Up Study were confirmed by medical records, histopathology, or pathologic reports. Controls for advanced adenoma cases had a negative colonoscopy (except for controls matched to cases with distal adenoma, which either had a negative sigmoidoscopy or colonoscopy exam).
Genotyping and quality control
Detailed information on genotyping and quality control procedures has been described [3, 34] and are available in S1 Text. Briefly, DNA from blood or buccal samples was genotyped using either Affymetrix (Gene Chip 10K, Mendel) (Affymetrix, Santa Clara, CA) or Illumina arrays (HumanHap550K, 610K, combined 300K and 240K, Human1M, HumanCytoSNP, HumanOmniExpress) (Illumina, Inc., San Diego, CA). Genotyped SNPs were excluded based on call rate (<98%), lack of Hardy-Weinberg Equilibrium in controls (P<1x10-4), and low minor allele frequency (MAF). All analyses were restricted to samples clustering with the Utah residents of Northern and Western European ancestry, using 1000 Genomes populations as reference, from the Centre d’etude du polymorphisme humain (CEPH) collection (CEU) population in principal component analysis [35].
Genotype imputation to 1000 Genomes Project
We imputed genotype data to increase the density of genetic variants. As the reference panel we used the haplotypes of 1,092 samples (all populations) from release version 2 of the 1000 Genomes Project Phase I (ftp://ftp-trace.ncbi.nih.gov/1000genomes/ftp/release/20110521) [36]. Combining reference data from all populations helps improve imputation accuracy of low-frequency variants [37]. The target panel comprised genome-wide genotype data obtained using the methods described above. The target panel was phased using Beagle [38], and the phased target panel was imputed to the 1000 Genomes reference panel using Minimac [39]. We used Rsq as the imputation quality measure for imputed SNPs [40]. For imputed SNPs, we required that variants with low MAFs had higher imputation quality: For SNPs with MAF>0.01, we excluded those with Rsq≤0.3; for MAFs of 0.005–0.01, we excluded Rsq<0.5; and for MAF<0.005, we excluded Rsq<0.99.
Statistical analysis
Variant selection
To determine genomic regions for fine-mapping, we identified 31 autosomal SNPs (index SNPs) in 22 loci previously associated with CRC risk in GWAS conducted in European ancestry individuals (Table B in S1 Text) [3–18]. Through fine-mapping, we aimed to (1) refine the location of potential functional candidate(s) tagged by these index SNPs and (2) identify novel or independent signals near these loci, the latter being hypothesis-generating. We thus defined a broad genomic region of interest as that spanning 250-kb upstream and downstream of each index SNP and evaluated all variants in this 500-kb interval.
Association testing
All statistical analyses were conducted centrally at the GECCO coordinating center on individual-level data to ensure a consistent analytical approach. Unless otherwise indicated, as appropriate, we adjusted for age at the reference time, sex, study center, smoking status (PHS only), batch effects (ASTERISK only: upon quality control there were slight variations in genotyping quality across batches, which were not observed in other studies), and the first three principal components from EIGENSTRAT [35] to account for population substructure.
For each study, we estimated the association between individual variants and colorectal tumor risk by calculating odds ratios (ORs) and 95% confidence intervals (CIs) using unconditional logistic regression assuming log-additive genetic effects. Each genotyped SNP was coded as 0, 1 or 2 copies of the variant allele. For imputed SNPs, we used the expected number of copies of the variant allele (the dosage), which gives unbiased effect estimates [41]. To combine study-specific estimates across studies, we obtained summary estimates using inverse-variance weighted fixed-effects meta-analysis. Colorectal cancer cases and their controls were analyzed separately from advanced adenoma cases and their controls before meta-analysis. We calculated heterogeneity P-values using Cochran's Q statistics [42]. Quantile-quantile (Q-Q) plots were used to assess whether the distribution of P-values was consistent with the null distribution (except for the extreme tail). In each region, we searched for additional independent association signals by testing each of the other variants conditioned on the top SNP in that region (i.e., 2 variants included in each model); variants are expected to be less correlated after conditioning on the top SNP. When testing for additional independent signals, we determined the P-value threshold for statistical significance by using the number of SNPs in each 500-kb region as the Bonferroni correction factor (α-level for a region = 0.05/# SNPs in that region). We used this approach to correct for multiple testing while also accounting for the knowledge that genetic variation in these regions is known to influence predisposition to CRC.
We used R (Version 2.15.1, R Foundation for Statistical Computing, Vienna, Austria) to conduct the statistical analysis, and LocusZoom [43] to visualize results. To determine the minimum detectable effect estimates in the present analysis, we estimated statistical power using Quanto Version 1.2.4 (http://hydra.usc.edu/gxe/).
Functional annotation using bioinformatics
Detailed information on functional annotation and various databases is provided in S1 Text. In brief, compared with variants that are either non-functional or not in linkage disequilibrium with the underlying functional variant(s), colorectal tumor association signals are expected to be strongest (show the smallest P-value) for the functional variant(s), or variants in high linkage disequilibrium with the functional variant(s). Thus we selected the following for bioinformatics follow-up: 1) the variant showing the strongest evidence for association (smallest P-value) in each region (i.e., top SNP), 2) the index SNP in each region, 3) among the top 10 variants with the smallest P-values in each region, those that were correlated (r2>0.5 in 1000 Genomes European populations) with the index SNP, and 4) any SNP completely correlated (r2 = 1 in 1000 Genomes European populations) with any SNP listed in parts 1–3. In addition, after performing conditional analyses that simultaneously included the index SNP(s) in multivariable models, we annotated SNPs showing P ≤ 5E-05 and any SNPs completely correlated with these.
We annotated the potential function of variants in coding regions using PolyPhen-2 [44]. For variants in non-coding regions, we used HaploReg [45, 46] and the University of California, Santa Cruz (UCSC) Genome Browser [47] to align each SNP to the reference genome and annotate them with multiple datasets generated from the Encyclopedia of DNA Elements (ENCODE) Project [48, 49] or the NIH Roadmap program on Epigenomics [50] as detailed in S1 Text. Annotation using these databases assumes that the disease-causing variant(s) affects disease by altering gene transcription through multiple regulatory mechanisms [48, 49]. Such mechanisms include indicators for regions that may influence transcriptional regulation of target genes, such as chromatin accessibility (open chromatin), histone modification, binding of regulatory proteins, and alteration of regulatory motifs [45, 51, 52]. Conservation across vertebrates can provide further evidence of biologically important regions [53, 54]. To identify variants showing any of these indicators of functional importance, we first queried HaploReg [45, 46], which provides an overview of available annotations. We further interrogated variants with any functional evidence using the UCSC Genome Browser [47] to examine signal enrichment in regions harboring these variants, which helps correct for false positive signals for each assay (https://sites.google.com/site/anshulkundaje/projects/idr). Specifically, we examined whether variants were located in functionally important regions using the following datasets compiled by HaploReg [46] or UCSC Genome Browser [47]: DNAse I hypersensitivity data in ENCODE cell lines, including two for CRC (HCT-116 and Caco-2), to assess open chromatin structure; ChIP-seq data in ENCODE cell lines as well as Roadmap data in normal colon and rectal tissues for histone enhancer or promoter modifications; ChIP-seq data in ENCODE cell lines to determine regions that bound to important regulatory proteins (e.g., promoters, enhancers, silencers, and insulators); change in log-odds score based on position weight matrices [45] to predict whether a sequence harboring either the reference or alternate allele would exhibit altered binding affinities for regulatory proteins; and PhastCons scores [53, 54] to predict genomic regions conserved across vertebrates. As intergenic variants often regulate the closest downstream gene [48, 49], we predicted the gene regulated by each variant based on proximity of each variant to a gene as well as the orientation (3’ or 5’) relative to the nearest end of the gene [45]. Recognizing that cis-regulatory elements can also skip the closest gene, in exploratory analyses we integrated expression quantitative trail locus (eQTL) analysis to identify other potential tissue-specific target genes from the Genotype-Tissue Expression (GTEx) database [46], GEUVADIS [55], and other recent studies [56–60] using HaploReg and the GTEx Portal. Further, we evaluated variants in potential splice sites using Genie [61].
The relative strength of functional candidates was determined based on the accumulation of evidence from each of these datasets. A priori, we defined a score to summarize the amount of functional evidence for each variant using the following algorithm: showed (+1) histone modification, (+1) open chromatin, (+1) protein binding, (+1) protein binding in the presence of open chromatin or histone modification, (+1) different patterns of histone modification in cancerous vs. noncancerous cell lines/ tissues, (+1) regulatory evidence in a CRC cell line (e.g. Caco-2 or HCT-116) or normal colon/rectal tissue, (+0.5) altered binding motif, and (+0.5) a conserved region across vertebrates. Thus, variants were assigned a maximum score of 7. Although not observed in our data, any variant in a coding region predicted by PolyPhen to be “possibly damaging” or “probably damaging” would have been assigned a score of 8 or 9, respectively. Variants in coding regions predicted by PolyPhen to be “benign” or “unknown” were scored as a non-coding regulatory variant, as DNA sequences can act as coding exons in one tissue and enhancers of nearby gene(s) in another [62]. Caution should be exercised when interpreting these scores as there is a degree of uncertainty when relating annotation data to SNP function. These data are based on transformed cell lines or tissues instead of living organisms, and regulatory mechanisms may vary temporally as well as across different types of cells or tissues. However, bioinformatics analysis is primarily useful for prioritizing a large number of variants for more onerous laboratory follow-up; to this end, we used these scores to create 3 categories that ranked the strength of functional evidence for each variant: score of 3–3.5 =“weak”, 4–4.5 =“moderate”, and ≥5 =“strong”.
Results
The mean age of the 26,211 participants was 64.2 years, ranging from 19 to 99 years (Table A in S1 Text). Two studies (HPFS, PHS) comprised only males, and 3 studies (NHS, PMH, WHI) only females. The proportion of females in the remaining studies ranged from 30.8% to 52.0%.
For the 31 previously reported CRC-related variants (index SNPs), 17 showed P-values ≤ 0.001, 22 showed P-values ≤ 0.01, and 27 showed P-values ≤ 0.05 (Table B in S1 Text). Further, ORs for 30 of 31 SNPs showed directions consistent with previous findings.
Across the 31 genomic regions encompassing index SNPs, there were on average 1,807 SNPs per 500-kb region, ranging from 967 to 2,364 SNPs per region. SNPs with the strongest evidence of CRC-associations may more likely be functional or strong proxies for functional candidates. To help refine regions harboring functional candidates, we identified the SNP showing the smallest P-value in each region (i.e., top SNP) (Table 1). The initial index SNP did not show the strongest association signal in any genomic region (Fig 1). For loci that harbored more than 1 index SNP, the regions encompassing each index SNP sometimes overlapped, yielding regions in which the top SNP was the same (e.g., at 1q41, the top SNP rs143030473 showed the smallest P-value in 2 regions, defined by index SNPs rs6687758 and rs6691170). This was observed in 1q41, 12p13.32, 14q22.2, and 15q13. Thus for the 31 regions studied there were 25 unique top SNPs (note 12p13.32 and 15q13 each contained 3 index SNPs); of these, 20 had an association with P-values ≤ 0.001 and all 25 showed P-values ≤ 0.01. For these 25 variants, the top SNP was correlated with an index SNP in European populations at r2 ≥ 0.8 for 8 SNPs, 0.6 ≤ r2 < 0.8 for 6 SNPs, 0.4 ≤ r2 < 0.6 for 4 SNPs, 0.2 ≤ r2 < 0.4 for 1 SNP, and r2 < 0.2 for the remaining 6 SNPs.
Table 1. Association results for variants showing the smallest P-values (top SNPs) in 31 regions surrounding previous GWAS-identified variants (index SNPs).
| Locus | Index SNP | Level a | # SNPs | Top SNP | Level a | Position b | Genetic | r2 with | Top SNP results | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| in | region | index | Ref/ | Ref | OR (95% CI) d,e | P | P-het | ||||||
| region | SNP c | other | allele | ||||||||||
| allele | freq | ||||||||||||
| 1q25.3 | rs10911251 | *** | 1886 | rs6669796 | *** | 183082825 | LAMC1 | 0.87 | G/C | 0.56 | 1.11 (1.07, 1.16) | 5.8E-07 | 6.0E-01 |
| 1q41 | rs6687758 | * | 1885 | rs143030473 | *** | 222161943 | DUSP10/CICP13 | <0.2 | C/T | 0.99 | 1.97 (1.35, 2.87) | 4.5E-04 | 9.2E-01 |
| rs6691170 | 2096 | rs143030473 | *** | 222161943 | DUSP10/CICP13 | <0.2 | C/T | 0.99 | 1.97 (1.35, 2.87) | 4.5E-04 | 9.2E-01 | ||
| 2q32.3 | rs11903757 | *** | 1536 | rs6731095 | *** | 192589442 | NABP1/SDPR | 1.00 | A/G | 0.84 | 0.89 (0.84, 0.95) | 2.3E-04 | 1.1E-01 |
| 3q26.2 | rs10936599 | 1651 | rs2421771 | ** | 169411370 | MECOM/MYNN | <0.2 | C/T | 0.96 | 1.21 (1.05, 1.40) | 7.5E-03 | 9.2E-01 | |
| 5q31.1 | rs647161 | ** | 1499 | rs2193941 | *** | 134469594 | PITX1/ H2AFY | 0.57 | G/A | 0.72 | 1.09 (1.05, 1.14) | 4.0E-05 | 9.2E-01 |
| 6p21 | rs1321311 | 2364 | rs13215272 | *** | 36589502 | SRSF3/CDKN1A | <0.2 | C/T | 0.73 | 1.10 (1.04, 1.15) | 5.3E-04 | 8.0E-02 | |
| 8q23.3 | rs16892766 | *** | 1432 | rs16888589 | *** | 117635602 | TRPS1/EIF3H | 0.92 | A/G | 0.91 | 0.80 (0.75, 0.86) | 3.3E-10 | 7.1E-01 |
| 8q24 | rs6983267 | *** | 2257 | rs7013278 | *** | 128414892 | SRRM1P1/POU5F1B/MYC | 0.40 | C/T | 0.66 | 0.88 (0.85, 0.92) | 7.8E-11 | 7.4E-01 |
| 9p24 | rs719725 | *** | 1907 | rs7875812 | *** | 6364533 | TPD52L3/UHRF2 | 1.00 | A/T | 0.63 | 1.09 (1.04, 1.13) | 7.9E-05 | 7.4E-01 |
| 10p14 | rs10795668 | * | 2363 | rs1537603 | ** | 8734295 | KRT8P16/TCEB1P3 | 0.45 | C/T | 0.50 | 1.06 (1.02, 1.10) | 1.2E-03 | 7.9E-01 |
| 11q13.4 | rs3824999 | *** | 1788 | rs72977282 | *** | 74300441 | LIPT2/POLD3 | 0.59 | T/A | 0.59 | 0.92 (0.88, 0.95) | 9.8E-06 | 8.1E-01 |
| 11q23 | rs3802842 | *** | 1830 | rs7130173 | *** | 111154072 | C11orf93 | 0.95 | C/A | 0.73 | 0.89 (0.85, 0.93) | 2.7E-08 | 3.8E-01 |
| 12p13.32 | rs10774214 | * | 1656 | rs3217874 | *** | 4400808 | CCND2 | <0.2 | C/T | 0.58 | 0.89 (0.85, 0.93) | 3.1E-08 | 7.8E-01 |
| rs3217810 | *** | 1571 | rs3217874 | *** | 4400808 | CCND2 | <0.2 | C/T | 0.58 | 0.89 (0.85, 0.93) | 3.1E-08 | 7.8E-01 | |
| rs3217901 | *** | 1539 | rs3217874 | *** | 4400808 | CCND2 | 0.65 | C/T | 0.58 | 0.89 (0.85, 0.93) | 3.1E-08 | 7.8E-01 | |
| 12q13.13 | rs11169552 | * | 1310 | rs7306677 | *** | 51205763 | ATF1 | <0.2 | C/T | 0.62 | 0.92 (0.89, 0.96) | 6.3E-05 | 3.4E-01 |
| rs7136702 | *** | 967 | rs11169524 | *** | 51089734 | DIP2B | 0.67 | T/A | 0.67 | 0.92 (0.88, 0.96) | 8.5E-05 | 5.3E-01 | |
| 12q24.21 | rs59336 | *** | 2072 | rs1427760 | *** | 115100714 | TBX5/TBX3 | 0.71 | T/C | 0.50 | 0.90 (0.87, 0.94) | 5.0E-07 | 2.5E-01 |
| 14q22.2 | rs1957636 | *** | 1613 | rs10130587 | *** | 54419110 | BMP4 | <0.2 | G/C | 0.63 | 0.89 (0.85, 0.93) | 4.1E-08 | 9.0E-01 |
| rs4444235 | *** | 1659 | rs10130587 | *** | 54419110 | BMP4 | 0.67 | G/C | 0.63 | 0.89 (0.85, 0.93) | 4.1E-08 | 9.0E-01 | |
| 15q13 | rs11632715 | ** | 1735 | rs2293582 | *** | 33010412 | GREM1 | 0.23 | G/A | 0.80 | 0.86 (0.82, 0.91) | 1.0E-09 | 3.8E-01 |
| rs16969681 | ** | 1692 | rs2293582 | *** | 33010412 | GREM1 | 0.22 | G/A | 0.80 | 0.86 (0.82, 0.91) | 1.0E-09 | 3.8E-01 | |
| rs4779584 | *** | 1701 | rs2293582 | *** | 33010412 | GREM1 | 0.71 | G/A | 0.80 | 0.86 (0.82, 0.91) | 1.0E-09 | 3.8E-01 | |
| 16q22.1 | rs9929218 | ** | 1867 | rs9932005 | ** | 68822019 | CDH1 | 0.63 | C/T | 0.76 | 1.08 (1.03, 1.13) | 1.2E-03 | 6.6E-01 |
| 18q21 | rs4939827 | *** | 1931 | rs2337113 | *** | 46452327 | SMAD7 | 0.91 | A/G | 0.55 | 1.13 (1.09, 1.18) | 1.0E-10 | 2.5E-01 |
| 19q13.1 | rs10411210 | * | 2307 | rs75414102 | ** | 33302424 | TDRD12/SLC7A9 | <0.2 | G/A | 0.99 | 0.51 (0.33, 0.79) | 2.6E-03 | 9.1E-01 |
| 20p12.3 | rs2423279 | 1901 | rs118184022 | ** | 7719449 | BMP2/HAO1 | <0.2 | C/T | 0.98 | 0.58 (0.41, 0.83) | 2.8E-03 | 9.4E-01 | |
| rs4813802 | *** | 1825 | rs6085662 | *** | 6698372 | FERMT1/BMP2 | 1.00 | G/C | 0.65 | 0.91 (0.87, 0.95) | 5.1E-06 | 3.0E-01 | |
| rs961253 | *** | 1990 | rs56083061 | *** | 6430696 | FERMT1/BMP2 | 0.28 | G/A | 0.79 | 0.90 (0.85, 0.94) | 5.6E-06 | 7.6E-01 | |
| 20q13.33 | rs4925386 | ** | 2173 | rs1760073 | *** | 60926106 | LAMA5 | 0.81 | G/A | 0.67 | 1.08 (1.04, 1.12) | 1.9E-04 | 8.8E-02 |
Abbreviations: SNP, single nucleotide polymorphism; Ref, reference; Freq, frequency; OR, odds ratio; CI, confidence interval; P-het, P value for test of heterogeneity across studies
aLevel of statistical evidence:
*SNP with P ≤ 0.05
**SNP with P ≤ 0.01
***SNP with P≤0.001. P-values are not adjusted for multiple testing as these data are intended to help refine previously identified CRC-related regions and highlight sets of variants warranting bioinformatics-based follow-up.
bBased on NCBI build 37 data.
cBased on 1000 Genomes Project data in European populations.
dAdjusted for age in years, sex, first 3 principal components, study center, batch (ASTERISK only), and smoking (PHS only).
eEstimate calculated using the log-additive genetic model for each additional reference allele.
Fig 1. Comparison of P-values for GWAS-identified variants (index SNPs) vs. variants with the smallest P-values (top SNPs) in 31 regions.
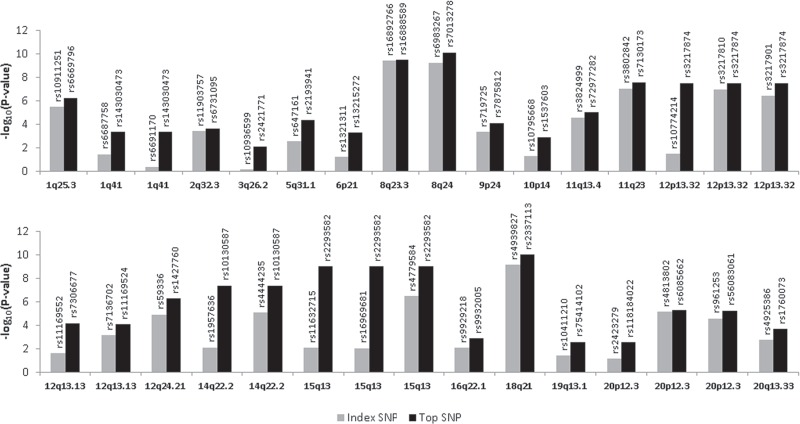
The height of each bar reflects the–log10 P-value of each SNP in our study population. A grey bar indicates the index SNP, and a black bar indicates the top SNP.
Variants carried forward for functional annotation spanned a median interval of 32-kb. We scored 21 variants in 12 loci as having “strong” functional evidence (Table 2, additional details in S1 Table). At 4 loci (8q24, 11q13.4, 19q13.1, 20p12.3) the index SNP was among the SNPs with the highest functional scores. All 21 candidates were located in regions that were non-coding (15 intronic and 6 intergenic) with open chromatin structure (i.e., accessible to regulatory factors). Twenty of 21 candidates (all except for 18q21/rs34007497) bound to multiple transcription factors. Fifteen variants were predicted to disrupt transcription factor binding. Several candidates showed different patterns of histone enhancer or promoter marks when comparing cancer cells vs. normal cells or tissues. Only 3 variants (8q24/rs6983267, 18q21/rs4939567, 20p12.3/rs4813802) were located in an evolutionarily conserved region, suggesting that most of the predicted regulatory regions may be dynamic through evolution.
Table 2. SNPs showing strong evidence for functional importance based on bioinformatics.
| Locus | Index SNP | Strong | Mean | r2 with | Predicted | Genomic | # Cell lines | Show histone regulatory marks e | Proteins | Altered binding | Evidence | ||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| functional | imput- | index | regulated | location of | showing | # Cancer/ | # Normal | Normal | bound e | motifs g | for | ||
| candidate | ation | SNP b | gene c | functional | open | progenitor | cell lines | colorectal | conserved | ||||
| r2 a | candidate | chromatin d | cell lines | tissues f | region h | ||||||||
| 5q31.1 | rs647161 | rs1366111 | 0.89 | 0.57 | PITX1 | intronic | 2 | 2 | — | 1, 2 | POL2,EGR1 | Zfx | 0 |
| 8q24 | rs6983267 | rs6983267 | 0.95 | index | MYC | intergenic | 1 CRC line | 1 | 2 | 2 | TCF4 (CRC line), | AP-1, Sox, TCF4 | 1 |
| P300, FOXA1, | |||||||||||||
| RXRA, SP1 | |||||||||||||
| 11q13.4 | rs3824999 | rs3824999 | 0.99 | index | POLD3 | intronic | 4 | — | 2 | 4 | JUND | AP-3, Evi-1, Mef2 | 0 |
| 11q23 | rs3802842 | rs7130173 | 0.95 | 0.93 | Unknown | intronic | 31 including | — | — | 2, 3 | RAD21, SMC3, | GSP1, SRF | 0 |
| 1 CRC line | YY1, CTCF | ||||||||||||
| 12p13.32 | rs3217901 | rs3217827 | 0.85 | 0.61 | CCND2 | intronic | 1 | — | — | 3 | RAD21, CTCF | — | 0 |
| 12q24.21 | rs59336 | rs71807 | 0.89 | 0.86 | TBX3 | intergenic | 1 | 2 | 4 | 1, 3 | BAF155, HAE2F1, | — | 0 |
| CTCF | |||||||||||||
| rs484443 | 0.91 | 0.90 | TBX3 | intronic | 8 | 1 | 2 | 2, 4 | P300,USF1 | RREB-1, Rad21 | 0 | ||
| 14q22.2 | rs4444235 | rs2071047 | 1.00 | 0.75 | BMP4 | intronic | 9 | 3 | 3 | 1–4 | TCF4 (CRC line) | BCL, HNF4, RXRA | 0 |
| rs10130587 | 0.74 | 0.67 | BMP4 | intronic | 2 | 3 | 3 | 1–4 | GATA3 | 5 motifs | 0 | ||
| rs35107139 | 0.82 | 0.64 | BMP4 | intronic | 2 | 3 | 3 | 1, 3, 4 | GATA3 | 12 motifs | 0 | ||
| 15q13 | rs4779584 | rs2293582 | 0.94 | 0.71 | GREM1 | intronic | 4 | 3 | 5 | 1–4 | POL2 | — | 0 |
| rs2293581 | 0.97 | 0.66 | GREM1 | intronic | 36 | 4 | 5 | 1–4 | SUZ12 | — | 0 | ||
| rs1406389 | 0.97 | 0.66 | GREM1 | intergenic | 12 | 3 | 4 | 1, 2, 4 | SUZ12 | Irf, SIX5 | 0 | ||
| 18q21 | rs4939827 | rs11874392 | 0.93 | 1.00 | SMAD7 | intronic | 4 | 3 | 1 | 1, 2, 4 | 11 proteins | — | 1 |
| rs4939567 | 0.91 | 1.00 | SMAD7 | intronic | 1 | 4 | 3 | 1–4 | MAFK | RREB-1, VDR_2 | 1 | ||
| rs34007497 | 0.91 | 0.57 | SMAD7 | intronic | 22 | 2 | 4 | 1–4 | CTCF,STAT1 | 4 motifs | 0 | ||
| 19q13.1 | rs10411210 | rs10411210 | 0.94 | index | RHPN2 | intronic | 48 including | 2 | 1 | 1, 2 | 17 proteins | Mef2, STAT | 1 |
| 1 CRC line | |||||||||||||
| 20p12.3 | rs961253 | rs966817 | 1.00 | 0.87 | BMP2 | intergenic | 18 | — | 1 | — | CJUN | Evi-1, FAC1, GLI | 1 |
| rs4813802 | rs4813802 | 0.86 | index | BMP2 | intergenic | 10 | 2 | 2 | 2 | 6 proteins | — | 1 | |
| rs6085661 | 0.89 | 0.76 | BMP2 | intergenic | 19 | 1 | — | 2 | 7 proteins | BCL, NRSF, VDR | 1 | ||
| 20q13.33 | rs4925386 | rs1741634 | 0.96 | 0.80 | LAMA5 | intronic | 7 | — | 2 | 1, 2 | GR | EWSR1-FLI1 | 0 |
aEstimated value of the squared correlation between imputed genotypes and true (unobserved) genotypes for the strong functional candidate, averaged across studies.
bBased on data from 1000 Genomes Pilot 1 CEU.
cBased on HaploReg data using proximity of each variant to a gene as well as the orientation (3’ or 5’) relative to the nearest end of the gene.
dBased on data from DNAse I hypersensitivity assays.
eENCODE ChIP-seq assay.
fBased on data from ChIP-seq assays in the Roadmap Epigenomics Project in the following normal (non-cancerous) tissues: (1) colon mucosa, (2) rectal mucosa, (3) colon smooth muscle, and (4) rectal smooth muscle.
gPosition weight matrix score between SNP alleles confers a change in log-odds (LOD) score > 5.
hBased on PhastCons scores: 1 = strong evidence of conserved element; 0 = no evidence of conserved element.
In each region, after conditioning on the top SNP and accounting for the number of tests, we did not observe any statistically significant SNPs.
Discussion
In this large study population of over 26,000 participants of European ancestry, we used high-density genetic data imputed from the 1000 Genomes Project to comprehensively fine-map genomic regions harboring 31 GWAS-identified CRC risk variants. In association tests, the index SNP did not show the smallest P-value in any genomic region. Using bioinformatics-based annotation to follow-up variants with the strongest association signals, we showed strong evidence for 21 functional candidates in 12 CRC-related loci. We observed limited evidence of additional independent CRC association signals within GWAS-identified regions.
Although the index SNP did not show the smallest P-value in any genomic region, all functional candidates were correlated with the index SNP (r2 of at least 0.57). Interestingly, however, the index SNP was a strong functional candidate in only 4 of the 12 loci harboring a strong functional candidate. Combined, these data from our association testing and functional annotation support the hypothesis that most GWAS-identified index SNPs are not the underlying functional variant, but may instead act as proxies of correlated variants with biological importance.
Eight previous studies have fine-mapped a limited number of GWAS-identified CRC loci in individuals of European ancestry [14, 17, 24–29]; these studies have reported 34 candidate SNPs showing functional evidence (summarized in Table C in S1 Text). In addition, 2 recent studies have comprehensively fine-mapped known CRC loci: Whiffin et al. [31] identified 4 additional candidates in 1q41, 15q13, 18q21, and 20q13.33 in European ancestry individuals (5,626 cases; 7,817 controls); Wang et al. [30] identified 1 additional candidate in 1q41 in African Americans (1,894 cases; 4,703 controls). Of these 39 reported candidates in 11 loci, 36 passed genotyping quality control in our study. In the present analysis, 16 of these SNPs had P-values ≤ 0.001, 5 had P-values >0.001 and ≤ 0.01, and 7 had P-values >0.01 and ≤ 0.05. Similar to our findings, only 5 of 39 previously reported functional candidates were GWAS-identified SNPs. We observed 3 exonic candidates out of an expanded list of 51 variants showing “weak”, “moderate”, or “strong” functional evidence (see S1 Table for an expanded list of functional candidates); similarly, only 2 previously reported candidates (rs706793, rs28626308) were in coding regions [24, 25]—highlighting the importance of non-coding effects on CRC [63].
To identify potential variants for laboratory follow-up, we compared all previously reported candidates (in 11 loci) with variants in the present analysis that showed “moderate” or “strong” functional evidence (Table C in S1 Text). In 5 loci (8q23.3, 8q24, 15q13, 18q21, and 19q13.1) our data confirmed previously reported functional candidates [25, 27, 29, 31]. In addition, in 11q23 and 16q22.1 we observed candidate(s) that highly correlated and were within 5-kb of a previously reported candidate variant [25, 28]. Fine-mapping can be limited in distinguishing which of several highly correlated SNPs located very close together is the true causal variant. Our candidates in these 2 loci, 11q23/rs7130173 and 16q22.1/rs9929218, showed stronger functional evidence compared with reported candidates, which either were not selected for functional annotation or showed less than “weak” functional evidence (scored less than 3). These data show that to avoid missing functional variation, laboratory studies should follow-up not just the strongest candidates, but also variants showing any evidence of biological importance that are very close and highly correlated. Our data did not show functional evidence for reported candidates in the remaining 4 loci. In 1q41 we did not identify a functional candidate; in 12q13.13 and 14q22.2 we predicted functional candidates that were >150-kb from those previously reported [14, 24]; and in 20q13.33 our candidates were >5-kb away and did not show high correlation with those previously reported (r2 = 0.59–0.60) [31]. In 3 of these loci (1q41, 12q13.13, 14q22.2) only 1 of 7 previously reported candidates showed P<0.05 in our study population, suggesting they may be false positives. In 20q13.33 the reported candidates, rs1741640 and rs2236202, were not selected for functional annotation in our study based on their P-values relative to other variants in the region. Taken together, these data support the utility of fine-mapping to reveal potential functional variation, but also highlight that these studies only serve as an initial step toward determining the underlying causal variant(s) that lead to disease. Results from bioinformatics-based annotation depend on various factors (e.g., queried variants, queried databases, choice of cell lines and tissues, uncertainty in interpreting data from qualitative assays, among others), which vary between studies. It is likely these differences in methodology and interpretation when annotating variants account in part for inconsistencies in results. Consequently, replication of fine-mapping findings is useful, and only targeted functional studies can provide more definitive evidence of SNP function [22, 23].
In our study, for instance, the 500-kb region containing rs6983267 (8q24) harbored 2,257 SNPs. Based on association testing, we narrowed this region to a 13-kb interval that included 7 correlated SNPs showing stronger association signals (Figure A panel A in S1 Text). After bioinformatics analysis, the best functional candidate was the index SNP rs6983267, which was predicted to alter the binding of TCF4 transcription factor. Consistent with this, Tuupanen et al. [27] showed in vitro and in vivo that rs6983267 resulted in differential TCF4 binding, which may result in enhanced responsiveness to Wnt signaling and a subsequent increase in risk. Further, several other laboratory experiments support the biological importance of this variant in CRC [64–66]. Similarly, the 500-kb region containing rs3802842 (11q23) harbored 1,830 SNPs. Association tests narrowed this region to an 18-kb interval that included 9 correlated SNPs for which we performed bioinformatics follow-up (Figure A panel B in S1 Text). Among these, rs7130173 showed strong regulatory evidence in our study. Consistent with these findings, Biancolella et al. [67] recently showed that the risk allele of rs7130173 reduced enhancer activity and resulted in reduced transcription factor binding affinity in CRC cells. A combination of fine-mapping and laboratory functional follow-up has also shown similar successes for other cancers and chronic diseases [23, 68, 69]. Taken together, these data suggest that by combining association testing and bioinformatics analysis, fine-mapping can reduce the size of relevant genomic regions and successfully prioritize candidates for molecular characterization, which greatly reduces the cost, time, and labor associated with testing a large number of variants in the laboratory.
In addition to confirming previous candidates, we suggest several novel candidates with strong functional evidence. These, located in 4 loci with previously reported functional candidates (12q13.13, 14q22.2, 15q13, 20q13.33) and 5 loci without any previously reported candidates (5q31.1, 11q13.4, 12p13.32, 12q24.21, 20p12.3), implicated genes expected to be involved in CRC development as well as those that were unexpected. For instance, duplication in the GREM1 (gremlin 1) promoter has been linked to hereditary mixed polyposis syndrome [70], suggesting it is a candidate gene for colorectal tumorigenesis. In our analysis rs2293582, an intronic SNP in GREM1 (15q13), showed the smallest P-value in this region and was among our best functional candidates. The region containing rs2293582 exhibited open chromatin and bound RNA Polymerase 2 in vivo (ENCODE tracks shown in Figure B in S1 Text). This region also showed strong promoter marks in colon cancer cell lines, but greatly reduced signals in normal colon and rectal tissues. These data suggest rs2293582 warrants experimental follow-up, along with two highly correlated variants within 1-kb, rs2293581 (r2 = 0.94) and a previously reported candidate rs1406389 (r2 = 0.94) [31], located in regions showing histone marks, open chromatin, and binding to the repressive transcription factor SUZ12 [71]. Fine-mapping can also help identify functional candidates that implicate unexpected genes for further functional study. LAMA5 (laminin, alpha 5), for instance, is involved in maintaining the extracellular matrix [72], which may not be expected to predict cancer risk. The SNP showing the smallest P-value in the 20q13.33/LAMA5 region, rs1760073, was completely correlated (r2 = 1) with the best functional candidate, rs1741634, which was located in an intron of LAMA5. The region containing rs1741634 exhibited open chromatin, bound the glucocorticoid receptor transcription factor, which has been implicated in cancer [73], and interestingly, was located in a region showing different enhancer marks in CRC cell lines vs. normal colon and rectal tissues (Figure C in S1 Text). In addition, Whiffin et al. [31] recently reported other functional candidates in this region. Thus, although unexpected, these data, along with those from GWAS showing associations with a variant in another laminin gene, LAMC1 (laminin, gamma 1) [3, 19], support the role of laminin proteins in colorectal carcinogenesis.
Particular strengths of this study included the large study population, high-density genetic data, as well as systematic approach to fine-mapping all GWAS-identified CRC risk variants; however, limitations should be noted. As we aimed to comprehensively investigate both common and less common genetic variation, we examined directly genotyped SNPs as well as SNPs imputed from the 1000 Genomes Project. Imputed genotypes can be called with varying accuracy, and we accounted for this using the genotype dosage, which have been shown to yield unbiased estimates [41]. However, lower imputation accuracy may attenuate the estimated significance of association signals [74, 75], and thus relative P-values for individual variants may not necessarily correspond to their functional importance. Accordingly, rather than only assessing the SNP showing the smallest P-value in each region we identified a set of SNPs showing stronger association signals for bioinformatics analysis, which enabled us to reduce considerably the number of potential functional SNPs per region and still be able to identify promising functional candidates. Even within our large study, limited statistical power may have accounted for the absence of less common independent association signals at known CRC susceptibility loci, particularly for regions where the initial GWAS showed weak effects. For common genetic variants (allele frequency = 20%), the present analysis had 80% power to detect a per-allele OR of 1.12; for less common variants (allele frequency = 1%), there was 80% power to detect a per-allele OR of 1.51 (Figure D in S1 Text). These estimates suggest that although larger populations are likely needed to detect weaker associations with less common variants, our data provided sufficient statistical power to detect less common SNPs with larger effect sizes.
In this large population, we comprehensively fine-mapped known common variants that predict CRC risk. We refined genomic regions harboring risk variants and proposed novel functional candidates, as well as confirmed several regions previously reported to contain functional variation. These findings support the utility of a systematic fine-mapping approach that integrates information from expanding publicly available databases to help refine regions surrounding GWAS-identified risk variants and identify a limited number of functional candidates. These insights may help establish a framework for follow-up laboratory studies, which are necessary to yield definitive evidence of functional SNPs that drive common genetic predisposition to CRC.
Supporting Information
(XLSX)
Describes in detail the study population and case/control definition; genotyping and quality control; as well as functional annotation using bioinformatics. Also includes Supplementary Tables A-C and Supplementary Figures A-D.
(DOCX)
Acknowledgments
ASTERISK: We are very grateful to Dr. Bruno Buecher without whom this project would not have existed. We also thank all those who agreed to participate in this study, including the patients and the healthy control persons, as well as all the physicians, technicians and students.
DACHS: We thank all participants and cooperating clinicians, and Ute Handte-Daub, Renate Hettler-Jensen, Utz Benscheid, Muhabbet Celik and Ursula Eilber for excellent technical assistance.
GECCO: The authors would like to thank all those at the GECCO Coordinating Center for helping bring together the data and making this project possible.
HPFS, NHS and PHS: We would like to acknowledge Patrice Soule and Hardeep Ranu of the Dana Farber Harvard Cancer Center High-Throughput Polymorphism Core who assisted in the genotyping for NHS, HPFS, and PHS under the supervision of Dr. Immaculata De Vivo and Dr. David Hunter, Qin (Carolyn) Guo and Lixue Zhu who assisted in programming for NHS and HPFS, and Haiyan Zhang who assisted in programming for the PHS. We would like to thank the participants and staff of the Nurses' Health Study and the Health Professionals Follow-Up Study, for their valuable contributions as well as the following state cancer registries for their help: AL, AZ, AR, CA, CO, CT, DE, FL, GA, ID, IL, IN, IA, KY, LA, ME, MD, MA, MI, NE, NH, NJ, NY, NC, ND, OH, OK, OR, PA, RI, SC, TN, TX, VA, WA, WY. In addition, this study was approved by the Connecticut Department of Public Health (DPH) Human Investigations Committee. Certain data used in this publication were obtained from the DPH. The authors assume full responsibility for analyses and interpretation of these data.
PLCO: The authors thank Drs. Christine Berg and Philip Prorok, Division of Cancer Prevention, National Cancer Institute, the Screening Center investigators and staff or the Prostate, Lung, Colorectal, and Ovarian (PLCO) Cancer Screening Trial, Mr. Tom Riley and staff, Information Management Services, Inc., Ms. Barbara O’Brien and staff, Westat, Inc., and Drs. Bill Kopp, Wen Shao, and staff, SAIC-Frederick. Most importantly, we acknowledge the study participants for their contributions to making this study possible.
PMH-CCFR: The authors would like to thank the study participants and staff of the Hormones and Colon Cancer study.
WHI: The authors thank the WHI investigators and staff for their dedication, and the study participants for making the program possible. A full listing of WHI investigators can be found at: https://cleo.whi.org/researchers/Documents%20%20Write%20a%20Paper/WHI%20Investigator%20Short%20List.pdf
Data Availability
For plco genetic data, the prostate and panscan study datasets can be accessed with appropriate approval through the dbgap online resource (http://cgems.cancer.gov/data/) accession numbers phs000207v.1p1 and phs000206.v3.p2, respectively, and the lung datasets from the dbgap website (http://www.ncbi.nlm.nih.gov/gap) through accession number phs000093 v2.p2. For the remaining gecco studies, genetic data is in the process of being uploaded to dbgap and will be available soon (accession number phs001078.v1.p1). While the data release is being finalized, the gecco coordinating center can assist with any data requests (contact: gecco@fredhutch.org).
Funding Statement
This work was supported by the following: GECCO: National Cancer Institute, National Institutes of Health, U.S. Department of Health and Human Services (U01 CA137088; R01 CA059045; U01 CA164930), M.D. is supported by grants R25 CA94880 and P30 CA008748 from the National Cancer Institute. ASTERISK: Hospital Clinical Research Program (PHRC) and supported by the Regional Council of Pays de la Loire, the Groupement des Entreprises Françaises dans la Lutte contre le Cancer (GEFLUC), the Association Anne de Bretagne Génétique and the Ligue Régionale Contre le Cancer (LRCC). COLO2&3: National Institutes of Health (R01 CA60987). CCFR: National Institutes of Health (RFA # CA-95-011) and through cooperative agreements with members of the Colon Cancer Family Registry and P.I.s. This genome wide scan was supported by the National Cancer Institute, National Institutes of Health by U01 CA122839. The content of this manuscript does not necessarily reflect the views or policies of the National Cancer Institute or any of the collaborating centers in the CFRs, nor does mention of trade names, commercial products, or organizations imply endorsement by the US Government or the CFR. The following Colon CFR centers contributed data to this manuscript and were supported by National Institutes of Health: Australasian Colorectal Cancer Family Registry (U01 CA097735), Ontario Registry for Studies of Familial Colorectal Cancer (U01 CA074783), and Seattle Colorectal Cancer Family Registry (U01 CA074794). DACHS: German Research Council (Deutsche Forschungsgemeinschaft, BR 1704/6-1, BR 1704/6-3, BR 1704/6-4 and CH 117/1-1), and the German Federal Ministry of Education and Research (01KH0404 and 01ER0814). DALS: National Institutes of Health (R01 CA48998 to M.L.S.); HPFS, NHS, and PHS: HPFS by the National Institutes of Health (P01 CA 055075, UM1 CA167552, R01 137178, and P50 CA 127003), NHS by the National Institutes of Health (R01 CA137178, P01 CA 087969 and P50 CA 127003,) and PHS by the National Institutes of Health (CA42182). A.T.C. is also supported by a Damon Runyon Clinical investigator Award and K24 DK098311. MEC: National Institutes of Health (R37 CA54281, P01 CA033619, and R01 CA63464). OFCCR: National Institutes of Health, through funding allocated to the Ontario Registry for Studies of Familial Colorectal Cancer (U01 CA074783); see CCFR section above. As subset of ARCTIC, OFCCR is supported by a GL2 grant from the Ontario Research Fund, the Canadian Institutes of Health Research, and the Cancer Risk Evaluation (CaRE) Program grant from the Canadian Cancer Society Research Institute. T.J.H. is a recipient of Senior Investigator Awards from the Ontario Institute for Cancer Research, through generous support from the Ontario Ministry of Research and Innovation. PLCO: Intramural Research Program of the Division of Cancer Epidemiology and Genetics and supported by contracts from the Division of Cancer Prevention, National Cancer Institute, NIH, DHHS. Additionally, a subset of control samples were genotyped as part of the Cancer Genetic Markers of Susceptibility (CGEMS) Prostate Cancer GWAS (Yeager, M et al. Nat Genet 2007 May;39(5):645-9), Colon CGEMS pancreatic cancer scan (PanScan) (Amundadottir, L et al. Nat Genet. 2009 Sep;41(9):986-90 and Petersen, GM et al Nat Genet. 2010 Mar;42(3):224-8), and the Lung Cancer and Smoking study. The prostate and PanScan study datasets were accessed with appropriate approval through the dbGaP online resource (http://cgems.cancer.gov/data/) accession numbers phs000207v.1p1 and phs000206.v3.p2, respectively, and the lung datasets were accessed from the dbGaP website (http://www.ncbi.nlm.nih.gov/gap) through accession number phs000093 v2.p2. Funding for the Lung Cancer and Smoking study was provided by National Institutes of Health (NIH), Genes, Environment and Health Initiative (GEI) Z01 CP 010200, NIH U01 HG004446, and NIH GEI U01 HG 004438. For the lung study, the GENEVA Coordinating Center provided assistance with genotype cleaning and general study coordination, and the Johns Hopkins University Center for Inherited Disease Research conducted genotyping. PMH-CCFR: National Institutes of Health (R01 CA076366 to P.A.N.). VITAL: National Institutes of Health (K05 CA154337). WHI: The WHI program is funded by the National Heart, Lung, and Blood Institute, National Institutes of Health, U.S. Department of Health and Human Services through contracts HHSN268201100046C, HHSN268201100001C, HHSN268201100002C, HHSN268201100003C, HHSN268201100004C, and HHSN271201100004C. CORECT: National Cancer Institute, National Institutes of Health under RFA # CA-09-002 (U19 CA148107). The content of this manuscript does not necessarily reflect the views or policies of the National Cancer Institute or any of the collaborating centers in CORECT, nor does mention of trade names, commercial products, or organizations imply endorsement by the US Government or CORECT. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.Lichtenstein P, Holm NV, Verkasalo PK, Iliadou A, Kaprio J, Koskenvuo M, et al. Environmental and heritable factors in the causation of cancer—analyses of cohorts of twins from Sweden, Denmark, and Finland. N Engl J Med. 2000;343(2):78–85. Epub 2000/07/13. 10.1056/NEJM200007133430201 . [DOI] [PubMed] [Google Scholar]
- 2.Dunlop MG, Tenesa A, Farrington SM, Ballereau S, Brewster DH, Koessler T, et al. Cumulative impact of common genetic variants and other risk factors on colorectal cancer risk in 42 103 individuals. Gut. 2013;62(6):871–81. Epub 2012/04/12. 10.1136/gutjnl-2011-300537 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Peters U, Jiao S, Schumacher FR, Hutter CM, Aragaki AK, Baron JA, et al. Identification of Genetic Susceptibility Loci for Colorectal Tumors in a Genome-wide Meta-analysis. Gastroenterology. 2012. Epub 2012/12/26. 10.1053/j.gastro.2012.12.020 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Houlston RS, Cheadle J, Dobbins SE, Tenesa A, Jones AM, Howarth K, et al. Meta-analysis of three genome-wide association studies identifies susceptibility loci for colorectal cancer at 1q41, 3q26.2, 12q13.13 and 20q13.33. Nature genetics. 2010;42(11):973–7. Epub 2010/10/26. 10.1038/ng.670 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jia WH, Zhang B, Matsuo K, Shin A, Xiang YB, Jee SH, et al. Genome-wide association analyses in east Asians identify new susceptibility loci for colorectal cancer. Nature genetics. 2012. Epub 2012/12/25. 10.1038/ng.2505 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dunlop MG, Dobbins SE, Farrington SM, Jones AM, Palles C, Whiffin N, et al. Common variation near CDKN1A, POLD3 and SHROOM2 influences colorectal cancer risk. Nature genetics. 2012;44(7):770–6. Epub 2012/05/29. 10.1038/ng.2293 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tomlinson IP, Webb E, Carvajal-Carmona L, Broderick P, Howarth K, Pittman AM, et al. A genome-wide association study identifies colorectal cancer susceptibility loci on chromosomes 10p14 and 8q23.3. Nature genetics. 2008;40(5):623–30. Epub 2008/04/01. 10.1038/ng.111 . [DOI] [PubMed] [Google Scholar]
- 8.Tomlinson I, Webb E, Carvajal-Carmona L, Broderick P, Kemp Z, Spain S, et al. A genome-wide association scan of tag SNPs identifies a susceptibility variant for colorectal cancer at 8q24.21. Nature genetics. 2007;39(8):984–8. Epub 2007/07/10. 10.1038/ng2085 . [DOI] [PubMed] [Google Scholar]
- 9.Zanke BW, Greenwood CM, Rangrej J, Kustra R, Tenesa A, Farrington SM, et al. Genome-wide association scan identifies a colorectal cancer susceptibility locus on chromosome 8q24. Nature genetics. 2007;39(8):989–94. Epub 2007/07/10. 10.1038/ng2089 . [DOI] [PubMed] [Google Scholar]
- 10.Haiman CA, Le Marchand L, Yamamato J, Stram DO, Sheng X, Kolonel LN, et al. A common genetic risk factor for colorectal and prostate cancer. Nature genetics. 2007;39(8):954–6. Epub 2007/07/10. 10.1038/ng2098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hutter CM, Slattery ML, Duggan DJ, Muehling J, Curtin K, Hsu L, et al. Characterization of the association between 8q24 and colon cancer: gene-environment exploration and meta-analysis. BMC Cancer. 2010;10:670 Epub 2010/12/07. 10.1186/1471-2407-10-670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kocarnik JD, Hutter CM, Slattery ML, Berndt SI, Hsu L, Duggan DJ, et al. Characterization of 9p24 risk locus and colorectal adenoma and cancer: gene-environment interaction and meta-analysis. Cancer Epidemiol Biomarkers Prev. 2010;19(12):3131–9. Epub 2010/10/28. 10.1158/1055-9965.EPI-10-0878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tenesa A, Farrington SM, Prendergast JG, Porteous ME, Walker M, Haq N, et al. Genome-wide association scan identifies a colorectal cancer susceptibility locus on 11q23 and replicates risk loci at 8q24 and 18q21. Nature genetics. 2008;40(5):631–7. Epub 2008/04/01. 10.1038/ng.133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tomlinson IP, Carvajal-Carmona LG, Dobbins SE, Tenesa A, Jones AM, Howarth K, et al. Multiple common susceptibility variants near BMP pathway loci GREM1, BMP4, and BMP2 explain part of the missing heritability of colorectal cancer. PLoS genetics. 2011;7(6):e1002105 Epub 2011/06/10. 10.1371/journal.pgen.1002105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Houlston RS, Webb E, Broderick P, Pittman AM, Di Bernardo MC, Lubbe S, et al. Meta-analysis of genome-wide association data identifies four new susceptibility loci for colorectal cancer. Nature genetics. 2008;40(12):1426–35. Epub 2008/11/18. 10.1038/ng.262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jaeger E, Webb E, Howarth K, Carvajal-Carmona L, Rowan A, Broderick P, et al. Common genetic variants at the CRAC1 (HMPS) locus on chromosome 15q13.3 influence colorectal cancer risk. Nature genetics. 2008;40(1):26–8. Epub 2007/12/18. 10.1038/ng.2007.41 . [DOI] [PubMed] [Google Scholar]
- 17.Broderick P, Carvajal-Carmona L, Pittman AM, Webb E, Howarth K, Rowan A, et al. A genome-wide association study shows that common alleles of SMAD7 influence colorectal cancer risk. Nature genetics. 2007;39(11):1315–7. Epub 2007/10/16. 10.1038/ng.2007.18 . [DOI] [PubMed] [Google Scholar]
- 18.Peters U, Hutter CM, Hsu L, Schumacher FR, Conti DV, Carlson CS, et al. Meta-analysis of new genome-wide association studies of colorectal cancer risk. Hum Genet. 2012;131(2):217–34. Epub 2011/07/16. 10.1007/s00439-011-1055-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Whiffin N, Hosking FJ, Farrington SM, Palles C, Dobbins SE, Zgaga L, et al. Identification of susceptibility loci for colorectal cancer in a genome-wide meta-analysis. Hum Mol Genet. 2014. Epub 2014/04/17. 10.1093/hmg/ddu177 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tenesa A, Dunlop MG. New insights into the aetiology of colorectal cancer from genome-wide association studies. Nat Rev Genet. 2009;10(6):353–8. Epub 2009/05/13. 10.1038/nrg2574 . [DOI] [PubMed] [Google Scholar]
- 21.Hirschhorn JN, Daly MJ. Genome-wide association studies for common diseases and complex traits. Nat Rev Genet. 2005;6(2):95–108. Epub 2005/02/18. 10.1038/nrg1521 . [DOI] [PubMed] [Google Scholar]
- 22.Freedman ML, Monteiro AN, Gayther SA, Coetzee GA, Risch A, Plass C, et al. Principles for the post-GWAS functional characterization of cancer risk loci. Nature genetics. 2011;43(6):513–8. Epub 2011/05/27. 10.1038/ng.840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lowe CE, Cooper JD, Brusko T, Walker NM, Smyth DJ, Bailey R, et al. Large-scale genetic fine mapping and genotype-phenotype associations implicate polymorphism in the IL2RA region in type 1 diabetes. Nature genetics. 2007;39(9):1074–82. Epub 2007/08/07. 10.1038/ng2102 . [DOI] [PubMed] [Google Scholar]
- 24.Spain SL, Carvajal-Carmona LG, Howarth KM, Jones AM, Su Z, Cazier JB, et al. Refinement of the associations between risk of colorectal cancer and polymorphisms on chromosomes 1q41 and 12q13.13. Hum Mol Genet. 2012;21(4):934–46. Epub 2011/11/15. 10.1093/hmg/ddr523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Carvajal-Carmona LG, Cazier JB, Jones AM, Howarth K, Broderick P, Pittman A, et al. Fine-mapping of colorectal cancer susceptibility loci at 8q23.3, 16q22.1 and 19q13.11: refinement of association signals and use of in silico analysis to suggest functional variation and unexpected candidate target genes. Hum Mol Genet. 2011;20(14):2879–88. Epub 2011/05/03. 10.1093/hmg/ddr190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pittman AM, Naranjo S, Jalava SE, Twiss P, Ma Y, Olver B, et al. Allelic variation at the 8q23.3 colorectal cancer risk locus functions as a cis-acting regulator of EIF3H. PLoS genetics. 2010;6(9):e1001126 Epub 2010/09/24. 10.1371/journal.pgen.1001126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tuupanen S, Turunen M, Lehtonen R, Hallikas O, Vanharanta S, Kivioja T, et al. The common colorectal cancer predisposition SNP rs6983267 at chromosome 8q24 confers potential to enhanced Wnt signaling. Nature genetics. 2009;41(8):885–90. Epub 2009/06/30. 10.1038/ng.406 . [DOI] [PubMed] [Google Scholar]
- 28.Pittman AM, Webb E, Carvajal-Carmona L, Howarth K, Di Bernardo MC, Broderick P, et al. Refinement of the basis and impact of common 11q23.1 variation to the risk of developing colorectal cancer. Hum Mol Genet. 2008;17(23):3720–7. Epub 2008/08/30. 10.1093/hmg/ddn267 . [DOI] [PubMed] [Google Scholar]
- 29.Pittman AM, Naranjo S, Webb E, Broderick P, Lips EH, van Wezel T, et al. The colorectal cancer risk at 18q21 is caused by a novel variant altering SMAD7 expression. Genome Res. 2009;19(6):987–93. Epub 2009/04/28. 10.1101/gr.092668.109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang H, Haiman CA, Burnett T, Fortini BK, Kolonel LN, Henderson BE, et al. Fine-mapping of Genome-wide Association Study-identified Risk Loci for Colorectal Cancer in African Americans. Hum Mol Genet. 2013. Epub 2013/07/16. 10.1093/hmg/ddt337 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Whiffin N, Dobbins SE, Hosking FJ, Palles C, Tenesa A, Wang Y, et al. Deciphering the genetic architecture of low-penetrance susceptibility to colorectal cancer. Hum Mol Genet. 2013. Epub 2013/08/02. 10.1093/hmg/ddt357 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chung CC, Ciampa J, Yeager M, Jacobs KB, Berndt SI, Hayes RB, et al. Fine mapping of a region of chromosome 11q13 reveals multiple independent loci associated with risk of prostate cancer. Hum Mol Genet. 2011;20(14):2869–78. Epub 2011/05/03. 10.1093/hmg/ddr189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Genomes Project C, Abecasis GR, Auton A, Brooks LD, DePristo MA, Durbin RM, et al. An integrated map of genetic variation from 1,092 human genomes. Nature. 2012;491(7422):56–65. Epub 2012/11/07. 10.1038/nature11632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hutter CM, Chang-Claude J, Slattery ML, Pflugeisen BM, Lin Y, Duggan D, et al. Characterization of gene-environment interactions for colorectal cancer susceptibility loci. Cancer Res. 2012;72(8):2036–44. Epub 2012/03/01. 10.1158/0008-5472.CAN-11-4067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Price AL, Patterson NJ, Plenge RM, Weinblatt ME, Shadick NA, Reich D. Principal components analysis corrects for stratification in genome-wide association studies. Nature genetics. 2006;38(8):904–9. Epub 2006/07/25. 10.1038/ng1847 . [DOI] [PubMed] [Google Scholar]
- 36.Genomes Project C, Abecasis GR, Altshuler D, Auton A, Brooks LD, Durbin RM, et al. A map of human genome variation from population-scale sequencing. Nature. 2010;467(7319):1061–73. Epub 2010/10/29. 10.1038/nature09534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Howie B, Marchini J, Stephens M. Genotype imputation with thousands of genomes. G3 (Bethesda). 2011;1(6):457–70. Epub 2012/03/03. 10.1534/g3.111.001198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Browning SR, Browning BL. Rapid and accurate haplotype phasing and missing-data inference for whole-genome association studies by use of localized haplotype clustering. Am J Hum Genet. 2007;81(5):1084–97. Epub 2007/10/10. 10.1086/521987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Howie B, Fuchsberger C, Stephens M, Marchini J, Abecasis GR. Fast and accurate genotype imputation in genome-wide association studies through pre-phasing. Nature genetics. 2012;44(8):955–9. Epub 2012/07/24. 10.1038/ng.2354 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li Y, Willer CJ, Ding J, Scheet P, Abecasis GR. MaCH: using sequence and genotype data to estimate haplotypes and unobserved genotypes. Genet Epidemiol. 2010;34(8):816–34. Epub 2010/11/09. 10.1002/gepi.20533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jiao S, Hsu L, Hutter CM, Peters U. The use of imputed values in the meta-analysis of genome-wide association studies. Genet Epidemiol. 2011;35(7):597–605. Epub 2011/07/20. 10.1002/gepi.20608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cochran WG. The combination of estimates from different experiments. Biometrics 1954;10:101–29. [Google Scholar]
- 43.Pruim RJ, Welch RP, Sanna S, Teslovich TM, Chines PS, Gliedt TP, et al. LocusZoom: regional visualization of genome-wide association scan results. Bioinformatics. 2010;26(18):2336–7. Epub 2010/07/17. 10.1093/bioinformatics/btq419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, et al. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7(4):248–9. Epub 2010/04/01. 10.1038/nmeth0410-248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ward LD, Kellis M. HaploReg: a resource for exploring chromatin states, conservation, and regulatory motif alterations within sets of genetically linked variants. Nucleic acids research. 2012;40(Database issue):D930–4. Epub 2011/11/09. 10.1093/nar/gkr917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ward LD, Kellis M. HaploReg v4: systematic mining of putative causal variants, cell types, regulators and target genes for human complex traits and disease. Nucleic acids research. 2016;44(D1):D877–81. 10.1093/nar/gkv1340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kent WJ, Sugnet CW, Furey TS, Roskin KM, Pringle TH, Zahler AM, et al. The human genome browser at UCSC. Genome Res. 2002;12(6):996–1006. Epub 2002/06/05. 10.1101/gr.229102 Article published online before print in May 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Consortium EP, Birney E, Stamatoyannopoulos JA, Dutta A, Guigo R, Gingeras TR, et al. Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature. 2007;447(7146):799–816. Epub 2007/06/16. 10.1038/nature05874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Consortium EP, Bernstein BE, Birney E, Dunham I, Green ED, Gunter C, et al. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489(7414):57–74. Epub 2012/09/08. 10.1038/nature11247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bernstein BE, Stamatoyannopoulos JA, Costello JF, Ren B, Milosavljevic A, Meissner A, et al. The NIH Roadmap Epigenomics Mapping Consortium. Nat Biotechnol. 2010;28(10):1045–8. Epub 2010/10/15. 10.1038/nbt1010-1045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Visel A, Rubin EM, Pennacchio LA. Genomic views of distant-acting enhancers. Nature. 2009;461(7261):199–205. Epub 2009/09/11. 10.1038/nature08451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Visel A, Blow MJ, Li Z, Zhang T, Akiyama JA, Holt A, et al. ChIP-seq accurately predicts tissue-specific activity of enhancers. Nature. 2009;457(7231):854–8. Epub 2009/02/13. 10.1038/nature07730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Siepel A, Bejerano G, Pedersen JS, Hinrichs AS, Hou M, Rosenbloom K, et al. Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Genome Res. 2005;15(8):1034–50. Epub 2005/07/19. 10.1101/gr.3715005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Pollard KS, Hubisz MJ, Rosenbloom KR, Siepel A. Detection of nonneutral substitution rates on mammalian phylogenies. Genome Res. 2010;20(1):110–21. Epub 2009/10/28. 10.1101/gr.097857.109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lappalainen T, Sammeth M, Friedlander MR, t Hoen PA, Monlong J, Rivas MA, et al. Transcriptome and genome sequencing uncovers functional variation in humans. Nature. 2013;501(7468):506–11. 10.1038/nature12531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Westra HJ, Peters MJ, Esko T, Yaghootkar H, Schurmann C, Kettunen J, et al. Systematic identification of trans eQTLs as putative drivers of known disease associations. Nature genetics. 2013;45(10):1238–43. 10.1038/ng.2756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Montgomery SB, Sammeth M, Gutierrez-Arcelus M, Lach RP, Ingle C, Nisbett J, et al. Transcriptome genetics using second generation sequencing in a Caucasian population. Nature. 2010;464(7289):773–7. 10.1038/nature08903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gibbs JR, van der Brug MP, Hernandez DG, Traynor BJ, Nalls MA, Lai SL, et al. Abundant quantitative trait loci exist for DNA methylation and gene expression in human brain. PLoS genetics. 2010;6(5):e1000952 10.1371/journal.pgen.1000952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Schadt EE, Molony C, Chudin E, Hao K, Yang X, Lum PY, et al. Mapping the genetic architecture of gene expression in human liver. PLoS biology. 2008;6(5):e107 10.1371/journal.pbio.0060107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Stranger BE, Nica AC, Forrest MS, Dimas A, Bird CP, Beazley C, et al. Population genomics of human gene expression. Nature genetics. 2007;39(10):1217–24. 10.1038/ng2142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Reese MG, Eeckman FH, Kulp D, Haussler D. Improved splice site detection in Genie. J Comput Biol. 1997;4(3):311–23. Epub 1997/10/01. . [DOI] [PubMed] [Google Scholar]
- 62.Birnbaum RY, Clowney EJ, Agamy O, Kim MJ, Zhao J, Yamanaka T, et al. Coding exons function as tissue-specific enhancers of nearby genes. Genome Res. 2012;22(6):1059–68. Epub 2012/03/24. 10.1101/gr.133546.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hindorff LA, Sethupathy P, Junkins HA, Ramos EM, Mehta JP, Collins FS, et al. Potential etiologic and functional implications of genome-wide association loci for human diseases and traits. Proc Natl Acad Sci U S A. 2009;106(23):9362–7. Epub 2009/05/29. 10.1073/pnas.0903103106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Pomerantz MM, Ahmadiyeh N, Jia L, Herman P, Verzi MP, Doddapaneni H, et al. The 8q24 cancer risk variant rs6983267 shows long-range interaction with MYC in colorectal cancer. Nature genetics. 2009;41(8):882–4. Epub 2009/06/30. 10.1038/ng.403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Sotelo J, Esposito D, Duhagon MA, Banfield K, Mehalko J, Liao H, et al. Long-range enhancers on 8q24 regulate c-Myc. Proc Natl Acad Sci U S A. 2010;107(7):3001–5. Epub 2010/02/06. 10.1073/pnas.0906067107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Sur IK, Hallikas O, Vaharautio A, Yan J, Turunen M, Enge M, et al. Mice lacking a Myc enhancer that includes human SNP rs6983267 are resistant to intestinal tumors. Science. 2012;338(6112):1360–3. Epub 2012/11/03. 10.1126/science.1228606 . [DOI] [PubMed] [Google Scholar]
- 67.Biancolella M, B KF, Tring S, Plummer SJ, Mendoza-Fandino GA, Hartiala J, et al. Identification and characterization of functional risk variants for colorectal cancer mapping to chromosome 11q23.1. Hum Mol Genet. 2013. Epub 2013/11/22. 10.1093/hmg/ddt584 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kote-Jarai Z, Saunders EJ, Leongamornlert DA, Tymrakiewicz M, Dadaev T, Jugurnauth-Little S, et al. Fine-mapping identifies multiple prostate cancer risk loci at 5p15, one of which associates with TERT expression. Hum Mol Genet. 2013;22(12):2520–8. Epub 2013/03/29. 10.1093/hmg/ddt086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Harley IT, Kaufman KM, Langefeld CD, Harley JB, Kelly JA. Genetic susceptibility to SLE: new insights from fine mapping and genome-wide association studies. Nat Rev Genet. 2009;10(5):285–90. Epub 2009/04/02. 10.1038/nrg2571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Jaeger E, Leedham S, Lewis A, Segditsas S, Becker M, Cuadrado PR, et al. Hereditary mixed polyposis syndrome is caused by a 40-kb upstream duplication that leads to increased and ectopic expression of the BMP antagonist GREM1. Nature genetics. 2012;44(6):699–703. Epub 2012/05/09. 10.1038/ng.2263 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Squazzo SL, O'Geen H, Komashko VM, Krig SR, Jin VX, Jang SW, et al. Suz12 binds to silenced regions of the genome in a cell-type-specific manner. Genome Res. 2006;16(7):890–900. Epub 2006/06/06. 10.1101/gr.5306606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Durkin ME, Loechel F, Mattei MG, Gilpin BJ, Albrechtsen R, Wewer UM. Tissue-specific expression of the human laminin alpha5-chain, and mapping of the gene to human chromosome 20q13.2–13.3 and to distal mouse chromosome 2 near the locus for the ragged (Ra) mutation. FEBS Lett. 1997;411(2–3):296–300. Epub 1997/07/14. . [DOI] [PubMed] [Google Scholar]
- 73.Yemelyanov A, Czwornog J, Chebotaev D, Karseladze A, Kulevitch E, Yang X, et al. Tumor suppressor activity of glucocorticoid receptor in the prostate. Oncogene. 2007;26(13):1885–96. Epub 2006/10/04. 10.1038/sj.onc.1209991 . [DOI] [PubMed] [Google Scholar]
- 74.Huang L, Wang C, Rosenberg NA. The relationship between imputation error and statistical power in genetic association studies in diverse populations. Am J Hum Genet. 2009;85(5):692–8. Epub 2009/10/27. 10.1016/j.ajhg.2009.09.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Zheng J, Li Y, Abecasis GR, Scheet P. A comparison of approaches to account for uncertainty in analysis of imputed genotypes. Genet Epidemiol. 2011;35(2):102–10. Epub 2011/01/22. 10.1002/gepi.20552 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(XLSX)
Describes in detail the study population and case/control definition; genotyping and quality control; as well as functional annotation using bioinformatics. Also includes Supplementary Tables A-C and Supplementary Figures A-D.
(DOCX)
Data Availability Statement
For plco genetic data, the prostate and panscan study datasets can be accessed with appropriate approval through the dbgap online resource (http://cgems.cancer.gov/data/) accession numbers phs000207v.1p1 and phs000206.v3.p2, respectively, and the lung datasets from the dbgap website (http://www.ncbi.nlm.nih.gov/gap) through accession number phs000093 v2.p2. For the remaining gecco studies, genetic data is in the process of being uploaded to dbgap and will be available soon (accession number phs001078.v1.p1). While the data release is being finalized, the gecco coordinating center can assist with any data requests (contact: gecco@fredhutch.org).