

Abstract
The archaeal minichromosomal maintenance (MCM) helicase from Sulfolobus solfataricus (SsoMCM) is a model for understanding structural and mechanistic aspects of DNA unwinding. Although interactions of the encircled DNA strand within the central channel provide an accepted mode for translocation, interactions with the excluded strand on the exterior surface have mostly been ignored with regard to DNA unwinding. We have previously proposed an extension of the traditional steric exclusion model of unwinding to also include significant contributions with the excluded strand during unwinding, termed steric exclusion and wrapping (SEW). The SEW model hypothesizes that the displaced single strand tracks along paths on the exterior surface of hexameric helicases to protect single-stranded DNA (ssDNA) and stabilize the complex in a forward unwinding mode. Using hydrogen/deuterium exchange monitored by Fourier transform ion cyclotron resonance MS, we have probed the binding sites for ssDNA, using multiple substrates targeting both the encircled and excluded strand interactions. In each experiment, we have obtained >98.7% sequence coverage of SsoMCM from >650 peptides (5–30 residues in length) and are able to identify interacting residues on both the interior and exterior of SsoMCM. Based on identified contacts, positively charged residues within the external waist region were mutated and shown to generally lower DNA unwinding without negatively affecting the ATP hydrolysis. The combined data globally identify binding sites for ssDNA during SsoMCM unwinding as well as validating the importance of the SEW model for hexameric helicase unwinding.
Keywords: archaea, ATPases associated with diverse cellular activities (AAA), DNA helicase, DNA replication, DNA-protein interaction, HDX-MS, MCM, steric exclusion wrapping
Introduction
DNA unwinding by hexameric replicative helicases is required for DNA replication elongation, providing the single-stranded DNA (ssDNA)5 templates for leading and lagging strand synthesis. These hexameric helicase complexes form ring-like structures that preferentially encircle one of the DNA strands, providing stability in DNA binding and enhancing processivity for unwinding long stretches of DNA. In the steric exclusion model for unwinding, the motor domains within the central channel translocate along the encircled ssDNA while physically excluding the complementary ssDNA on the exterior. Although DNA unwinding by helicases can be effectively measured in gel-based and fluorescence assays, the specific molecular interactions with DNA are poorly described. Previously, we have proposed that the excluded strand is not a passive component of unwinding; rather, it will interact along specific exterior paths on the hexameric helicase surface to both stabilize the complex and promote forward unwinding (1). Although structures of hexameric helicases that include small stretches of ssDNA have been solved recently (2, 3), the interactions are constrained to central channel residues, and no molecular information is available at the duplex region or with respect to the excluded strand.
Hexameric DNA replication helicases are known to exist across the three domains of life, including viruses. Although their overall structural toroidal features are conserved, their amino acid sequences are not, and they are generally classified into three families by their unwinding polarity and specific protein folds (4, 5). Bacteria and many phages are members of the superfamily 4 helicases, which include RecA motor domains and 5′–3′ unwinding polarity. In contrast, eukaryotic viruses from superfamily 3 (i.e. E1 and SV40 Large T) and eukaryotic and archaeal superfamily 6 minichromosomal maintenance (MCM) helicases are members of the AAA+ (ATPase associated with various cellular activities) enzymes and translocate 3′–5′.
The eukaryotic heterohexameric MCM2–7 helicase interacts with and unwinds DNA as a part of a larger GINS-Cdc45-MCM2–7 (CMG) complex that also includes Cdc45 and GINS components (6, 7). Interactions with ssDNA within this complex have been noted on the exterior, specifically with the Cdc45 subunit (8). Recently, a high resolution electron microscopy structure of the yeast MCM2–7 complex shows twisted and tilted double hexamers on DNA, but the conformations of the individual hexamers are symmetrical, and contacts with DNA are constrained to the central channel (9). The archaeal replicative MCM helicase generally exists as a homohexamer that shares homology with each of the six sequence-distinctive subunits in eukarya (MCM2 to MCM7) and is essential for replication initiation and elongation (10). Archaeal MCM helicases from Sulfolobus solfataricus (SsoMCM) and Methanothermobacter thermoautotrophicus serve as simplified models for understanding mechanisms of ATP hydrolysis, DNA binding, and unwinding for more complex eukaryotic hexameric DNA replication helicases (11). Crystal structures of archaeal MCMs with the N-terminal domain (NTD) alone (12, 13) as well as nearly full-length structures with both the NTD and AAA+ domain (14, 15) have been solved and show similar symmetry and subunit arrangements. It is interesting to note that an EM structure of M. thermoautotrophicus MCMs has identified a binding site for DNA on the external surface of the helicase, possibly implicated as a prebound engaged state prior to loading and encircling onto DNA at replication origins (16).
We previously elucidated the unwinding mechanisms for the SsoMCM helicase and found specific and required interactions with the excluded ssDNA (1, 17). Specific electrostatic mutations on the exterior surface disrupted the excluded strand interactions and reduced unwinding rates. These results advanced the traditional steric exclusion unwinding model to include external surface interactions with the excluded strand and designated a new helicase unwinding model termed steric exclusion and wrapping (SEW). Although we identified specific sites of interaction with the excluded strand through mutagenesis, a more comprehensive and global description of the external binding path is needed to validate and better describe the SEW model.
Using hydrogen/deuterium exchange mass spectrometry (HDX-MS), we are able to identify specific contacts with both the encircled and excluded strands and the SsoMCM homohexamer in solution. The benefit of using HDX-MS to probe protein/DNA interactions is that it can measure H/D exchange correlated with binding interfaces in the native solution state without any perturbations (18, 19). HDX-MS detects protein backbone amide proton exchanges that are accessible to solvent and can identify regions that are protected upon ligand binding or through conformational changes (20, 21) as validated by comparison with HDX exchange observed by proton NMR (22). Hydrogen atoms on the side chains or on the N and C termini of the peptide also undergo H/D exchange. However, at these rapidly exchanging sites, incorporated deuteriums back-exchange by H2O in the mobile phase during HPLC separation. Hence, the mass increment is solely due to the deuterium exchange of the backbone amide hydrogens (23).
Ultrahigh resolution Fourier transform ion cyclotron resonance MS (24), improved instrument automation (25–27), and efficient protein digestion (28) allow for detection of specific ssDNA interactions with single subunits within a redundant MCM homohexamer. Using HDX-MS, we are able to probe interfacial residues within the SsoMCM hexamer from the monomeric x-ray structure (14) and define a more global interaction path of the excluded strand with the exterior surface, thus validating the SEW model as a bona fide unwinding mechanism.
Experimental Procedures
Materials
ATP was obtained from Invitrogen. Optikinase was purchased from U. S. Biochemical Corp. All other materials were from commercial sources and were analytical grade or better. Oligonucleotides were purchased from IDT Corp. (Coralville, IA) and gel-purified as described previously (1). [γ-32P]ATP was purchased from PerkinElmer Life Sciences. Helicase buffer is used in all assays consisting of 125 mm potassium acetate, 25 mm Tris acetate (pH 7.5), and 10 mm magnesium acetate.
SsoMCM Cloning and Purification
SsoMCM R33A, K111A, K111A/R113A, K129A, and R226A were created using a standard QuikChange protocol (Agilent, Santa Clara, CA) with KAPA HiFi DNA polymerase (KAPA Biosystems, Woburn, MA). Primer sequences included silent mutations for unique restriction site screening. Mutations were confirmed using the DNA Sequencing Facility at the University of Texas (Austin, TX). WT, the point mutants, and previously cloned K323A/R440A were purified as described (1). Briefly, autoinduced SsoMCM was heat-treated at 70 °C for 20 min, and the supernatant was applied to MonoQ, heparin, and S-200 gel filtration columns (GE Healthcare) to isolate the purified hexameric species.
Hydrogen/Deuterium Exchange
HDX was performed as described previously (29). Stock solutions of 9 μm SsoMCM were prepared in helicase buffer either alone or in the presence of 3′-tail DNA or with 3′-tail/5′-tail DNA in an equivalent molar ratio (1:1). Similar buffer conditions were applied for the preparation of the corresponding D2O buffers. HDX experiments were optimized and automated with an HTC Pal autosampler (Eksigent Technologies, Dublin, CA). 5 μl of SsoMCM was mixed with 45 μl of corresponding buffer in D2O to initiate each H/D exchange period. For the blank control, the initial dilution was made in H2O buffer. Reactions were performed in triplicate at 1–2 °C to reduce back-exchange for HDX incubation periods of 0.5, 1, 2, 4, 8, 15, 30, 60, 120, and 240 min, each followed by acid quench and proteolysis for 3 min. Each sample was quenched by rapid mixing with 25 μl of 200 mm tris(2-carboxyethyl)phosphine, 8 m urea in 1.0% formic acid and 25 μl of a 40% saturated protease type XIII (Sigma-Aldrich) solution in 1.0% formic acid (final pH ∼2.3). On-line LC electrospray ionization 14.5-tesla Fourier transform ion cyclotron resonance MS with an LTQ Velos (Thermo Fisher Scientific, San Jose, CA) front end yielded high mass resolving power (m/Δm50% > 200,000 at m/z 400) (30). There were 772 peptides for apo-SsoMCM, 722 peptides for SsoMCM 3′-tail, and 676 peptides for SsoMCM 3′-tail/5′-tail. Data were analyzed by an in-house Predator software package (26). The measured deuterium uptake and associated S.D. values were recorded for each incubation period and fitted by a maximum entropy method (MEM) (31). The average relative deuterium uptake difference (ARDD) between free and complexed protein was calculated by the following equation,
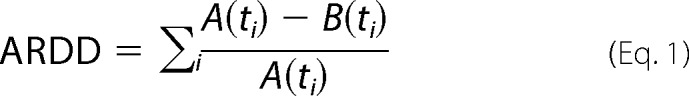 |
in which A is the deuterium uptake for sample A (apo-SsoMCM) at a specified time (ti), and B (SsoMCM/DNA) is the deuterium uptake for sample B at a specified time (ti) (32).
A focused analysis of each peptide was also performed to more accurately reveal highly sequence localized changes in solvent accessibility that can be obfuscated by the averaging of multiple sequence overlapped peptides of varying length as is done in the global analysis. This direct comparison of identical peptides between experimental conditions can often uncover localized changes that may otherwise be overlooked. Potentially poorer quality data with S.D. values >0.5 were excluded for this analysis. This procedure reduced the data to 574 peptides for each of the three experimental data sets. We then assigned values to each peptide by calculating the midpoint of the peptide (first and last amino acid number) to align the peptides semisequentially. If the assigned value was the same for two different peptides, the peptide with the lower first amino acid number was listed first. For those peptides with two or more charges, we averaged the values for each incubation period, parsing the data to 497 peptides. The three data sets were then compared at 10 incubation periods. Only those peptides that yielded values for each incubation period were retained, reducing the number of peptides to 238. The data were then plotted as relative deuterium uptake versus assigned peptide number for the butterfly plots. Differences between free and complexed protein were determined based on significant differences in absolute H/D exchange as well as exponential rate constants.
DNA Unwinding Assays
Unwinding assays were performed as described previously (1). Briefly, Optikinase was used to 32P-label the 5′-end of the duplex. SsoMCM variants (2.4 μm monomer, 400 nm hexamer) were incubated with 15 nm 5′-radiolabeled DNA for 5 min at 60 °C and initiated with 2 mm ATP. Reactions were quenched with an equal volume of glycerol quench (0.5% (w/v) SDS, 50% (v/v) glycerol, 0.1% (w/v) bromphenol blue, 100 mm EDTA, pH 8.0, and 300 nm trap ssDNA). Quenched reactions were stored on ice until the addition of 4 μl of Proteinase K 20 mg/ml (Thermo Fisher Scientific) and incubation at 37 °C for 1 h for digestion. DNA was resolved on the 16% acrylamide gels with 0.1% SDS to remove any remaining bound protein. Gels were then phosphorimaged with a Storm 820 phosphor imager (GE Healthcare) and quantified for the unwinding rate.
ATPase Assays
SsoMCM variants (2 μm) were incubated in the absence or presence of unlabeled forked DNA as described previously (33). Briefly, 30-μl reactions were incubated at 60 °C for 5 min, and ATP was added to initiate the reaction. Samples were quenched at 5, 10, and 15 min after initiation in equal volumes of 0.7 m formic acid. A total of 0.8 μl of quenched reaction was spotted on Millipore TLC PEI Cellulose F, allowed to dry, resolved in 0.6 m potassium phosphate (pH 3.5), phosphorimaged, and quantified for the ATPase rate (pmol/min).
Results
HDX-MS Analysis of SsoMCM Homohexamer
We used HDX-MS to study the SsoMCM hexamer solution conformation and the ssDNA binding locations. Digestion with protease XIII yielded >574 overlapping peptides covering 98.7% of the sequence for SsoMCM (Fig. 1, A and B). Deuterium uptake (deuteration level, percentage) was measured for three experimental conditions: 1) apo-SsoMCM, 2) SsoMCM with 3′-tail DNA, and 3) SsoMCM with 3′-tail/5′-tail fork DNA (Fig. 1, C–F). Examination of the apo-SsoMCM data reveals significant correlation with solvent-exposed regions within a previously developed hexameric model (6-fold symmetry) of the monomeric SsoMCM crystal structure (Protein Data Bank code 3F9V) (Fig. 2) (14). There is one other nearly full-length crystal structure of a hexameric archaeal MCM that consists of a hybrid of S. solfataricus (residues 7–269) and Pyrococcus furiosus (residues 257–361 and 729–965) (Protein Data bank code 4POG) (15). Although the two structures have similar folds and interactions between subunits, the A-subdomain in the hybrid structure is displaced outward compared with 3F9V, resulting in even more solvent-exposed surface between the A-subdomain and the C-subdomain as well as between subunits, which is less consistent with our HDX data. Therefore, we shall primarily correlate our HDX-MS data with the hexameric model, 3F9V.
FIGURE 1.

Protein sequence coverage, experimental schematic, and HDX kinetic data. A, SsoMCM primary sequence, motifs, and domains. A, A-subdomain; B, B-subdomain; C, C-subdomain; ZBD, zinc-binding domain; N-C, N-terminal to C-terminal linker; WA, Walker A; WB, Walker B; S1, sensor 1; RF, arginine finger; S2, sensor 2; WH, winged helix. B, protease XIII digestion of apo-SsoMCM (576 peptides, 5–30 residues in length) was characterized, providing 98.7% sequence coverage. C, hexameric model and monomeric structure of SsoMCM (Protein Data Bank code 3F9V) highlighting the domains and motifs. D, schematic of the three experimental conditions for the HDX-MS experiments comparing MCM alone (SsoMCM Apo) (○), MCM and 3′-tail (□), or MCM and 3′-tail/5′-tail (♢) DNA substrates. E, ARDD (free minus bound) averaged over all HDX incubation periods. F, heat maps for SsoMCM under the three conditions showing the deuterium uptake level (as a percentage) calculated by ARDD.
FIGURE 2.

SsoMCM apo-hexamer HDX kinetic data. A, HDX kinetic data color-coded by deuteration level (percentage) onto the primary sequence after a 120-s incubation in D2O. The deuteration level was also mapped on the SsoMCM monomeric structure (Protein Data Bank code 3F9V) (B), a cutaway of the hexamer showing the interior channel (C), and the hexameric model showing the external surface (D). Protected regions i, ii, and iii are highlighted by pink circles indicating solvent-exposed patches that have low deuterium uptake levels.
Analysis of the relative deuterium uptake for the apo-structure alone and binning the values at each residue show regions (i, ii, and iii) that are solvent-exposed in the hexamer model, with low deuterium uptake (Fig. 2, B–D). We compared the HDX data with both hydrophobic and electrostatic surfaces for regions i–iii (Fig. 3). Region i is consistent with a hydrophobic patch on the N-terminal domain. This apparent inconsistency can be explained as the segment that interacts with the first 1–6 residues that are absent from the x-ray structure.
FIGURE 3.

Deuterium uptake, hydrophobic, and electrostatic surfaces for SsoMCM. The relative deuterium uptake values from the HDX experiments along with the calculated hydrophobicity and electrostatic values were mapped onto the structure of SsoMCM (Protein Data Bank code 3F9V) for comparison of regions i, ii, and iii.
Region ii exists in a combined hydrophobic and electrostatic pocket within the interface between subunits (residues 108–115 with 219–226). Our HDX data seem to indicate a tighter compaction between subunits in this area than represented in the model. Given that the hexameric structure for SsoMCM is modeled based on 6-fold symmetry from a monomeric crystal structure (14), precise intersubunit contacts containing specific hydrophobic patches may be poorly represented by the model. Analysis of a helical filamentous SsoMCM structure (34) shows a tighter compaction in this area (region ii) between subunits (data not shown), but it is arranged in a much larger oligomeric structure and shifted slightly between subunits, which is not consistent with our hexameric solution state. It is possible that the solution state of hexameric SsoMCM adopts an intermediary structure between 6-fold symmetry and helical filament that protects region ii from significant deuterium exchange.
The third apparent surface-exposed area with minimal deuterium uptake (region iii) is in a hydrophobic patch in the C-terminal domain and consists of residues 328–334, 472–473, and 562–568. Region iii is in a hydrophobic pocket at the base of the structure (Fig. 3) composed of the α6–α8 helix region (14). Because this is the only other region that shows significant deviation between the hexameric model and our HDX data, we propose that the difference is due to the flexible C-terminal helix-turn-helix (HTH) domain interacting at this site. The HTH domain is absent from all available x-ray structural data and is typically removed for crystallization trials because of its flexibility; however, a recent NMR structure of the HTH domain alone has been determined and docked onto the nearly full-length x-ray structure at the α6–α8 helices (35). Unlike other data supporting this hypothesis, our HDX experiments include all residues of SsoMCM and can therefore directly identify protected regions unobserved in other structurally modified experiments. Our results most likely indicate that the HTH domain is indeed binding directly to the α6–α8 helix region or otherwise causing some conformational rearrangement at that site, transmitting DNA binding information through to the ATPase site.
Deuterium Uptake Kinetic Differences: SsoMCM Bound to DNA Substrates
To detect possible DNA binding surfaces, we performed HDX-MS experiments for SsoMCM bound to 3′-tail DNA or fork DNA with a 3′-tail/5′-tail. The difference in deuteration level for each DNA binding experiment relative to apo-SsoMCM is averaged over time for every residue (Equation 1). Even if the ssDNA binds the SsoMCM surface stably, the experimental HDX data are difficult to interpret because each subunit is identical. Thus, very small differences contributed from DNA-bound subunits may be lost in averaging due to contributions from the unbound and unperturbed subunits. In fact, using ARDD, we were able to detect significant perturbations only for the 3′-tail/5′-tail substrate relative to the apo-SsoMCM experiment. Instead, for the 3′-tail substrate alone, we analyzed relative fractional exchange for each peptide along with the corresponding S.E. values (36). Regions that do not absolutely mirror one another have differences in relative deuterium uptake or the H/D exchange rate constant indicative of either structural changes or DNA binding. “Relative fractional exchange” describes the deuterium uptake for each peptide segment. For example, if 5-deuterium incorporation is observed for a peptide with 10 exchangeable amide hydrogens, the “relative fractional exchange” for that peptide is 0.5. ARDD describes the difference in deuterium incorporation under two distinctive conditions. For example, if 5-deuterium uptake is observed for a peptide from the apoprotein, and that same peptide incorporates 4 deuteriums after binding of DNA, ARDD for that peptide is calculated to be −20%. The relative fractional exchange butterfly plots are able to identify HDX differences within identical peptides between experimental conditions for a more focused analysis.
Butterfly plots showing the relative fractional exchange for apo- versus 3′-tail DNA identify altered deuterium uptakes for specific peptides (Table 1), mapped onto the crystal structure (Fig. 4). Peptide 22 provides an example control peptide where no significant deuterium uptake differences are noted between conditions consistent with no binding perturbation at that site (Fig. 4B). The regions of reduced deuteration (shades of blue) exhibit putative locations in which the 3′-tail binds SsoMCM. Surprisingly, few of these locations are in the interior channel of the SsoMCM hexamer. Peptide 56 (residues 183–194) (Fig. 4C) makes some contacts on the interior, and residues within this region have been identified previously as part of an MCM single-stranded DNA binding motif (MSSB) (2). Another peptide 153 (residues 425–440) (Fig. 4D) spans residues associated with a C-terminal hairpin within the pre-sensor 1 (PS1) region that is involved in DNA binding (33). The remaining peptides map to external surface locations on the SsoMCM hexamer (Fig. 4, F–H). It is known that SsoMCM binds and unwinds 3′-tail DNA with a slightly reduced affinity and reduced rate compared with fork DNA (1). The detection of deuteration changes on the exterior surface of SsoMCM could highlight an initial engaged DNA binding state for the helicase that is a precursor to loading. External regions of reduced deuteration occur at the extreme C terminus (residues 590–599, α8-helix) near where the ssDNA-dsDNA junction is proposed to bind (33) and where the HTH domain is proposed to interact (35). The HTH domain itself shows no significant changes upon DNA binding. Other regions of reduced deuteration include the α/β-α linker region primarily within the α-region of the C-subdomain (residues 522–534) and up through the waist, which is clearly on the exterior surface. Both of these regions have not been well characterized through mutagenesis, but it is possible that the α/β-α linker and/or the α-region are altered upon DNA binding, possibly through sensing DNA from the HTH domain. Alternatively, direct binding of the 3′-tail to these external regions is an intriguing conformational predecessor to a fully loaded MCM hexamer on DNA.
TABLE 1.
Peptides with significant deuteration differences
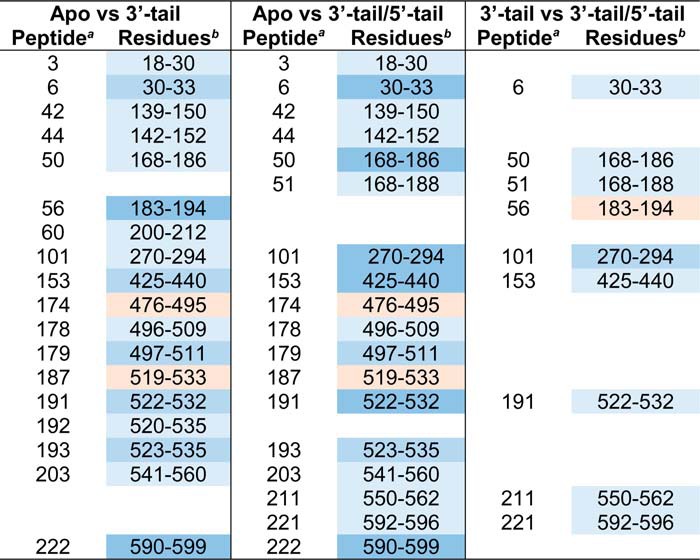
FIGURE 4.

HDX of apo-SsoMCM versus SsoMCM with 3′-tail DNA substrate. A, butterfly plot of SsoMCM (○, positive values) and SsoMCM and 3′-tail DNA (□, negative values) relative fractional exchange versus peptide number. *, peptides with significant differences. B–E, deuterium incorporation during the H/D exchange period for four representative peptides, including one with no significant change (B) and three others (C–E) with significant differences between the two conditions (○, apo, blue; □, 3′-tail, pink). Time course deuterium incorporation levels were generated by an MEM fitting method as described under “Experimental Procedures.” Peptide regions with significant deuterium uptake differences are mapped on the surface of SsoMCM, highlighting monomer (F), the interior channel (cutaway of top two subunits) (G), and the exterior surface (H).
Next, we compared apo- versus 3′-tail/5′-tail (fork) DNA and again identified peptides with altered deuterium uptake (Table 1 and Fig. 5). Generally, these regions were identical to those from the 3′-tail only population but were generally enhanced, allowing for easier identification, probably representing a more stably bound complex. Specifically, perturbation at peptide 153 was enhanced with the 3′-tail/5′-tail DNA, which includes residue Arg440, which we previously identified as a critical residue for exterior 5′-tail binding (1), as well as most of the PS1-hp (residues 424–439), which has also been implicated in DNA binding within the central channel (14, 33). The PS1-hp is generally located within the interior channel and may become more engaged with the encircled strand once MCM is externally stabilized by a fork substrate. In addition, the regions around peptide 211 (residues 550–562) (Fig. 5E) and peptide 222 (residues 590–599), which encompass the α/β-α linker and α-region, are enhanced. These areas seem to define an external ssDNA binding path from the DNA duplex region, through the CTD, and up to the waist of the SsoMCM hexamer. A comparison of SsoMCM bound to the 3′-tail or the 3′-tail/5-tail (Fig. 6) generally confirms the binding locations determined in the previous two plots and better highlights specific interactions on the exterior surface.
FIGURE 5.

HDX of apo-SsoMCM versus SsoMCM with 3′-tail/5′-tail forked DNA substrate. A, butterfly plot for SsoMCM (○, positive values) and SsoMCM and 3′-tail/5′-tail DNA (♢, negative values) relative fractional exchange versus peptide number. *, peptides with significant differences. B–E, deuterium incorporation during the H/D exchange period for four representative peptides, including one with no significant change (B) and three others (C–E) with significant differences between the two conditions (○, apo, blue; ♢, 3′-tail/5′-tail, green). Time course deuterium incorporation levels were generated by an MEM fitting method as described under “Experimental Procedures.” F–H, peptide regions with significant deuterium uptake differences are mapped on the surface of SsoMCM, highlighting monomer (F), the interior channel (cutaway of top two subunits) (G), and the exterior surface (H).
FIGURE 6.

HDX of SsoMCM with 3′-tail DNA versus SsoMCM with 3′-tail/5′-tail forked DNA substrate. A, butterfly plot for SsoMCM and 3′-tail DNA (□, positive values) and SsoMCM and 3′-tail/5′-tail DNA (♢, negative values) relative fractional exchange versus peptide number. *, peptides with significant differences. B–E, deuterium incorporation during the H/D exchange period for four representative peptides, including one with no significant change (B) and three others (C–E) with significant differences between the two conditions (□, 3′-tail, pink; ♢, 3′-tail/5′-tail, green). Time course deuterium incorporation levels were generated by an MEM fitting method as described under “Experimental Procedures.” Peptide regions with significant deuterium uptake differences are mapped on the surface of SsoMCM, highlighting monomer (F), the interior channel (cutaway of top two subunits) (G), and the exterior surface (H). Error bars, S.E.
Analysis of the cumulative HDX data between apo- and the 3′-tail/5′-tail substrate that averages all peptides based on the ARDD method (Equation 1) is also generally consistent with this more selective analysis, but the degree of change between experiments is smaller due to averaging. Here, similar protected regions were identified, including residues within the α/β-α linker and α-region (Fig. 7F). This method also found a region of decreased ARDD (residues 224–250) corresponding to the N-terminal DNA-binding hairpin (NHP) (12, 33). It is interesting to note that the amino acids 423–440 are associated with increased average deuteration when DNA is present over that of the apo-structure. This result is opposite to what was identified with the butterfly plots, in which peptide 153 (containing residues 425–440) shows decreased deuteration. It may be that DNA binding to one or two of the six subunits has a stabilizing effect on either the exterior surface or through the interior PS1 motif, whereas on average, the other four or five unbound subunits are more dynamic. Another possibility is that the peptides cover slightly different regions in the two analysis methods, where individual peptides are compared with averaging of multiple peptides across the same region. Nevertheless, the cumulative data suggest a longitudinal external binding path for the 5′-tail across the C-terminal domain and into the waist of SsoMCM.
FIGURE 7.

ARDD of WT SsoMCM versus K323A/R440A bound to DNA. A–E, representative deuterium uptake profiles for SsoMCM (K323A/R440A) peptides with significant differences between apo- and 3′-tail DNA substrate. Time course deuterium incorporation levels were generated by an MEM fitting method, as described under “Experimental Procedures.” F and G, ARDD between apo-WT SsoMCM (F) and K323A/R440A (G) and 3′-tail DNA bound and mapped onto the surface. Red residues indicate the positions of Lys323 and Arg440. A putative external ssDNA binding path is altered between WT and K323A/R440A, as indicated in yellow. Error bars, S.E.
External Surface Electrostatic Mutations Alter Deuterium Uptake Kinetics for MCM Binding to Forked DNA
Previously, we have shown that electrostatic mutations (K323A/R440A) in the waist region of SsoMCM alter the dynamics and interactions with the excluded strand and decrease its unwinding ability (1). Although HDX-MS experiments with fork DNA and K323A/R440A were attempted multiple times, unexplained precipitation during the experiment resulted in an incomplete time course for HDX kinetics. Instead, we compared the ARDD for SsoMCM (K323A/R440A) (apo- versus 3′-tail DNA) (Fig. 7, A–E) with that above for WT SsoMCM. There were obvious changes in the external binding sites for DNA for SsoMCM (K323A/R440A), as indicated by the presence of additional peptides with reduced ARDD (109–125 and 571–578) relative to WT (Fig. 7, F versus G). In addition, peptides containing residues 18–30 and 501–509 shown above to have reduced ARDD upon WT binding to DNA are unchanged when SsoMCM (K323A/R440A) is bound to DNA. This altered HDX profile for the mutant SsoMCM (K323A/R440A) suggests that the DNA binding path on the external surface has been altered within the C-terminal domain and is absent in the N-terminal domain (Fig. 7G).
Further SsoMCM External Mutations Alter DNA Unwinding Ability
Based on the external DNA interactions that we have identified by HDX-MS, we chose to test further electrostatic surface waist SsoMCM mutations (K111A/R113A, K129A, and R226A) on DNA unwinding and ATPase activities. According to the SsoMCM hexamer model and the HDX data, each of the mutation positions has a slightly different environment within the same general location (Fig. 8A). Residues Lys111 and Arg113 encompass a positively charged area within the waist, adjacent to Lys129 and slightly more toward the N-terminal domain than Lys323 and Arg440 identified previously to disrupt unwinding when mutated (1). Lys111 is not conserved throughout the MCM family, whereas Arg113 is conserved (Fig. 8C). Lys129 is a nonconserved positively charged residue (adjacent to Arg226), which is not as solvent-exposed as the others and is used as a control mutation. Arg226 is a conserved solvent-exposed residue identified from the ARDD analysis which is protected from deuterium exchange upon DNA binding.
FIGURE 8.
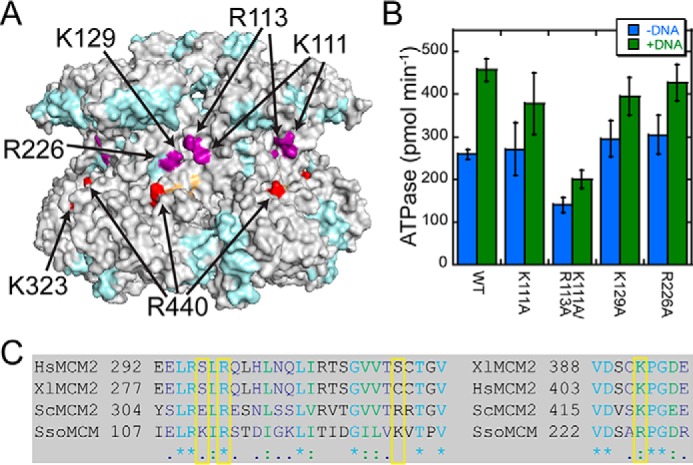
Exterior waist mutants of SsoMCM. A, position of alanine waist mutants (purple, this study; red, K323A/R440A) mapped onto the ARDD map for apo- versus 3′-tail/5′-tail DNA. B, plot of the ATPase rates performed in the absence (blue) and presence (green) of fork DNA. Error bars, S.E. C, conservation of waist mutations (boxed yellow) from S. solfataricus (Sso), Saccharomyces cerevisiae (Sc), Xenopus laevis (Xl), and Homo sapiens (Hs).
The ATPase activity for all of the mutants is stimulated in the presence of DNA (∼1.4-fold) similar to wild type (1.8-fold) (Fig. 8B and Table 2) and consistent with previously published data showing typical DNA-dependent ATPase stimulation of 1.2–2.0-fold (14, 33, 37–39). The double mutant (K111A/R113A) has an overall reduced ATPase rate (0.5–0.6) compared with WT and the other mutants. This decrease in ATP hydrolysis seems most likely the result of mutating both Lys111 and Arg113, because K111A has normal ATPase activity. However, the stimulation in the ATPase rate with DNA for K111A/R113A suggests that binding to DNA is not perturbed.
TABLE 2.
SsoMCM kinetic parameters; ATPase and unwinding
| SsoMCM | Unwindinga | ATPaseb |
Oligomeric statec | |
|---|---|---|---|---|
| Without DNA | With DNA | |||
| s−1 | pmol min−1 | |||
| WT | 1.18 ± 0.06 | 259 ± 11 | 456 ± 27 | Hexamer |
| K323A/R440Ad | 0.14 ± 0.05 | NDe | NDe | Hexamer |
| K111A | NDe | 271 ± 61 | 377 ± 72 | Hexamer |
| K111A/R113A | 0.11 ± 0.07 | 140 ± 17 | 200 ± 21 | Hexamer |
| K129A | 1.08 ± 0.16 | 295 ± 42 | 395 ± 44 | Hexamer |
| R226A | 0.54 ± 0.05 | 305 ± 46 | 426 ± 43 | Hexamer |
a Measured as fraction unwound per second.
b MCM concentration is 2 μm.
c As determined by gel filtration.
d From Ref. 1.
e ND, not determined.
Although the DNA-dependent ATPase stimulation observed for the mutants is largely unperturbed, the unwinding ability of the mutants is perturbed. DNA unwinding rates of both mutants containing conserved residues, Arg113 and Arg226, are significantly reduced relative to wild type (Fig. 9 and Table 2). K111A/R113A had barely detectable unwinding, similar to the K323A/R440A mutant (1), and R226A had a 2-fold decrease in the unwinding rate relative to wild type. The almost complete loss in DNA unwinding for K111A/R113A can only be partially explained by the modestly decreased ATPase rate and is more likely the result of a loss in exterior DNA binding in combination with reduced ATPase activity. The results for K111A/R113A are very similar to those for K323A/R440A, shown previously to disrupt exterior interactions and eliminate unwinding (1). The control nonconserved K129A mutation has unwinding activity indistinguishable from that of wild type. Therefore, mutation of conserved positively charged surface residues identified within the waist of SsoMCM has severe effects on external surface DNA interactions that translate to reduced unwinding activity.
FIGURE 9.
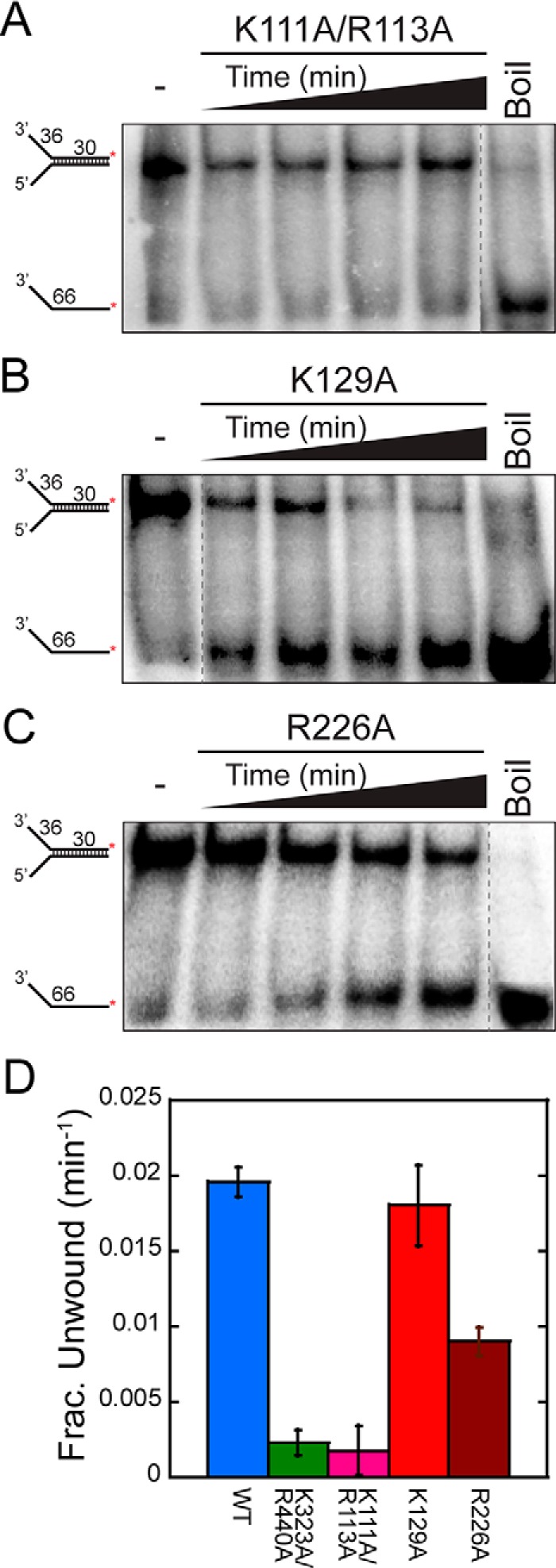
DNA unwinding activity for exterior waist mutants. Representative DNA unwinding time courses for SsoMCM mutants K111A/R113A (A), K129A (B), and R226A (C) (at 2.4 μm) from 10 to 30 min. Shown is fork DNA with a 30-base duplex and 32P-labeled at the 5′-end of the translocating strand. D, plots of the unwinding rates for the waist mutants (K323A/R440A (green), K111A/R113A (pink), K129A (red), and R226A (russet)) compared with wild type (blue) quantified as the average from at least three separate experiments. Error bars, S.E. from at least three unwinding experiments.
Discussion
Using a global experimental approach, we can now validate the SEW model of unwinding for the hexameric SsoMCM DNA replication helicase. High resolution HDX-MS monitored the deuterium exchange kinetic differences, comparing unbound and bound SsoMCM with two different DNA substrates in solution. It is interesting to note that the HDX profiles were generally similar for a 3′-tail only substrate and the 3′-tail/5′-tail substrate, indicating both internal and external binding paths. The 3′-tail DNA was detected primarily on the exterior surface along the proposed external binding path, demonstrating a probable intermediate state of DNA engagement or a nonproductive binding mode. However, the forked substrate (3′-tail/5′-tail) generally reduced the deuterium exchange rate further relative to the 3′-tail only DNA, strengthening the identification of both the internal channel and external surface ssDNA binding paths and stabilizing the forked DNA-SsoMCM complex.
The symmetrical SsoMCM hexameric model (14) as a whole is a good representation of the oligomeric structure detected by HDX-MS. The HDX data were able to determine the probable binding surfaces of missing residues and domains from the x-ray structure. Discrepancies between our HDX data and the x-ray structure are also noted in the A-subdomains, which are known to adopt different conformations depending on the species and nucleotide bound state, and mutations within that site can modulate its position (15, 16, 34, 40). This protruding A-subdomain may be one area where significant dynamics or conformational change occurs in solution to enforce contacts between subunits or engage DNA binding along the outside surface.
Within the interior channel, two β-hairpins (NHP and PS1-hp) and another single-stranded binding element (MSSB) showed changes in deuteration kinetics upon DNA binding. The NHP and the MSSB motif are located in the NTD, whereas the PS1-hp is located in the CTD. All three motifs have been shown previously to be important for DNA binding and unwinding (2, 33). The other peptides containing the PS1-hp traverse from the interior to the exterior surface, making it difficult to conclude exactly which region shows the most change. Both residues Lys430 (PS1-hp, internal DNA binding) (33) and Arg440 (external ssDNA interaction) (1) are contained within this peptide. Lys430 has been shown previously to be important for DNA binding and unwinding, whereas Arg440 is implicated as an external ssDNA binding site. Decreased deuterium exchange is also noted for Lys194 within the MSSB motif, especially for the 3′-tail substrate. It is interesting to note that perturbation at this site is not seen with the fork 3′-tail/5′-tail DNA substrate. Binding of fork DNA may more precisely position the duplex region at the CTD and prevent nonspecific binding of ssDNA internally at the NTD in the opposite orientation. Although the length of ssDNA (30 bases) is long enough to traverse the central channel, the exact path is unknown and might include spiraling or interactions that preclude binding to the NTD MSSB when in the preferred orientation. More than likely, the six identical NHPs within the central channel exchange binding of the encircled strand, making detection of specific binding difficult using this analytical method. Moreover, HDX data mapped onto the apo-hexameric structure of SsoMCM show that solvent exchange within the central channel is as rapid as on the external surface. Therefore, although we expected to find significant protection within the central channel upon DNA binding, in hindsight, the core is large enough to accommodate significant solvent as well as facile exchange and binding of the encircled strand with multiple subunits within close proximity.
Another possible explanation for detecting primarily exterior binding surfaces is that increased temperature is required to induce conformational changes within the hexamer or promote DNA encircling through thermal energy for this thermophilic SsoMCM. Because our HDX experiments were performed near 0 °C, thermally induced DNA binding would not be detected. DNA unwinding experiments for SsoMCM are generally performed at 60 °C and with ATP, at which encircling can be induced. However, DNA binding experiments, including EMSA and anisotropy, performed at room temperature show no significant differences at higher temperatures (40–60 °C).6 Of course, binding experiments only quantify total binding and cannot differentiate between DNA encircling and external surface binding.
Except for polarity, there is no molecular difference in 3′-tail DNA versus 5′-tail DNA. An external interaction will be isolated from near neighbor interactions with other subunits based on atomic distance. On the contrary, binding of the 3′-tail within the interior channel places it in close proximity to at least six identical DNA binding hairpins (33) that may easily exchange contact with the 3′-tail, blurring deuterium uptake detection. Binding to fork DNA would properly position the SsoMCM helicase with optimal polarity and exterior stabilizing interactions.
Although it is always possible that unknown allosteric conformational changes in the hexamer account for the differential HDX profiles measured when DNA is bound, there are at least three arguments against this. First, previous EM structures show no significant conformational changes when DNA is included except for overall stabilization of the hexamer (16, 41). Second, no nucleotide was used in these HDX experiments to prevent conformational changes leading to unwinding. This limitation may also account for little detected binding within the central channel. It may be that the observed protection pattern for ssDNA will change when the helicases enters a translocation competent state; however, that would be extremely difficult to interpret using HDX-MS. Finally, all significant HDX changes occur on the surface, and no significant interfacial changes between subunits are noted.
The exterior binding site of SsoMCM for ssDNA has been shown previously to include at least basic residues Lys323 and Arg440 (1); however, it is likely that more residues are involved in DNA binding, both along the waist and along the longitudinal exterior surface, as indicated by the HDX data. It is interesting to note that HDX experiments performed on SsoMCM (K323A/R440A) show a change in the external DNA binding path from the CTD and up through the waist. Moreover, protection seen on the NTD with WT SsoMCM disappears when K323A/R440A is used. This change in the binding path, as well as reduced contact in the NTD, is consistent with the changes in single molecule FRET ssDNA interactions seen for this mutant (1). It also suggests that multiple contacts exist along the exterior length of SsoMCM, providing significant binding stability and contributing substantially to the mechanism of unwinding.
Additional conserved positively charged residues Arg113 and Arg226 within the waist region have now also been identified as not only being protected from deuterium exchange (in the case of Arg226) but also shown to be essential for efficient DNA unwinding activity. Because both of these residues reside within the A-subdomain of SsoMCM, external interactions with ssDNA are now validated up through the CTD and into the NTD. Arg113 and Arg226 (in the NTD) combined with Lys323 and Arg440 (in the CTD) now provide unequivocal functional evidence for excluded strand interactions with the large waist region of the hexamer. The exact path and specific regions identified in the HDX ARDD method from the CTD up to the waist region still need to be validated biochemically. For example, further external surface mutations within the N-C linker (residues 270–294) and the α/β-α linker (residues 498–515 and 522–533) show reduced HDX in the presence of DNA. Presumably, a collective external path of moderate electrostatic charge from the extreme CTD, where dsDNA is separated, up through the waist and into the NTD will be required to maintain strand separation and promote the forward action of the helicase during unwinding.
Since the SEW model was first proposed for SsoMCM (1), external excluded strand contacts with DNA have been revealed for other helicases and shown to be important for unwinding. Even monomeric superfamily 1b helicase, Dda, and superfamily 2 helicase, HCV NS3, make contacts with the excluded strand at the fork and during unwinding (42, 43). Of course, other well studied recombination helicase nucleases, RecBCD and AddAB, have been known to interact and translocate on both strands with opposite polarity to separate dsDNA, digest ssDNA, and identify recombination hot spots (44–46). In addition to archaeal MCM, there are other hexameric helicases for which contact with the excluded strand provides some stability to unwinding and replication at the fork, including T7 gp4 (47), Escherichia coli DnaB (48), and within the eukaryotic CMG complex (8, 49).
It is interesting that this external mechanistic contact seems to be shared from monomeric to hexameric helicases across all forms of life but surprisingly has not been thoroughly characterized previously. Instead, a sole focus on contacts within the interior channel or with the translocating strand has limited our understanding of the complete DNA unwinding mechanism. In fact, the work presented here suggests that external surface contacts are probably very important to the mechanism, as evidenced by nearly complete loss of unwinding activity with mutation of certain external surface residues, while imparting essentially no allosteric change to the inner channel solvent accessibility or ATPase activity. Recently, a new model of unwinding has been proposed for eukaryotic CMG that includes a “pumpjack” motion that involves linear translocation along DNA and a large ATPase-induced conformational change that propels the complex forward (50). In this model, exterior interactions with the excluded lagging strand are proposed to stabilize forward unwinding steps consistent with our SEW model.
The external DNA contacts identified for the SEW unwinding model may also contribute during the helicase loading process. The mechanism for loading of hexameric helicase onto and encircling of DNA is still unknown, but it most likely requires origin-binding proteins and helicase accessory proteins to distort the origin of replication and pry open the hexameric helicase (51–53). Maintaining contact with the excluded strand while encircling the translocating strand would be more effective in sustaining an open dsDNA bubble for loading helicases. Additional work will be required to determine whether the external interactions detected here are also utilized for loading and activation of archaeal MCM and other hexameric replication helicases or whether mutations at these sites reduce loading at origins. The diversity and length of contacts on the exterior surface of hexameric helicases with the excluded strand may also be an opportunistic target for inhibition of loading and unwinding across domains.
Author Contributions
B. W. G. and M. A. T. designed the experiments. B. W. G., K. L. D., and D. O. performed cloning and prepared the proteins. N. L. Y., A. G. M., and M. A. T. designed the HDX-MS experiments, which were performed by Y. T. C. T. T. performed the ATPase experiments. K. L. D. and D. O. performed the unwinding experiments. M. A. T., B. W. G., and N. L. Y. wrote the manuscript. All authors reviewed the results and approved the final version of the manuscript.
Acknowledgments
HDX experiments were performed at the National High Magnetic Field Laboratory (Tallahassee, FL). We thank Sean Carney for helpful conversations. We thank the Molecular Bioscience Core at Baylor University for access to equipment.
This work was supported by American Cancer Society Research Scholar Grant RSG-11-049-01-DMC (to M. A. T.) and National Science Foundation Division of Materials Research Grant DMR-11-57490 (to A. G. M.). The authors declare that they have no conflicts of interest with the contents of this article.
M. A. Trakselis, unpublished observations.
- ssDNA
- single-stranded DNA
- MCM
- minichromosomal maintenance
- SsoMCM
- S. solfataricus MCM
- MSSB
- MCM single-stranded binding motif
- NHP
- N-terminal hairpin
- NTD
- N-terminal domain
- CTD
- C-terminal domain
- SEW
- steric exclusion and wrapping
- HDX
- hydrogen-deuterium exchange
- H/D
- hydrogen/deuterium
- MEM
- maximum entropy method
- ARDD
- average relative deuterium uptake difference
- HTH
- helix-turn-helix
- PS1
- pre-sensor 1
- CMG
- GINS-Cdc45-MCM2–7.
References
- 1.Graham B. W., Schauer G. D., Leuba S. H., and Trakselis M. A. (2011) Steric exclusion and wrapping of the excluded DNA strand occurs along discrete external binding paths during MCM helicase unwinding. Nucleic Acids Res. 39, 6585–6595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Froelich C. A., Kang S., Epling L. B., Bell S. P., and Enemark E. J. (2014) A conserved MCM single-stranded DNA binding element is essential for replication initiation. Elife 3, e01993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Enemark E. J., and Joshua-Tor L. (2006) Mechanism of DNA translocation in a replicative hexameric helicase. Nature 442, 270–275 [DOI] [PubMed] [Google Scholar]
- 4.Berger J. M. (2008) SnapShot: Nucleic acid helicases and translocases. Cell 134, 888–888.e1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Iyer L. M., Leipe D. D., Koonin E. V., and Aravind L. (2004) Evolutionary history and higher order classification of AAA+ ATPases. J. Struct. Biol. 146, 11–31 [DOI] [PubMed] [Google Scholar]
- 6.Moyer S. E., Lewis P. W., and Botchan M. R. (2006) Isolation of the Cdc45/Mcm2–7/GINS (CMG) complex, a candidate for the eukaryotic DNA replication fork helicase. Proc. Natl. Acad. Sci. U.S.A. 103, 10236–10241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Petojevic T., Pesavento J. J., Costa A., Liang J., Wang Z., Berger J. M., and Botchan M. R. (2015) Cdc45 (cell division cycle protein 45) guards the gate of the eukaryote replisome helicase stabilizing leading strand engagement. Proc. Natl. Acad. Sci. U.S.A. 112, E249–E258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Costa A., Renault L., Swuec P., Petojevic T., Pesavento J. J., Ilves I., MacLellan-Gibson K., Fleck R. A., Botchan M. R., and Berger J. M. (2014) DNA binding polarity, dimerization, and ATPase ring remodeling in the CMG helicase of the eukaryotic replisome. Elife 3, e03273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li N., Zhai Y., Zhang Y., Li W., Yang M., Lei J., Tye B. K., and Gao N. (2015) Structure of the eukaryotic MCM complex at 3.8 Å. Nature 524, 186–191 [DOI] [PubMed] [Google Scholar]
- 10.Barry E. R., and Bell S. D. (2006) DNA replication in the archaea. Microbiol. Mol. Biol. Rev. 70, 876–887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sakakibara N., Kelman L. M., and Kelman Z. (2009) Unwinding the structure and function of the archaeal MCM helicase. Mol. Microbiol. 72, 286–296 [DOI] [PubMed] [Google Scholar]
- 12.Fletcher R. J., Bishop B. E., Leon R. P., Sclafani R. A., Ogata C. M., and Chen X. S. (2003) The structure and function of MCM from archaeal M. thermoautotrophicum. Nat. Struct. Biol. 10, 160–167 [DOI] [PubMed] [Google Scholar]
- 13.Liu W., Pucci B., Rossi M., Pisani F. M., and Ladenstein R. (2008) Structural analysis of the Sulfolobus solfataricus MCM protein N-terminal domain. Nucleic Acids Res. 36, 3235–3243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Brewster A. S., Wang G., Yu X., Greenleaf W. B., Carazo J. M., Tjajadi M., Klein M. G., and Chen X. S. (2008) Crystal structure of a near-full-length archaeal MCM: functional insights for an AAA+ hexameric helicase. Proc. Natl. Acad. Sci. U.S.A. 105, 20191–20196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Miller J. M., Arachea B. T., Epling L. B., and Enemark E. J. (2014) Analysis of the crystal structure of an active MCM hexamer. Elife 3, e03433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Costa A., van Duinen G., Medagli B., Chong J., Sakakibara N., Kelman Z., Nair S. K., Patwardhan A., and Onesti S. (2008) Cryo-electron microscopy reveals a novel DNA-binding site on the MCM helicase. EMBO J. 27, 2250–2258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rothenberg E., Trakselis M. A., Bell S. D., and Ha T. (2007) MCM forked substrate specificity involves dynamic interaction with the 5′-tail. J. Biol. Chem. 282, 34229–34234 [DOI] [PubMed] [Google Scholar]
- 18.L ísal J., Kainov D. E., Lam T. T., Emmett M. R., Wei H., Gottlieb P., Marshall A. G., and Tuma R. (2006) Interaction of packaging motor with the polymerase complex of dsRNA bacteriophage. Virology 351, 73–79 [DOI] [PubMed] [Google Scholar]
- 19.Nevin P., Engen J. R., and Beuning P. J. (2015) Steric gate residues of Y-family DNA polymerases DinB and pol κ are crucial for dNTP-induced conformational change. DNA Repair 29, 65–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang Z., and Smith D. L. (1993) Determination of amide hydrogen exchange by mass spectrometry: a new tool for protein structure elucidation. Protein Sci. 2, 522–531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pirrone G. F., Iacob R. E., and Engen J. R. (2015) Applications of hydrogen/deuterium exchange MS from 2012 to 2014. Anal. Chem. 87, 99–118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang Z., Li W., Li M., Logan T. M., Guan S., and Marshall A. G. (1997) Higher-order structure and dynamics of FK506-binding protein probed by backbone amide hydrogen/deuterium exchange and electrospray ionization Fourier transform ion cyclotron resonance mass spectrometry. in Techniques in Protein Chemistry (Marshak D. R., ed) pp. 703–713, Academic Press, San Diego [Google Scholar]
- 23.Smith D. L., Deng Y., and Zhang Z. (1997) Probing the non-covalent structure of proteins by amide hydrogen exchange and mass spectrometry. J. Mass Spectrom. 32, 135–146 [DOI] [PubMed] [Google Scholar]
- 24.Marshall A. G., Hendrickson C. L., and Jackson G. S. (1998) Fourier transform ion cyclotron resonance mass spectrometry: a primer. Mass Spectrom. Rev. 17, 1–35 [DOI] [PubMed] [Google Scholar]
- 25.Kazazic S., Zhang H. M., Schaub T. M., Emmett M. R., Hendrickson C. L., Blakney G. T., and Marshall A. G. (2010) Automated data reduction for hydrogen/deuterium exchange experiments, enabled by high-resolution Fourier transform ion cyclotron resonance mass spectrometry. J. Am. Soc. Mass. Spectrom. 21, 550–558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Blakney G. T., Hendrickson C. L., and Marshall A. G. (2011) Predator data station: a fast data acquisition system for advanced FT-ICR MS experiments. Int. J. Mass Spectrom. 306, 246–252 [Google Scholar]
- 27.Zhang H. M., Bou-Assaf G. M., Emmett M. R., and Marshall A. G. (2009) Fast reversed-phase liquid chromatography to reduce back exchange and increase throughput in H/D exchange monitored by FT-ICR mass spectrometry. J. Am. Soc. Mass Spectrom. 20, 520–524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang H. M., Kazazic S., Schaub T. M., Tipton J. D., Emmett M. R., and Marshall A. G. (2008) Enhanced digestion efficiency, peptide ionization efficiency, and sequence resolution for protein hydrogen/deuterium exchange monitored by Fourier transform ion cyclotron resonance mass spectrometry. Anal. Chem. 80, 9034–9041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang Q., Chen J., Kuwajima K., Zhang H. M., Xian F., Young N. L., and Marshall A. G. (2013) Nucleotide-induced conformational changes of tetradecameric GroEL mapped by H/D exchange monitored by FT-ICR mass spectrometry. Sci. Rep. 3, 1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schaub T. M., Hendrickson C. L., Horning S., Quinn J. P., Senko M. W., and Marshall A. G. (2008) High-performance mass spectrometry: Fourier transform ion cyclotron resonance at 14.5 tesla. Anal. Chem. 80, 3985–3990 [DOI] [PubMed] [Google Scholar]
- 31.Zhang Z., Li W., Logan T. M., Li M., and Marshall A. G. (1997) Human recombinant [C22A] FK506-binding protein amide hydrogen exchange rates from mass spectrometry match and extend those from NMR. Protein Sci. 6, 2203–2217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhang Q., Willison L. N., Tripathi P., Sathe S. K., Roux K. H., Emmett M. R., Blakney G. T., Zhang H. M., and Marshall A. G. (2011) Epitope mapping of a 95 kDa antigen in complex with antibody by solution-phase amide backbone hydrogen/deuterium exchange monitored by Fourier transform ion cyclotron resonance mass spectrometry. Anal. Chem. 83, 7129–7136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.McGeoch A. T., Trakselis M. A., Laskey R. A., and Bell S. D. (2005) Organization of the archaeal MCM complex on DNA and implications for the helicase mechanism. Nat. Struct. Mol. Biol. 12, 756–762 [DOI] [PubMed] [Google Scholar]
- 34.Slaymaker I. M., Fu Y., Toso D. B., Ranatunga N., Brewster A., Forsburg S. L., Zhou Z. H., and Chen X. S. (2013) Mini-chromosome maintenance complexes form a filament to remodel DNA structure and topology. Nucleic Acids Res. 41, 3446–3456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wiedemann C., Szambowska A., Häfner S., Ohlenschläger O., Gührs K. H., and Görlach M. (2015) Structure and regulatory role of the C-terminal winged helix domain of the archaeal minichromosome maintenance complex. Nucleic Acids Res. 43, 2958–2967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Houde D., Berkowitz S. A., and Engen J. R. (2011) The utility of hydrogen/deuterium exchange mass spectrometry in biopharmaceutical comparability studies. J. Pharm. Sci. 100, 2071–2086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Moreau M. J., McGeoch A. T., Lowe A. R., Itzhaki L. S., and Bell S. D. (2007) ATPase site architecture and helicase mechanism of an archaeal MCM. Mol. Cell 28, 304–314 [DOI] [PubMed] [Google Scholar]
- 38.Liew L. P., and Bell S. D. (2011) The interplay of DNA binding, ATP hydrolysis and helicase activities of the archaeal MCM helicase. Biochem. J. 436, 409–414 [DOI] [PubMed] [Google Scholar]
- 39.Bauer R. J., Graham B. W., and Trakselis M. A. (2013) Novel interaction of the bacterial-like DnaG primase with the MCM helicase in archaea. J. Mol. Biol. 425, 1259–1273 [DOI] [PubMed] [Google Scholar]
- 40.Fletcher R. J., and Chen X. S. (2006) Biochemical activities of the BOB1 mutant in Methanobacterium thermoautotrophicum MCM. Biochemistry 45, 462–467 [DOI] [PubMed] [Google Scholar]
- 41.Costa A., Pape T., van Heel M., Brick P., Patwardhan A., and Onesti S. (2006) Structural studies of the archaeal MCM complex in different functional states. J. Struct. Biol. 156, 210–219 [DOI] [PubMed] [Google Scholar]
- 42.Aarattuthodiyil S., Byrd A. K., and Raney K. D. (2014) Simultaneous binding to the tracking strand, displaced strand and the duplex of a DNA fork enhances unwinding by Dda helicase. Nucleic Acids Res. 42, 11707–11720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Reynolds K. A., Cameron C. E., and Raney K. D. (2015) Melting of duplex DNA in the absence of ATP by the NS3 helicase domain through specific interaction with a single-strand/double-strand junction. Biochemistry 54, 4248–4258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Spies M., Amitani I., Baskin R. J., and Kowalczykowski S. C. (2007) RecBCD enzyme switches lead motor subunits in response to chi recognition. Cell 131, 694–705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Singleton M. R., Dillingham M. S., Gaudier M., Kowalczykowski S. C., and Wigley D. B. (2004) Crystal structure of RecBCD enzyme reveals a machine for processing DNA breaks. Nature 432, 187–193 [DOI] [PubMed] [Google Scholar]
- 46.Krajewski W. W., Fu X., Wilkinson M., Cronin N. B., Dillingham M. S., and Wigley D. B. (2014) Structural basis for translocation by AddAB helicase-nuclease and its arrest at χ sites. Nature 508, 416–419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Satapathy A. K., Kulczyk A. W., Ghosh S., van Oijen A. M., and Richardson C. C. (2011) Coupling dTTP hydrolysis with DNA unwinding by the DNA helicase of bacteriophage T7. J. Biol. Chem. 286, 34468–34478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Manhart C. M., and McHenry C. S. (2015) Identification of subunit binding positions on a model fork and displacements that occur during sequential assembly of the Escherichia coli primosome. J. Biol. Chem. 290, 10828–10839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Costa A., Ilves I., Tamberg N., Petojevic T., Nogales E., Botchan M. R., and Berger J. M. (2011) The structural basis for MCM2–7 helicase activation by GINS and Cdc45. Nat. Struct. Mol. Biol. 18, 471–477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yuan Z., Bai L., Sun J., Georgescu R., Liu J., O'Donnell M. E., and Li H. (2016) Structure of the eukaryotic replicative CMG helicase suggests a pumpjack motion for translocation. Nat. Struct. Mol. Biol. 23, 217–224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Arias-Palomo E., O'Shea V. L., Hood I. V., and Berger J. M. (2013) The bacterial DnaC helicase loader is a DnaB ring breaker. Cell 153, 438–448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Duderstadt K. E., and Berger J. M. (2013) A structural framework for replication origin opening by AAA+ initiation factors. Curr. Opin. Struct. Biol. 23, 144–153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Riera A., Tognetti S., and Speck C. (2014) Helicase loading: how to build a MCM2–7 double-hexamer. Semin. Cell Dev. Biol. 30, 104–109 [DOI] [PubMed] [Google Scholar]