

Abstract
Phospholipase Cϵ (PLCϵ), an effector of Ras and Rap small GTPases, plays a crucial role in inflammation by augmenting proinflammatory cytokine expression. This proinflammatory function of PLCϵ is implicated in its facilitative role in tumor promotion and progression during skin and colorectal carcinogenesis, although their direct link remains to be established. Moreover, the molecular mechanism underlying these functions of PLCϵ remains unknown except that PKD works downstream of PLCϵ. Here we show by employing the colitis-induced colorectal carcinogenesis model, where ApcMin/+ mice are administered with dextran sulfate sodium, that PLCϵ knock-out alleviates the colitis and suppresses the following tumorigenesis concomitant with marked attenuation of proinflammatory cytokine expression. In human colon epithelial Caco2 cells, TNF-α induces sustained expression of proinflammatory molecules and sustained activation of nuclear factor-κB (NF-κB) and PKD, the late phases of which are suppressed by not only siRNA-mediated PLCϵ knockdown but also treatment with a lysophosphatidic acid (LPA) receptor antagonist. Also, LPA stimulation induces these events in an early time course, suggesting that LPA mediates TNF-α signaling in an autocrine manner. Moreover, PLCϵ knockdown results in inhibition of phosphorylation of IκB by ribosomal S6 kinase (RSK) but not by IκB kinases. Subcellular fractionation suggests that enhanced phosphorylation of a scaffolding protein, PEA15 (phosphoprotein enriched in astrocytes 15), downstream of the PLCϵ-PKD axis causes sustained cytoplasmic localization of phosphorylated RSK, thereby facilitating IκB phosphorylation in the cytoplasm. These results suggest the crucial role of the TNF-α-LPA-LPA receptor-PLCϵ-PKD-PEA15-RSK-IκB-NF-κB pathway in facilitating inflammation and inflammation-associated carcinogenesis in the colon.
Keywords: inflammation, NF-kappa B (NF-KB), Phospholipase C, S6 kinase, tumor necrosis factor (TNF), lysophosphatidic acid
Introduction
Phosphatidylinositol-specific PLCs3 catalyze the hydrolysis of phosphatidylinositol 4,5-bisphosphate into two vital second messengers, inositol 1,4,5-trisphosphate and diacylglycerol, which induce release of Ca2+ from the intracellular stores and activation of diacylglycerol target proteins, including PKC isoforms, respectively. Thirteen PLC isoforms were identified in mammals and divided into six classes, β, δ, γ, ϵ, η, and ζ, based on the structural similarity (1). PLCϵ was first identified as an effector of Ras small GTPases (2, 3), and further studies revealed that it is also activated by other small GTPases, such as Rap1, Rap2, and Rho, as well as by heterotrimeric G protein Gα12, Gα13, and β1γ2 subunits (4–7). Stimulation of G protein-coupled receptors with their ligands, such as LPA, S1P, and thrombin, induces PLCϵ activation (6, 7). PLCϵ is expressed in non-immune cells, such as epithelial cells and fibroblasts of various tissues, but not in immune cells, such as lymphocytes, granulocytes, macrophages, and dendritic cells (8).
By employing genetically modified mice for PLCϵ, we demonstrated that PLCϵ plays a crucial role in inflammation in various tissues; mice homozygous for an allele devoid of the lipase activity (PLCϵΔX/ΔX mice) exhibited markedly attenuated inflammatory responses in various animal models, including the phorbol ester-induced dermatitis, contact dermatitis, and bronchial asthma models (8–11), and, moreover, transgenic mice overexpressing PLCϵ in the skin keratinocytes spontaneously developed chronic skin inflammation resembling human psoriasis (12). Concurrently, PLCϵΔX/ΔX mice exhibited marked resistance to tumor formation in the two-stage skin chemical carcinogenesis using a phorbol ester as a promoter as well as to the de novo intestinal carcinogenesis on the ApcMin/+ (adenomatous polyposis coli) background, which were associated with attenuation of tumor-associated inflammation exemplified by reduced expression of proinflammatory cytokines (13, 14). These findings suggested that PLCϵ plays a role in tumor promotion through augmentation of inflammatory responses. Recently, strong support for the crucial role of PLCϵ in human carcinogenesis came from genome-wide association studies, which identified PLCϵ (PLCE1) as a predisposing gene for gastric and esophageal carcinomas (15, 16). However, the direct link between the proinflammatory and tumor-promoting functions of PLCϵ awaits further verification by using an animal model of inflammation-induced carcinogenesis.
Recent studies indicated that PLCϵ activation augments the NF-κB-dependent expression of inflammation-associated genes, particularly those of proinflammatory cytokines in cultured cells (17, 18). In addition, it was reported that PKD, a serine/threonine kinase also called atypical PKCμ, which had been known to mediate NF-κB activation triggered by various stimuli, such as LPA (19, 20), was activated downstream of PLCϵ (18, 21, 22), although underlying molecular mechanisms remain poorly understood. NF-κB binds as a dimer of p65 RelA and p50 to a consensus DNA sequence on the promoters and enhancers of the target genes and regulates their transcription. NF-κB activation is tightly controlled by several factors. Under a resting condition, NF-κB is associated with IκB and exists as an inactive form in the cytoplasm. Upon stimulation by extracellular ligands, such as TNF-α, IκB is phosphorylated by IκB kinases at its specific serine residues, including Ser-32, and targeted for ubiquitination and subsequent proteasome degradation, thereby allowing NF-κB to undergo nuclear translocation and activate the target gene expression (23, 24). There are several proteins functioning as IκB kinases, such as IKK complex, consisting of IKKα, IKKβ, and IKKγ (25); p90 RSK (26, 27); and CK2 (28).
RSK family proteins are serine/threonine kinases and activated through phosphorylation by ERK1/2 and PDK1 (phosphoinositide-dependent kinase 1) in the cytoplasm. Thr-573 of RSK, located in the activation loop of the C-terminal kinase domain, is phosphorylated by ERK, leading to autophosphorylation of Ser-380 in the linker region (29). Subsequently, RSK is fully activated through phosphorylation by PDK1. Activated RSK is located in both the cytoplasm and nuclei and phosphorylates its substrates, such as IκB (26, 27), in the cytoplasm and c-Fos (30) and CREB (31) in the nuclei. The subcellular localization of RSK is reported to be regulated by a scaffold protein, PEA15 (phosphoprotein enriched in astrocytes 15), which directly associates with RSK and holds it in the cytoplasm, based on the observation that overexpression of PEA15 prevented nuclear translocation of RSK (32). Moreover, overexpression of a phosphorylation-mimicking mutant, S104D, of PEA15 failed to elevate the CREB promoter activity, suggesting that the phosphorylation of PEA15 might regulate the RSK localization (33).
In this study, we further investigate the relationship between the proinflammatory and tumor-promoting functions of PLCϵ by employing the colitis-induced colorectal carcinogenesis model. Further, we analyze the molecular mechanism by which PLCϵ augments the NF-κB-dependent expression of inflammation-associated genes, demonstrating that the PLCϵ-PKD axis, activated by LPA receptor engagement, augments NF-κB activation by causing sustained cytoplasmic localization of RSK via phosphorylation of PEA15 and thereby facilitating its phosphorylation of IκB in the cytoplasm.
Experimental Procedures
Materials
The following antibodies were obtained from Cell Signaling Technologies: rabbit anti-p65 RelA mAb (catalog no. 8242), rabbit anti-PKD Ab (catalog no. 2052), rabbit anti-phospho-PKD (Ser-916) Ab (catalog no. 2051), rabbit anti-RSK mAb (catalog no. 5528), rabbit anti-phospho-RSK (Ser-380) mAb (catalog no. 11989), rabbit anti-IκB Ab (catalog no. 9242), rabbit anti-phospho-IκB (Ser-32) mAb (catalog no. 2859), rabbit anti-ERK Ab (catalog no. 9102), rabbit anti-phospho-ERK (Thr-202/Tyr-204) Ab (catalog no. 9101), rabbit anti-phospho-IKKα/β (Ser-176/180) mAb (catalog no. 2697), rabbit anti-PEA15 Ab (catalog no. 2780), rabbit anti-phospho-PEA15 (Ser-104) Ab (catalog no. 2776), mouse anti-HA tag mAb (catalog no. 2367), and anti-α-tubulin (catalog no. 3873) Ab. The following antibodies were commercially obtained: rabbit anti-PLCϵ Ab (HPA015597, Sigma), mouse anti-actin mAb (MAB1501R, Chemicon), rabbit anti-IKKα/β Ab (sc-7607, Santa Cruz Biotechnology, Inc.), rabbit anti-TBP Ab (sc-273, Santa Cruz Biotechnology), rabbit anti-FLAG-tag Ab (PM020, MBL), goat anti-CCL2 (CC chemokine ligand 2) Ab (sc-1785, Santa Cruz Biotechnology), and goat anti-CXCL2 (chemokine CXC motif ligand 2) Ab (AF-452-NA, R&D Systems). An anti-PLCϵ antibody raised against the C-terminal peptide of mouse PLCϵ was generated in house (34). Recombinant TNF-α (300-01A) was obtained from Peprotech. The following chemicals were commercially obtained: 1-oleoyl-2-hydroxy-sn-glycero-3-phosphate (LPA, Avanti), tetradecyl phosphonate (LPA receptor antagonist, Cayman Chemical Co.), JTE013 (S1P receptor antagonist, Cayman Chemical Co.), CID755673 (PKD inhibitor, Millipore), CID2011756 (PKD inhibitor, Tocris Bioscience), U0126 (MEK inhibitor, Calbiochem), Trametinib (MEK inhibitor, Selleckchem), BI-D1870 (RSK inhibitor, Enzo Life Technologies), MG-132 (proteasome inhibitor, Calbiochem), and TBB (CK2 inhibitor, Sigma). Protein G-Sepharose was obtained from GE Healthcare. Chemicals were used according to the manufacturers' recommendations.
Plasmids
The full-length cDNA of human PLCϵ was purchased as Flex ORF clones (Promega) and cloned into pFLAG-CMV2 (Sigma) for expression as FLAG fusions. The lipase-dead human PLCϵ mutant, PLCϵΔX, was generated by PCR-mediated mutagenesis to delete amino acids 1391–1541. pCMV-HA-PEA15 was constructed by inserting the human PEA15 cDNA, obtained by RT-PCR using Caco2 cell mRNA as a template, into pCMV-HA (Clontech). pCMV-HA-PEA15S104A and pCMV-HA-PEA15S104D were derived from it by site-directed mutagenesis. The primers used are listed in Table 1.
TABLE 1.
Primers used for RT-PCR/qRT-PCR analyses, cDNA cloning, and mutagenesis
| Gene | Forward primer (from 5′ to 3′) | Reverse primer (from 5′ to 3′) | Purpose | Species |
|---|---|---|---|---|
| Tnf-α | tgatcggtccccaaagg | ggtctgggccatagaactga | qRT-PCR | Mouse |
| Cox-2 | tgtacaagcagtggcaaagg | gcagccatttccttctctcc | qRT-PCR | Mouse |
| Cxcl1 | acccaaaccgaagtcatagc | tggggacaccttttagcatc | qRT-PCR | Mouse |
| Cxcl2 | agtttgccttgaccctgaag | ctttggttcttccgttgagg | qRT-PCR | Mouse |
| Ccl2 | ttgtcaccaagctcaagagaga | gaggtggttgtggaaaaggtag | qRT-PCR | Mouse |
| Cxcr2 | atgccctctattctgccagat | gtgctccggttgtataagatgac | qRT-PCR | Mouse |
| Ccr2 | atccacggcatactatcaacatc | caaggctcaccatcatcgtag | qRT-PCR | Mouse |
| Cd68 | cttcacgatgacacctacag | tcttggactagtagcagtgc | qRT-PCR | Mouse |
| CXCL1 | agggaattcaccccaagaac | caccagtgagcttcctcctc | RT-PCR | Human |
| COX-2 | gcaaattgctggcagggttg | agggcttcagcataaagcgt | RT-PCR | Human |
| CCL20 | gcgcaaatccaaaacagact | caagtccagtgaggcacaaa | RT-PCR | Human |
| PLCϵ | gctcttcagcggattattggaa | tcataccgtccatcctcctgata | RT-PCR | Human |
| CCL2 | ccccagtcacctgctgttat | tggaatcctgaacccacttc | qRT-PCR | Human |
| CXCL8 | cttggcagccttcctgattt | ttctttagcactccttggcaaaa | qRT-PCR | Human |
| PLCϵ | tccagaagagggatacatgg | tcataccgtccatcctcctgata | qRT-PCR | Human |
| β-Actin | atgaagatcaagatcattgctcctc | acatctgctggaaggtggacag | RT-PCR/qRT-PCR | Mouse/human |
| GAPDH | gaaggtgaaggtcggagtc | gaagatggtgatgggatttc | qRT-PCR | Human |
| PEA15 | aaagaattcccgctgagtacgggaccctc | aaaaggtacctcaggccttcttcggtggg | PCR Cloning | Human |
| PEA15 | atccccgccgccaagaagtacaaagacatta | cttggcggcggggatacgggttagcttg | S104A mutant construction | Human |
| PEA15 | atccccgacgccaagaagtacaaagacatta | cttggcgtcggggatacgggttagcttg | S104D mutant construction | Human |
| PLCϵ | aatgttctcctgtgactcatcattttttgaggcaa | aagctaaaagcccatcagacgccagtggatatctt | ΔX mutant construction | Human |
Cell Culture
A human colon cancer epithelial cell line Caco2 was purchased from the RIKEN cell bank (RBRC-RCB0988) and maintained in 5% CO2 at 37 °C in modified Eagle's minimum essential medium supplemented with 20% fetal bovine serum, non-essential amino acids, and 100 μg/ml penicillin-streptomycin. Cells were washed with PBS and serum-starved for 3 h before subjecting to various experiments. Cells were pretreated with inhibitors and antagonists before stimulation by ligands, such as TNF-α and LPA.
Transfection of siRNAs and Plasmids
Caco2 cells (8 × 106 cells) were transfected with Stealth siRNA (Life Technologies, Inc.) targeting PLCϵ (HSS181915, siRNA 1) or its control (12935-400) by electroporation using GenePulser (Bio-Rad) as described before (8). Another siRNA (5′-GCCAAAUAUUCCUACAGCAUCCUGA-3′, siRNA 2) was also used to target PLCϵ as described (35). Transfection of plasmids into Caco2 cells was performed by using Lipofectamine3000 (Life Technologies). 14–16 h after transfection, cells were subjected to various experiments after exchange to fresh medium.
Subcellular Fractionation
Subcellular fractionation was carried out as described (36). Briefly, Caco2 cells were harvested using a scraper in ice-cold PBS, and, after centrifugation, the cell pellet was resuspended in a lysis buffer (10 mm Hepes, pH 7.9, 10 mm KCl, 1.5 mm MgCl2, 0.1 mm EDTA, 0.25% (v/v) Nonidet P-40, and protease inhibitor mixture (Nacalai Tesque)). After centrifugation at 1,000 × g for 10 min, the supernatant was used as the nuclear fraction. The pellet was resuspended in a nuclear extraction buffer (20 mm Hepes, pH 7.9, 420 mm NaCl, 1.5 mm MgCl2, 0.1 mm EDTA, 1 mm DTT, 25% (v/v) glycerol, and protease inhibitor mixture) and incubated for 30 min, and, after centrifugation at 21,500 × g for 10 min, the supernatant was used as the cytoplasmic fraction.
Immunoblotting and Immunoprecipitation
Cells were lysed in a lysis buffer (20 mm Tris-HCl, pH 7.4, 250 mm NaCl, 3 mm EDTA, 0.5% (v/v) Nonidet P-40, and protease inhibitor mixture). The lysates were centrifuged at 21,500 × g for 10 min, and the supernatants were used for immunoblotting and immunoprecipitation. Immunoblotting was performed by the standard procedure followed by acquisition of immunoreactive signals on the blots using ImageQuant LAS4000mini (GE Healthcare). The intensities of the immunoreactive signals were quantitated using ImageQuant TL (GE Healthcare).
RT-PCR and qRT-PCR
Total cellular RNA isolation, cDNA synthesis, RT-PCR, and qRT-PCR were performed as described previously (37). The relative mRNA level of each transcript was determined by the ΔΔCt method with β-actin or GAPDH as a reference gene. The primers used are listed in Table 1.
Animals
ApcMin/+ mice, purchased from the Jackson Laboratory (Bar Harbor, ME), were bred with PLCϵΔX/ΔX mice (14), which had been back-crossed to C57BL/6JJcl mice (CLEA Japan, Tokyo, Japan) for at least 8 generations. The breeding strategy was ultimately designed to generate ApcMin/+ mice with the PLCϵ+/+ or PLCϵΔX/ΔX background. The genotypes of PLCϵ (13) and Apc (see the Jackson Laboratory Web site) were determined by PCR of the tail DNAs. All of the animals were maintained in the animal facility of Kobe University Graduate School of Medicine, and the use and care of the animals were reviewed and approved by the Institutional Animal Care and Use Committee of Kobe University.
Induction of Colitis and Colitis-induced Colorectal Cancer
To induce colitis, drinking water containing 2.5% (w/v) DSS (molecular weight = 36,000–50,000; Wako Pure Chemical, Osaka, Japan), dissolved in tap water, was orally administered to 8-week-old mice for 5 days (38, 39). After that, the drinking water was substituted by tap water, and the mice were maintained for 16 days to develop colon tumors.
Histochemical Analyses
After sacrificing, the colons were removed from mice, washed with PBS, serially sectioned in the longitudinal direction, and embedded in paraffin. After that, the sections were subjected to H&E staining (14). MPO staining was performed on frozen sections (10 μm thick) as described (40).
Histopathological Classification of Tumors
Tumors in H&E-stained sections were observed under a microscope and classified into early and late adenomas and adenocarcinomas according to the histopathological criteria recommended for the study of mouse models of intestinal cancer as described (41). The classification was carried out by a pathologist blinded in regard to the mouse genotypes.
Immunohistochemistry
The whole colon was rolled up in a “Swiss roll” configuration and embedded in OCT compound for sectioning. After fixation with 4% paraformaldehyde, the sections (10 μm thick) were subjected to immunostaining as described (11, 14). Images were taken with an Olympus FSX100 microscope and Olympus FSX100-BSW software.
Statistical Analysis
Data are expressed as the mean ± S.D. An unpaired Student's t test was performed for determination of p values. In cases where the p values were <0.05, differences were considered to be statistically significant. All of the data were obtained from at least three independent experiments.
Results
Role of PLCϵ in Inflammation-induced Carcinogenesis
We employed the ApcMin/+ mouse colitis-induced colorectal carcinogenesis model, where tumor development is dependent on the DSS-induced colon inflammation (38, 39). At first, PLCϵ+/+ and PLCϵΔX/ΔX mice were orally administered with 2.5% DSS for 5 days to induce acute colitis, the severity of which was evaluated by the body weight loss and histological examination. Compared with PLCϵ+/+ mice, the reduction in the body weight was attenuated in PLCϵΔX/ΔX mice (Fig. 1A). Likewise, mucosal erosion and leukocyte infiltration were alleviated in PLCϵΔX/ΔX mice (Fig. 1B). In this model, infiltration of neutrophils expressing CXCR2 (CXC chemokine receptor 2) and macrophages expressing CCR2 (CC chemokine receptor 2) is known to play critical roles in the pathogenesis of colitis (42, 43). Indeed, we observed marked increases in the numbers of leukocytes and macrophages in the lesional area as detected by MPO staining and immunostaining for CD68, respectively (Fig. 1C). Marked elevation of CXCR2 and CCR2 mRNAs was observed in the DSS-administered colons of PLCϵ+/+ mice (Fig. 1D). In contrast, the colons of PLCϵΔX/ΔX mice exhibited markedly attenuated responses to DSS administration. Moreover, the expression of other inflammation-associated genes, such as Tnf-α, Cox-2, Cxcl1, Cxcl2, and Ccl2, showed a substantial increase after DSS administration in PLCϵ+/+ mice, which was also markedly suppressed in PLCϵΔX/ΔX mice (Fig. 1D). Intriguingly, CXCL2 and CCL2, the ligands for CXCR2 and CCR2, respectively, were abundantly expressed after DSS administration in the colon epithelial cells expressing PLCϵ, and this expression was markedly suppressed in PLCϵΔX/ΔX mice (Fig. 1, E and F). These results suggested that PLCϵ might augment colon inflammation by facilitating the secretion from the colon epithelial cells of proinflammatory factors, such as CXCL2 and CCL2, which recruit neutrophils and macrophages, respectively.
FIGURE 1.

Roles of PLCϵ in colitis and colitis-induced carcinogenesis. Acute colitis was induced in ApcMin/+ mice carrying various PLCϵ backgrounds by oral administration of DSS for 5 days, and the colons were collected at day 0 and 5 (3 animals/group). A, results of the daily body weight measurements of PLCϵ+/+ and PLCϵΔX/ΔX mice. B, the longitudinal sections of the colons of PLCϵ+/+ (top panels) and PLCϵΔX/ΔX mice (bottom panels) with (right) or without (left) DSS administration were subjected to H&E staining. C, infiltrating neutrophils (left panels) and macrophages were detected by MPO staining and immunostaining for CD68, respectively, in the colon sections of PLCϵ+/+ and PLCϵΔX/ΔX mice with or without DSS administration. The bar graphs on the bottom show the numbers of MPO-positive neutrophils (left) and CD68-positive macrophages (right) per inflamed region (mm2). D, The mRNA levels of the indicated proteins in the colons of PLCϵ+/+ and PLCϵΔX/ΔX mice with or without DSS administration were quantified by qRT-PCR and normalized to the β-actin mRNA level. E, the colon sections of PLCϵ+/+ mice administered with DSS were subjected to immunofluorescence staining for CXCL2 or CCL2 (red) and PLCϵ (green) and DAPI staining (blue). Magnified images of the boxed regions of the left panels are shown in the right panels. F, CCL2 and CXCL2 were detected by immunofluorescence staining with the anti-CCL2 and anti-CXCL2 antibodies, respectively (red), in the colon sections of PLCϵ+/+ and PLCϵΔX/ΔX mice with or without DSS administration. The sections were also stained with DAPI (blue). Representative data are shown in B, C, E, and F. G, colorectal tumors were observed on the 16th day after DSS administration (6 animals/group). Shown are the total numbers of tumors/mouse (left) and the numbers/mouse (middle) and the percentages (right) of tumors classified into low grade and high grade adenomas and adenocarcinomas in PLCϵ+/+ and PLCϵΔX/ΔX mice. The data are shown as the mean ± S.D. (error bars). *, p < 0.05. Scale bars, 25 μm.
We next analyzed the effects of the PLCϵ genotypes on the colitis-induced colorectal carcinogenesis of ApcMin/+ mice (38, 39). As a result, PLCϵΔX/ΔX mice exhibited substantial reduction in the total numbers of colon tumors observed on the 16th day after DSS administration, compared with PLCϵ+/+ mice (Fig. 1G). Histopathological classification of the tumors revealed that the ratios of low grade and high grade adenomas and adenocarcinomas were ∼40, 30, and 30%, respectively, in PLCϵ+/+ mice. In contrast, >80% of the tumors were low grade adenomas and essentially no adenocarcinoma was formed in PLCϵΔX/ΔX mice. These results taken together gave further support for our notion that PLCϵ facilitates tumor promotion and malignant progression by augmenting inflammation.
Role of PLCϵ in NF-κB-dependent Proinflammatory Gene Expression
The molecular mechanism underlying the action of PLCϵ on the expression of proinflammatory factors was analyzed by using a human colon epithelial cell line, Caco2. Because PLCϵ had been implicated in the induction of the inflammation-associated genes in response to TNF-α stimulation (8, 11, 17), we first examined the effects of the siRNA-mediated knockdown of PLCϵ on TNF-α-induced expression of these genes (Fig. 2). In Caco2 cells transfected with an siRNA control, the expression of CXCL8 and CCL2 was elevated in 1–2 h after TNF-α stimulation (Fig. 2A). The transfection of two distinct siRNAs against PLCϵ, siRNA 1 and siRNA 2, which efficiently knocked down PLCϵ expression (Fig. 2B), inhibited the expression of both CXCL8 and CCL2 significantly at the 2-h time point but not effectively at the 1-h time point (Fig. 2A). In addition, the expression of COX-2 showed sustained elevation in 1–2 h, as was the case with CXCL8 (Fig. 2C). The expression of CCL20 was mainly elevated in 2 h only, whereas expression of CXCL1 was mainly elevated in 1 h only. In these cases, too, the transfection of siRNA 1 inhibited the expression at the 2-h time point of COX-2 and CCL20 (Fig. 2C).
FIGURE 2.
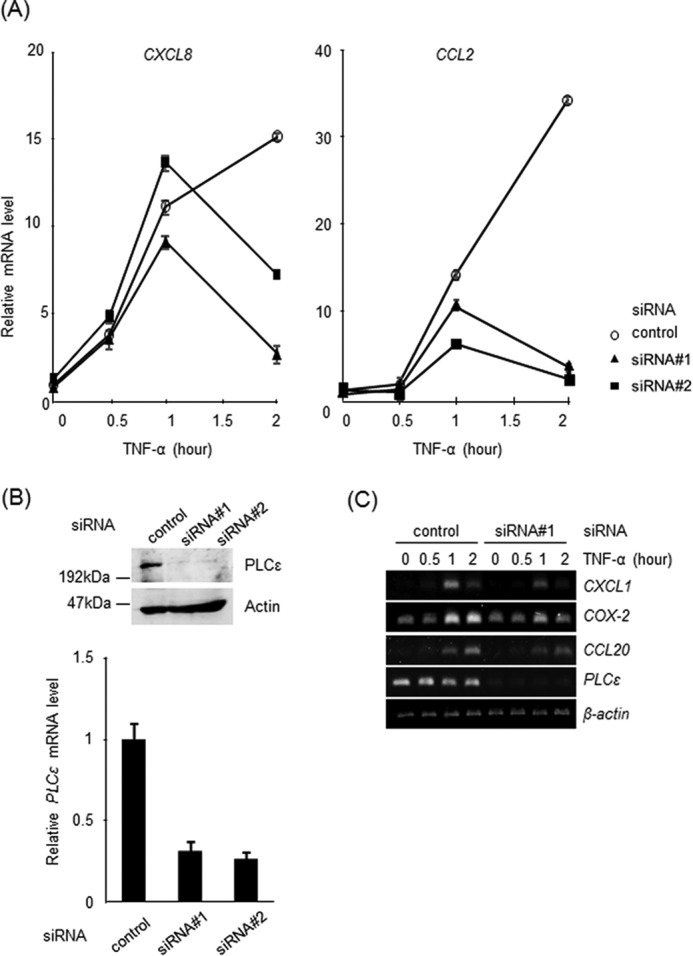
Effects of PLCϵ knockdown on induction of the inflammation-associated genes by TNF-α stimulation. Caco2 cells were transfected with siRNA 1 or siRNA 2 targeting PLCϵ or a control siRNA and cultured for 38–42 h. A, cells were serum-starved for 3 h and stimulated with 20 ng/ml TNF-α for the indicated times. The mRNA levels of CXCL8 (left) and CCL2 (right) were measured by qRT-PCR and normalized to the GAPDH mRNA level. B, PLCϵ and actin in the cell lysates were detected by immunoblotting with the anti-PLCϵ and anti-actin Abs, respectively. The PLCϵ mRNA levels were measured by qRT-PCR and normalized to the GAPDH mRNA level. C, the mRNA levels of the indicated genes were analyzed in cells treated as described in A by semiquantitative RT-PCR. The β-actin mRNA was used as a control. Three experiments performed independently yielded equivalent results.
TNF-α-induced expression of the inflammation-associated genes, shown here to be regulated by PLCϵ, was known to be mediated by the NF-κB pathway (44). This led us to examine the effects of PLCϵ knockdown on the nuclear translocation of p65 (Fig. 3). Nuclear accumulation of p65 was detected in 30 and 90 min after TNF-α stimulation in Caco2 cells transfected with the control siRNA (Fig. 3A). Transfection of the two distinct PLCϵ siRNAs inhibited the p65 nuclear accumulation at the time point of 90 min but not 30 min. The Ser-32 phosphorylation of IκB, which is required for its degradation and subsequent p65 nuclear translocation (23, 24), was observed in a biphasic manner, peaking at 10 and 90 min after TNF-α stimulation in cells transfected with the control siRNA (Fig. 3B). Transfection of the two PLCϵ siRNAs lowered the peak at 90 min but not that at 10 min. The p65 nuclear localization observed at 30 min seemed to result from the IκB phosphorylation at 10 min, taking account of the time lag between the two events. These results suggested that PLCϵ might be involved in the late phase of the TNF-α-induced NF-κB activation.
FIGURE 3.

Effects of PLCϵ knockdown on NF-κB activation induced by TNF-α stimulation. A, Caco2 cells transfected with siRNA 1 or siRNA 2 targeting PLCϵ or the control siRNA were cultured for 38–42 h, serum-starved for 3 h, and stimulated with 20 ng/ml TNF-α for the indicated times. The cell lysates were subjected to subcellular fractionation, and the resulting nuclear and cytoplasmic fractions were subjected to immunoblotting with the anti-p65 RelA Ab. TBP and α-tubulin were used as markers for the nuclear and the cytoplasmic fractions, respectively. The numbers below the immunoblots indicate -fold changes of the immunoreactive signals over those at 0 min after TNF-α stimulation of the control cells. The averages of the intensities of the immunoreactive signals of p65 in the nuclear fractions at 90 min of at least three independent experiments are expressed as the mean ± S.D. (error bars) in arbitrary units (AU) with p values (bottom). B, Caco2 cells were transfected with the indicated siRNAs as described in A, serum-starved for 3 h, treated with 10 μm MG-132 for 30 min, and subsequently stimulated with 20 ng/ml TNF-α for the indicated times. IκB phosphorylated at Ser-32, total IκB, PLCϵ, and actin in the cell lysates were detected by immunoblotting with the anti-phospho-IκB, anti-IκB, anti-PLCϵ, and anti-actin Abs, respectively. The numbers below the immunoblots indicate -fold changes of the phospho-IκB signals divided by the total IκB signals over that at 0 min after TNF-α stimulation of the control cells. The averages of the intensities of immunoreactive signals of phospho-IκB at 90 min of at least three independent experiments are expressed as the mean ± S.D. in arbitrary units with p values (right). At least three experiments performed independently yielded equivalent results.
Roles of PKD and LPA Receptor in PLCϵ-dependent NF-κB Activation
We next examined the role of PKD downstream of PLCϵ. Treatment of Caco2 cells with the two distinct PKD inhibitors, CID755673 and CID2011756, inhibited the IκB phosphorylation at 90 min but not at 10 min as well as the p65 nuclear translocation at 90 min but not at 30 min after TNF-α stimulation (Fig. 4, A and B). These results were very similar to those obtained with the siRNA-mediated knockdown of PLCϵ (Fig. 3). Moreover, the Ser-916 phosphorylation of PKD induced by TNF-α stimulation was suppressed by transfection of the two PLCϵ siRNAs (Fig. 4C), suggesting that PKD might function downstream of PLCϵ for regulation of the NF-κB activation.
FIGURE 4.

Role of PKD in TNF-α-induced NF-κB activation. A, Caco2 cells were serum-starved for 3 h, treated with 10 μg/ml CID755673 or 50 μm CID2011756 in combination with 10 μm MG-132 for 30 min, and subsequently stimulated with 20 ng/ml TNF-α for the indicated times. IκB phosphorylation was measured as described in the legend to Fig. 3B. The averages of the intensities of the immunoreactive signals of phospho-IκB at 90 min of three independent experiments are expressed as the mean ± S.D. (error bars) in arbitrary units (AU) with p values (right). B, Caco2 cells were treated as described in A except that MG-132 treatment was omitted. Nuclear and cytoplasmic localization of p65 was measured as described in the Fig. 3A legend. TBP and α-tubulin were used as markers for the nuclear and the cytoplasmic fractions, respectively. The averages of the intensities of the immunoreactive signals of p65 in the nuclear fractions at 90 min of three independent experiments are expressed as the mean ± S.D. in arbitrary units with p values (right). C, Caco2 cells transfected with the indicated siRNAs were cultured for 38–42 h, serum-starved for 3 h, and stimulated with 20 ng/ml TNF-α for the indicated times. PKD phosphorylated at Ser-916, total PKD, PLCϵ, and actin in the cell lysates were detected by immunoblotting with the anti-phospho-PKD, anti-PKD, anti-PLCϵ, and anti-actin Abs, respectively. The numbers below the immunoblots indicate -fold changes of the phospho-PKD signals divided by the total PKD signals over that at 0 min after TNF-α stimulation of the control cells. The averages of the intensities of the immunoreactive signals of phospho-IκB at 90 min of three independent experiments are expressed as the mean ± S.D. in arbitrary units with p values (right). Three experiments performed independently yielded equivalent results.
The delayed action of PLCϵ upon the TNF-α-induced NF-κB activation led us to think of the possibility that another humoral factor might mediate the activation of PLCϵ induced by TNF-α stimulation. It was reported that TNF-α stimulation of cells induced the production of LPA and S1P (45, 46), which were known to activate PLCϵ (6, 7). This prompted us to examine the effects of the antagonists of the receptors for LPA and S1P on the TNF-α-induced cytokine expression and NF-κB activation (Fig. 5). The expression of CXCL8 was abrogated by treatment of cells with the LPA receptor antagonist tetradecyl phosphonate (Fig. 5A). In contrast, treatment with the S1P receptor antagonist JTE013 seemed to have little effect on the CXCL8 expression at the 2-h time point, although it unexpectedly exhibited a stimulatory effect even in the absence of TNF-α (Fig. 5A). Presently, we do not have any idea for the mechanism underlying this stimulatory effect. Tetradecyl phosphonate inhibited the IκB phosphorylation at 90 min but not at 10 min as well as the Ser-916 phosphorylation of PKD at 90 min after TNF-α stimulation (Fig. 5B). Furthermore, LPA stimulation of Caco2 cells induced both the IκB phosphorylation and PKD phosphorylation in an early time course of 30 min, which was abrogated by the siRNA-mediated knockdown of PLCϵ (Fig. 5C). These results strongly suggested that TNF-α stimulation induces LPA production and subsequent activation of LPA receptors in an autocrine manner, thereby leading to activation of the PLCϵ-PKD axis.
FIGURE 5.

Role of LPA receptor activation in PLCϵ-dependent IκB phosphorylation. A, Caco2 cells were serum-starved for 3 h, treated with 15 μg/ml tetradecyl phosphonate or 10 μg/ml JTE013 for 30 min, and subsequently stimulated with 20 ng/ml TNF-α for the indicated times. The CXCL8 mRNA levels were measured by qRT-PCR with the GAPDH mRNA as an internal control. B, Caco2 cells were treated as described in A except that 10 μm MG-132 treatment for 30 min was included. IκB phosphorylation and PKD phosphorylation were measured as described in the legends to Figs. 3B and 4C, respectively. Actin was detected by the anti-actin Ab. The averages of the intensities of the immunoreactive signals of the indicated phosphoproteins at 90 min of three independent experiments are expressed as the mean ± S.D. (error bars) in arbitrary units (AU) with p values (right). C, Caco2 cells transfected with the indicated siRNAs were cultured for 38–42 h, serum-starved for 3 h, and stimulated with 50 μm LPA for the indicated times. IκB phosphorylation and PKD phosphorylation were measured as described in the legends to Fig. 3B and 4C, respectively. PLCϵ and actin were detected by immunoblotting with the anti-PLCϵ and anti-actin Abs, respectively. The averages of the intensities of the immunoreactive signals of the indicated phosphoproteins at 30 min of three independent experiments are expressed as the mean ± S.D. in arbitrary units with p values (right). Three experiments performed independently yielded equivalent results.
Role of RSK in PLCϵ-dependent NF-κB Activation
We next analyzed the molecular mechanism by which PLCϵ induced the IκB phosphorylation, depending on the stimulation by TNF-α or LPA. To this end, we tested the involvement of three kinases, IKK, RSK, and CK2, which had been known to phosphorylate IκB at Ser-32 (25–28). The phosphorylation of IKKα/β at Ser-176 and Ser-180, which is responsible for IKK activation (47, 48), was induced in 10 min but not later after TNF-α stimulation (Fig. 6A). This early IKK phosphorylation was unaffected by the siRNA-mediated PLCϵ knockdown and likely to account for the early phase of the IκB phosphorylation directly caused by the TNF-α receptor signaling (Fig. 3B). Because no increase in phosphorylation was observed after 10 min, it is unlikely that IKK is involved in the PLCϵ-dependent IκB phosphorylation. This led us to test RSK and CK2. The TNF-α-induced CXCL8 expression was suppressed by treatment with the RSK inhibitor BI-D1870 but not the CK2 inhibitor TBB (Fig. 6B). In Caco2 cells transfected with the control siRNA, TNF-α stimulation induced elevation of RSK phosphorylation at Ser-380 in 90 min, which was diminished in cells transfected with the PLCϵ siRNA (Fig. 6C) or pretreated with CID755673 and CID2011756 (Fig. 6D). Moreover, the RSK Ser-380 phosphorylation induced in 10–30 min after LPA stimulation was abrogated by the siRNA-mediated PLCϵ knockdown (Fig. 6E). In either case, phosphorylation of ERK1/2 at Thr-202 and Tyr-204 was elevated in a similar time course with the RSK phosphorylation and unaffected by the PLCϵ knockdown. Furthermore, pretreatment with the MEK inhibitors, U0126 (49) and trametinib (50), or the RSK inhibitor BI-D1870 abrogated the IκB phosphorylation at 90 min after TNF-α stimulation (Fig. 6, F and G). The two MEK inhibitors almost completely abolished the RSK phosphorylation, confirming that ERK activation is necessary for the Ser-380 phosphorylation and activation of RSK (29). These results taken together indicated that the PLCϵ-PKD axis facilitates the phosphorylation and activation of RSK by ERK, leading to the phosphorylation and activation of IκB by RSK. The ERK activation did not seem to be regulated by it.
FIGURE 6.

Role of RSK in PLCϵ-dependent IκB phosphorylation. A, Caco2 cells transfected with the indicated siRNAs were cultured for 38–42 h, serum-starved for 3 h, and stimulated with 20 ng/ml TNF-α for the indicated times. IKKα/β phosphorylated at Ser-176 and Ser-180, total IKKα/β, and actin in the cell lysates were detected by immunoblotting with the anti-phospho-IKKα/β, anti-IKKα/β, and anti-actin Abs, respectively. The numbers below the immunoblots indicate -fold changes of the phospho-IKK signals divided by the total IKK signals over that at 0 min after TNF-α stimulation of the control cells. The averages of the intensities of the immunoreactive signals of phospho-IKKα/β at 90 min of three independent experiments are expressed as the mean ± S.D. (error bars) in arbitrary units (AU) with p values (right). B, Caco2 cells were serum-starved for 3 h, treated with 10 μg/ml BI-D1870 or 28 μg/ml TBB for 30 min, and stimulated with 20 ng/ml TNF-α for the indicated times. The CXCL8 mRNA levels were measured as described in the legend to Fig. 5A. C, Caco2 cells were treated with the indicated siRNAs and stimulated with TNF-α as described in A. RSK phosphorylated at Ser-916 and total RSK in the cell lysates were detected by immunoblotting with the anti-phospho-RSK and anti-RSK Abs, respectively. ERK1/2 phosphorylated at Thr-202 and Tyr-204 and total ERK1/2 were detected by immunoblotting with the anti-phospho-ERK and anti-ERK Abs, respectively. PLCϵ and actin were detected by immunoblotting with the anti-PLCϵ and anti-actin Abs, respectively. The numbers below the immunoblots indicate -fold changes of the signals of the phosphorylated proteins divided by the signals of the total proteins over those at 0 min after TNF-α stimulation of the control cells. The averages of the intensities of the immunoreactive signals of the indicated phosphoproteins at 90 min of three independent experiments are expressed as the mean ± S.D. in arbitrary units with p values (bottom). D, Caco2 cells were serum-starved for 3 h, treated with 10 μg/ml CID755673 or 50 μm CID2011756 for 30 min, and stimulated with 20 ng/ml TNF-α for the indicated times. The phosphorylations of RSK and ERK1/2 were measured as described in C. The averages of the intensities of the immunoreactive signals of the indicated phosphoproteins at 90 min of three independent experiments are expressed as the mean ± S.D. in arbitrary units with p values (bottom). E, Caco2 cells were serum-starved for 3 h, treated with the indicated siRNAs, and stimulated with 50 μm LPA for the indicated times. The phosphorylations of RSK and ERK1/2 were measured as described in C. The averages of the intensities of the immunoreactive signals of the indicated phosphoproteins at 30 min of three independent experiments are expressed as the mean ± S.D. in arbitrary units with p values (bottom). F, Caco2 cells were serum-starved for 3 h, treated with 10 μg/ml U0126 or 10 μm trametinib in combination with 10 μm MG-132 for 30 min, and stimulated with 20 ng/ml TNF-α for the indicated times. IκB phosphorylation was measured as described in the legend to Fig. 3B, and the phosphorylations of RSK and ERK1/2 were measured as described in C. The averages of the intensities of the immunoreactive signals of the indicated phosphoproteins at 90 min of three independent experiments are expressed as the mean ± S.D. in arbitrary units with p values (bottom). G, Caco2 cells were serum-starved for 3 h, treated with 10 μg/ml BI-D1870 and 10 μm MG-132 for 30 min, and stimulated with 20 ng/ml TNF-α for the indicated times. IκB phosphorylation was measured as described in the Fig. 3B legend. Three experiments performed independently yielded equivalent results. The averages of the intensities of the immunoreactive signals of phospho-IκB at 90 min of three independent experiments are expressed as the mean ± S.D. in arbitrary units with p values (right).
Molecular Mechanism for RSK Regulation by PLCϵ
RSK, activated through phosphorylation by ERK and PDK1 in the cytoplasm, was located in the cytoplasm and the nuclei, where it phosphorylates IκB and CREB, respectively (26–31), suggesting that blocking the nuclear translocation of RSK might enhance IκB phosphorylation. Thus, we examined whether PLCϵ affects the subcellular localization of RSK. In control Caco2 cells, the amount of the phosphorylated RSK located in the cytoplasm was increased in 90 min after TNF-α stimulation (Fig. 7A). Notably, transfection of the two distinct PLCϵ siRNAs markedly reduced the amount of the phosphorylated RSK in the cytoplasm at 90 min, which was accompanied by its increase in the nuclei. Likewise, treatment with CID755673 or CID2011756 also resulted in disappearance from the cytoplasm and nuclear accumulation of the phosphorylated RSK at 90 min (Fig. 7B). Furthermore, overexpression of PLCϵ, but not the lipase-dead mutant, PLCϵΔX, increased the amount of the phosphorylated RSK located in the cytoplasm, decreased the amount of IκB in the cytoplasm, and increased the nuclear translocation of p65 upon stimulation by a low dose of LPA (Fig. 7C), suggesting that the lipase activity of PLCϵ was involved. These phenomena were suppressed by treatment with CID755673. These results taken together indicated that activation of the PLCϵ-PKD axis causes preferential localization of the phosphorylated RSK in the cytoplasm, leading to enhancement of the IκB phosphorylation and nuclear translocation of p65.
FIGURE 7.

Roles of PLCϵ and PKD in cytoplasmic localization of phosphorylated RSK. A, Caco2 cells transfected with the indicated siRNAs were cultured for 38–42 h, serum-starved for 3 h, and stimulated with 20 ng/ml TNF-α for the indicated times. The cell lysates were subjected to subcellular fractionation, and the resulting nuclear and cytoplasmic fractions were subjected to immunoblotting with the anti-phospho-RSK Ab. TBP and α-tubulin were used as markers for the nuclear and the cytoplasmic fractions, respectively. The averages of the intensities of the immunoreactive signals of phospho-RSK in the cytoplasmic and nuclear fractions at 90 min of three independent experiments are expressed as the mean ± S.D. (error bars) in arbitrary units (AU) with p values (bottom). B, Caco2 cells were serum-starved for 3 h, pretreated with 10 μg/ml CID755673 or 50 μm CID2011756 for 30 min, and stimulated with 20 ng/ml TNF-α for the indicated times. Subcellular localization of phosphorylated RSK was examined as described in A. The averages of the intensities of the immunoreactive signals of phospho-RSK in the cytoplasmic and nuclear fractions at 90 min of three independent experiments are expressed as the mean ± S.D. in arbitrary units with p values (bottom). C, Caco2 cells were transfected with pFLAG-CMV2-PLCϵ or pFLAG-CMV2-PLCϵΔX and cultured for 14–18 h. Subsequently, the cells were serum-starved for 14–18 h, treated with or without 10 μg/ml CID755673 for 15 min, and stimulated with 20 μm LPA for 30 min. Subcellular localization of phosphorylated RSK was examined as described in A. Subcellular localization of p65 was measured as described in the legend to Fig. 3A. IκB was detected by immunoblotting with the anti-IκB Ab, and its levels are shown as -fold changes over those in the control cells. The averages of the intensities of the immunoreactive signals of phospho-RSK and IκB in the cytoplasmic fractions and p65 in the nuclear fractions obtained from LPA-stimulated cells of three independent experiments are expressed as the means ± S.D. in arbitrary units with p values (right). D, Caco2 cells were co-transfected with pFLAG-CMV2-PLCϵ and pCMV-HA-PEA15, treated with or without CID755673, and stimulated with LPA as described in C. The cell lysates were immunoprecipitated (IP) with the anti-HA Ab and protein G-Sepharose. The resulting immunoprecipitates were subjected to immunoblotting with anti-phospho-RSK, anti-RSK, and anti-HA Abs. The numbers below the immunoblots show -fold changes of the levels of phosphorylated RSK and total RSK over those in the control cells. The averages of the intensities of the immunoreactive signals of phospho-RSK and total RSK of the immunoprecipitates obtained from LPA-stimulated cells of three independent experiments are expressed as the mean ± S.D. in arbitrary units with p values (right). E, phosphorylation of PEA15 at Ser-104 was detected in cells described in D by immunoblotting with anti-phospho-PEA15 and anti-PEA15 antibodies. The numbers below the immunoblots indicate -fold changes of the phospho-PEA15 signals divided by the total PEA15 signals over that in the control cells. The averages of the intensities of the immunoreactive signals of phospho-PEA15 obtained from LPA-stimulated cells of three independent experiments are expressed as the means ± S.D. in arbitrary units with p values (right). F and G, cells were transfected with either pCMV-HA-PEA15, pCMV-HA-PEA15S104A, or pCMV-HA-PEA15S104D in combination with pFLAG-CMV2-PLCϵ and stimulated with LPA as described in C. F, immunoprecipitation with the anti-HA antibody and immunoblotting for detection of phosphorylated RSK, total RSK, and HA-PEA15 were performed as described in D. G, subcellular localization of phosphorylated RSK was examined as described in A. The averages of the intensities of the immunoreactive signals of phospho-RSK and total RSK in the immunoprecipitates (F) and phospho-RSK in the cytoplasmic fractions (G) of three independent experiments are expressed as the mean ± S.D. in arbitrary units with p values (bottom). Three experiments performed independently yielded equivalent results.
We next analyzed the molecular mechanism by which PLCϵ causes preferential cytoplasmic localization of RSK. We focused on PEA15, a scaffold protein for RSK in the cytoplasm, which had been reported to regulate subcellular localization of RSK, depending on the phosphorylation state (32, 33). RSK could be co-immunoprecipitated with PEA15 in Caco2 cells stimulated with LPA, which was markedly enhanced by PLCϵ overexpression (Fig. 7D). The association of RSK with PEA15 seemed independent of its Ser-380 phosphorylation. Moreover, LPA stimulation elevated the phosphorylation of PEA15 at Ser-104, a putative PKC target site implicated in enhancing the cytoplasmic localization of RSK, which was also enhanced by PLCϵ overexpression (33) (Fig. 7E). In either case, the phenomenon was effectively inhibited by treatment with CID755673. These results suggested that the Ser-104 phosphorylation of PEA15, induced by the activated PLCϵ-PKD axis, might enhance its association with the phosphorylated RSK and facilitate its cytoplasmic localization. To test this hypothesis, we analyzed the effects of the mutations, S104A and S104D, of PEA15. The phosphorylation-mimicking mutant S104D efficiently associated with RSK, including the phosphorylated one, even in the absence of LPA stimulation, whereas the S104A mutant failed to associate with RSK (Fig. 7F). Moreover, overexpression of the S104D mutant enhanced the cytoplasmic localization of the phosphorylated RSK in the absence of LPA stimulation, whereas that of the S104A mutant showed inhibition of the LPA-induced cytoplasmic localization (Fig. 7G). These results further supported the crucial role of the Ser-104 phosphorylation of PEA15, mediated by the PLCϵ-PKD axis, in holding the phosphorylated RSK in the cytoplasm.
Discussion
In this study, we have shown that PLCϵ augments inflammatory reactions by facilitating the proinflammatory cytokine expression in the colon administered with DSS. By employing the colitis-induced colorectal carcinogenesis model, we obtained further support for our hypothesis that this proinflammatory function of PLCϵ is responsible for its crucial role in facilitating tumor promotion and malignant progression. PLCϵΔX/ΔX mice showed markedly attenuated responses to DSS administration, represented by reduced expression of the proinflammatory molecules, such as TNF-α, COX-2, CXCL1, CXCL2, and CCL2 (Fig. 1D), all of which had been implicated in the pathogenesis of DSS-induced colitis and human inflammatory bowel diseases (42, 43, 51–58). Although this result indicated the involvement of PLCϵ in augmenting the expression of the proinflammatory molecules, it was difficult to distinguish whether the observed increase of the proinflammatory molecules was the direct consequence of the PLCϵ activation or the secondary phenomenon accompanying the infiltration of immune inflammatory cells. In this regard, it is of particular interest that CXCR2 ligands, CXCL1 and CXCL2, and a CCR2 ligand, CCL2, were overexpressed in the colon epithelial cells of DSS-administered PLCϵ+/+ mice (Fig. 1, E and F). Because PLCϵ is not expressed in immune cells (8), it was likely that the epithelial expression of these chemokines represents one of the direct consequences of the PLCϵ activation. Because mice carrying knock-out of CXCR2 or CCR2 had been shown to become less susceptible to DSS-induced colitis (42, 43, 51), these results suggested a mechanism whereby PLCϵ might augment colon inflammation by facilitating the production from the colon epithelial cells of proinflammatory chemokines, such as CXCL2 and CCL2, which recruit neutrophils and macrophages, respectively, to the inflamed sites.
By taking our results obtained with the human colon epithelial cell line Caco2 together, we propose the following model for the molecular mechanism by which PLCϵ augments the expression of proinflammatory molecules (Fig. 8). TNF-α stimulation induces the early phase of NF-κB activation through phosphorylation and activation of IκB by IKK, leading to the expression of the inflammation-associated genes, such as CXCL1, CXCL8, and COX-2, observed in 1 h (Fig. 2, A and C). Although this phase of the inflammation-associated gene expression seems to be partially abrogated by PLCϵ knockdown, we presently have no mechanistic explanation except that the PKD-RSK-IκB-NF-κB pathway is not involved. Alternatively, the gene expression observed at the 1-h time point might be significantly contributed by that in the late phase, which will be explained below. In parallel with the early phase of the NF-κB activation, TNF-α stimulation enhances the production and secretion of LPA, which induces activation of PLCϵ through engagement of its receptors in an autocrine manner. PLCϵ produces diacylglycerol and thereby induces PKC activation, leading to the activation of PKD through direct binding of diacylglycerol to its C1 domain and phosphorylation by PKC (59). Subsequently, the activated PKD induces phosphorylation of PEA15 at Ser-104. The phosphorylated PEA15 binds to RSK activated through phosphorylation by ERK, which is also activated by the LPA receptor engagement (Fig. 6, C–E). The activated RSK, held in the cytoplasm by association with the phosphorylated PEA15, phosphorylates IκB and thereby causes the late phase of the TNF-α-induced NF-κB activation, leading to the expression of CXCL8, COX-2, CCL2, and CCL20. CXCL8, whose counterpart is nonexistent in mice, is a ligand of CXCR2, causes neutrophil recruitment, and hence is regarded as a functional homologue of mouse CXCL1 and CXCL2 (60–62). Thus, the PLCϵ-dependent induction of CXCL8 observed in Caco2 cells may functionally substitute for CXCL2, which is induced in mouse colon epithelial cells in a PLCϵ-dependent manner after DSS administration. The delayed kinetics of the PLCϵ-dependent cytokine expression in response to TNF-α stimulation was observed before in dermal fibroblasts and epidermal keratinocytes, which led us to propose that unknown humoral factors might mediate activation of PLCϵ in an autocrine manner (8). This is consistent with our present observation. The results taken together indicated that the differential actions of IKK and RSK on IκB phosphorylation account for the sustained NF-κB activation and the sustained expression of proinflammatory molecules, which seem to be involved in augmentation of inflammatory reactions and promotion of inflammation-associated carcinogenesis. Several questions, including the following, remain to be addressed experimentally: whether TNF-α stimulation of cells really induces the increased production of LPA as reported (45), by what mechanism PLCϵ is activated downstream of the LPA receptors, and what kinase is directly responsible for the Ser-104 phosphorylation of PEA15.
FIGURE 8.
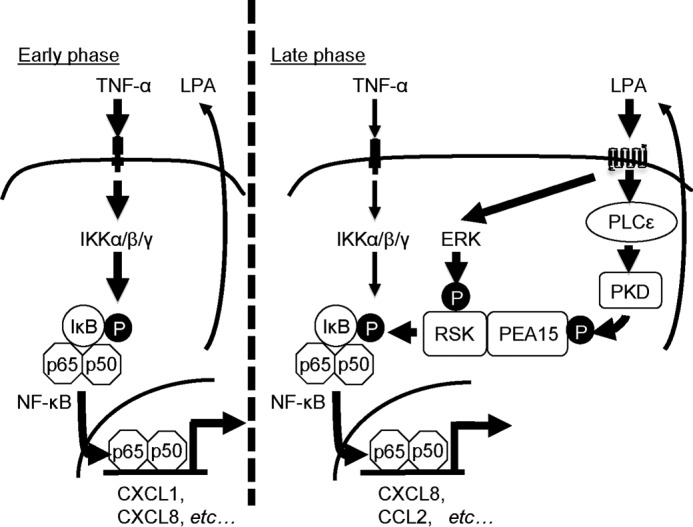
A model for TNF-α-induced expression of the inflammation-associated genes mediated by PLCϵ. In Caco2 cells, TNF-α stimulation induces the early phase of NF-κB activation via the canonical NF-κB pathway involving IKK, leading to the expression of the inflammation-associated genes (left). Concomitantly, it induces the late phase of NF-κB activation via enhancing the production and secretion of LPA, which activates the PLCϵ-PKD axis in an autocrine manner and leads to phosphorylation of PEA15. The phosphorylated PEA15 binds to the activated RSK and holds it in the cytoplasm, thereby inducing IκB phosphorylation.
The results of our present study may give a new mechanistic insight into not only the pathogenesis of human inflammatory bowel diseases and colorectal carcinogenesis but also of other inflammatory diseases and inflammation-associated carcinogenesis. Moreover, our study suggests that PLCϵ may become a good candidate target for the development of anti-inflammatory and cancer-preventing drugs.
Author Contributions
M. W., H. E., and T. K. designed the study and analyzed the data. M. W. conducted most of the experiments. M. L. and A. E. conducted a part of the experiments. S. K. conducted the histopathological classification. T. K. conceived the idea for the project and wrote the paper with M. W. All of the authors reviewed the results and approved the final version of the manuscript.
Acknowledgments
We thank all of the members of our laboratory for helpful suggestions and discussions.
This work was supported by JSPS KAKENHI Grants 23390071 and 24590379 and in part by Global COE Program A08 and the Project for Development of Innovative Research on Cancer Therapeutics from MEXT. The authors declare that they have no conflicts of interest with the contents of this article.
- PLC
- phospholipase C
- LPA
- lysophosphatidic acid
- S1P
- sphingosine-1-phosphate
- NF-κB
- nuclear factor-κB
- IKK
- IκB kinase
- RSK
- ribosomal S6 kinase
- CK2
- casein kinase 2
- CREB
- cyclic AMP-response element-binding protein
- TBP
- TATA-binding protein
- qRT-PCR
- quantitative RT-PCR
- DSS
- dextran sodium sulfate
- MPO
- myeloperoxidase
- Ab
- antibody.
References
- 1.Suh P. G., Park J. I., Manzoli L., Cocco L., Peak J. C., Katan M., Fukami K., Kataoka T., Yun S., and Ryu S. H. (2008) Multiple roles of phosphoinositide-specific phospholipase C isozymes. BMB Rep. 41, 415–434 [DOI] [PubMed] [Google Scholar]
- 2.Song C., Hu C. D., Masago M., Kariyai K., Yamawaki-Kataoka Y., Shibatohge M., Wu D., Satoh T., and Kataoka T. (2001) Regulation of a novel human phospholipase C, PLCϵ, through membrane targeting by Ras. J. Biol. Chem. 276, 2752–2757 [DOI] [PubMed] [Google Scholar]
- 3.Kelley G. G., Reks S. E., Ondrako J. M., and Smrcka A. V. (2001) Phospholipase Cϵ: a novel Ras effector. EMBO J. 20, 743–754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kelley G. G., Reks S. E., and Smrcka A. V. (2004) Hormonal regulation of phospholipase Cϵ through distinct and overlapping pathways involving G12 and Ras family G-proteins. Biochem. J. 378, 129–139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jin T. G., Satoh T., Liao Y., Song C., Gao X., Kariya K., Hu C. D., and Kataoka T. (2001) Role of the CDC25 homology domain of phospholipase Cϵ in amplification of Rap1-dependent signaling. J. Biol. Chem. 276, 30301–30307 [DOI] [PubMed] [Google Scholar]
- 6.Seifert J. P., Wing M. R., Snyder J. T., Gershburg S., Sondek J., and Harden T. K. (2004) RhoA activates purified phospholipase C-ϵ by a guanine nucleotide-dependent mechanism. J. Biol. Chem. 279, 47992–47997 [DOI] [PubMed] [Google Scholar]
- 7.Hains M. D., Wing M. R., Maddileti S., Siderovski D. P., and Harden T. K. (2006) Gα12/13- and Rho-dependent activation of phospholipase C-ϵ by lysophosphatidic acid and thrombin receptors. Mol. Pharmacol. 69, 2068–2075 [DOI] [PubMed] [Google Scholar]
- 8.Hu L., Edamatsu H., Takenaka N., Ikuta S., and Kataoka T. (2010) Crucial role of phospholipase Cϵ in induction of local skin inflammatory reactions in the elicitation stage of allergic contact hypersensitivity. J. Immunol. 184, 993–1002 [DOI] [PubMed] [Google Scholar]
- 9.Ikuta S., Edamatsu H., Li M., Hu L., and Kataoka T. (2008) Crucial role of phospholipase Cϵ in skin inflammation induced by tumor-promoting phorbol ester. Cancer Res. 68, 64–72 [DOI] [PubMed] [Google Scholar]
- 10.Oka M., Edamatsu H., Kunisada M., Hu L., Takenaka N., Sakaguchi M., Kataoka T., and Nishigori C. (2011) Phospholipase Cϵ has a crucial role in ultraviolet B-induced neutrophil-associated skin inflammation by regulating the expression of CXCL1/KC. Lab. Invest. 91, 711–718 [DOI] [PubMed] [Google Scholar]
- 11.Nagano T., Edamatsu H., Kobayashi K., Takenaka N., Yamamoto M., Sasaki N., Nishimura Y., and Kataoka T. (2014) Phospholipase Cϵ, an effector of Ras and Rap small GTPases, is required for airway inflammatory response in a mouse model of bronchial asthma. PLoS One 9, e108373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Takenaka N., Edamatsu H., Suzuki N., Saito H., Inoue Y., Oka M., Hu L., and Kataoka T. (2011) Overexpression of phospholipase Cϵ in keratinocytes upregulates cytokine expression and causes dermatitis with acanthosis and T-cell infiltration. Eur. J. Immunol. 41, 202–213 [DOI] [PubMed] [Google Scholar]
- 13.Bai Y., Edamatsu H., Maeda S., Saito H., Suzuki N., Satoh T., and Kataoka T. (2004) Crucial role of phospholipase Cϵ in chemical carcinogen-induced skin tumor development. Cancer Res. 64, 8808–8810 [DOI] [PubMed] [Google Scholar]
- 14.Li M., Edamatsu H., Kitazawa R., Kitazawa S., and Kataoka T. (2009) Phospholipase Cϵ promotes intestinal tumorigenesis of ApcMin/+ mice through augmentation of inflammation and angiogenesis. Carcinogenesis 30, 1424–1432 [DOI] [PubMed] [Google Scholar]
- 15.Abnet C. C., Freedman N. D., Hu N., Wang Z., Yu K., Shu X. O., Yuan J. M., Zheng W., Dawsey S. M., Dong L. M., Lee M. P., Ding T., Qiao Y. L., Gao Y. T., Koh W. P., et al. (2010) A shared susceptibility locus in PLCE1 at 10q23 for gastric adenocarcinoma and esophageal squamous cell carcinoma. Nat. Genet. 42, 764–767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang L. D., Zhou F. Y., Li X. M., Sun L. D., Song X., Jin Y., Li J. M., Kong G. Q., Qi H., Cui J., Zhang L. Q., Yang J. Z., Li J. L., Li X. C., Ren J. L., et al. (2010) Genome-wide association study of esophageal squamous cell carcinoma in Chinese subjects identifies susceptibility loci at PLCE1 and C20orf54. Nat. Genet. 42, 759–763 [DOI] [PubMed] [Google Scholar]
- 17.Harada Y., Edamatsu H., and Kataoka T. (2011) PLCϵ cooperates with the NF-κB pathway to augment TNFα-stimulated CCL2/MCP1 expression in human keratinocyte. Biochem. Biophys. Res. Commun. 414, 106–111 [DOI] [PubMed] [Google Scholar]
- 18.Dusaban S. S., Purcell N. H., Rockenstein E., Masliah E., Cho M. K., Smrcka A. V., and Brown J. H. (2013) Phospholipase Cϵ links G protein-coupled receptor activation to inflammatory astrocytic responses. Proc. Natl. Acad. Sci. U.S.A. 110, 3609–3614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chiu T. T., Leung W. Y., Moyer M. P., Strieter R. M., and Rozengurt E. (2007) Protein kinase D2 mediates lysophosphatidic acid-induced interleukin 8 production in nontransformed human colonic epithelial cells through NF-κB. Am. J. Physiol. Cell Physiol. 292, C767–C777 [DOI] [PubMed] [Google Scholar]
- 20.Storz P., and Toker A. (2003) Protein kinase D mediates a stress-induced NF-κB activation and survival pathway. EMBO J. 22, 109–120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang L., Malik S., Pang J., Wang H., Park K. M., Yule D. I., Blaxall B. C., and Smrcka A. V. (2013) Phospholipase Cϵ hydrolyzes perinuclear phosphatidylinositol 4-phosphate to regulate cardiac hypertrophy. Cell 153, 216–227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dusaban S. S., Kunkel M. T., Smrcka A. V., and Brown J. H. (2015) Thrombin promotes sustained signaling and inflammatory gene expression through the CDC25 and Ras-associating domains of phospholipase Cϵ. J. Biol. Chem. 290, 26776–26783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wullaert A., Bonnet M. C., and Pasparakis M. (2011) NF-κB in the regulation of epithelial homeostasis and inflammation. Cell Res. 21, 146–158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pasparakis M. (2012) Role of NF-κB in epithelial biology. Immunol. Rev. 246, 346–358 [DOI] [PubMed] [Google Scholar]
- 25.Mercurio F., Zhu H., Murray B. W., Shevchenko A., Bennett B. L., Li J., Young D. B., Barbosa M., Mann M., Manning A., and Rao A. (1997) IKK-1 and IKK-2: cytokine-activated IκB kinases essential for NF-κB activation. Science 278, 860–866 [DOI] [PubMed] [Google Scholar]
- 26.Schouten G. J., Vertegaal A. C., Whiteside S. T., Israël A., Toebes M., Dorsman J. C., van der Eb A. J., and Zantema A. (1997) IκBα is a target for the mitogen-activated 90 kDa ribosomal S6 kinase. EMBO J. 16, 3133–3144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Peng C., Cho Y. Y., Zhu F., Xu Y. M., Wen W., Ma W. Y., Bode A. M., and Dong Z. (2010) RSK2 mediates NF-κB activity through the phosphorylation of IκBα in the TNF-R1 pathway. FASEB J. 24, 3490–3509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Taylor J. A., Bren G. D., Pennington K. N., Trushin S. A., Asin S., and Paya C. V. (1999) Serine 32 and serine 36 of IκBα are directly phosphorylated by protein kinase CKII in vitro. J. Mol. Biol. 290, 839–850 [DOI] [PubMed] [Google Scholar]
- 29.Anjum R., and Blenis J. (2008) The RSK family of kinases: emerging roles in cellular signalling. Nat. Rev. Mol. Cell Biol. 9, 747–758 [DOI] [PubMed] [Google Scholar]
- 30.Chen R. H., Abate C., and Blenis J. (1993) Phosphorylation of the c-Fos transrepression domain by mitogen-activated protein kinase and 90-kDa ribosomal S6 kinase. Proc. Natl. Acad. Sci. U.S.A. 90, 10952–10956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Xing J., Ginty D. D., and Greenberg M. E. (1996) Coupling of the RAS-MAPK pathway to gene activation by RSK2, a growth factor-regulated CREB kinase. Science 273, 959–963 [DOI] [PubMed] [Google Scholar]
- 32.Vaidyanathan H., and Ramos J. W. (2003) RSK2 activity is regulated by its interaction with PEA-15. J. Biol. Chem. 278, 32367–32372 [DOI] [PubMed] [Google Scholar]
- 33.Vaidyanathan H., Opoku-Ansah J., Pastorino S., Renganathan H., Matter M. L., and Ramos J. W. (2007) ERK MAP kinase is targeted to RSK2 by the phosphoprotein PEA-15. Proc. Natl. Acad. Sci. U.S.A. 104, 19837–19842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wu D., Tadano M., Edamatsu H., Masago-Toda M., Yamawaki-Kataoka Y., Terashima T., Mizoguchi A., Minami Y., Satoh T., and Kataoka T. (2003) Neuronal lineage-specific induction of phospholipase Cϵ expression in the developing mouse brain. Eur. J. Neurosci. 17, 1571–1580 [DOI] [PubMed] [Google Scholar]
- 35.Kelley G. G., Kaproth-Joslin K. A., Reks S. E., Smrcka A. V., and Wojcikiewicz R. J. (2006) G-protein-coupled receptor agonists activate endogenous phospholipase Cϵ and phospholipase Cβ3 in a temporally distinct manner. J. Biol. Chem. 281, 2639–2648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhang L., Li N., Caicedo R., and Neu J. (2005) Alive and dead Lactobacillus rhamnosus GG decrease tumor necrosis factor-α-induced interleukin-8 production in Caco-2 cells. Gastroenterology 135, 1752–1756 [DOI] [PubMed] [Google Scholar]
- 37.Tadano M., Edamatsu H., Minamisawa S., Yokoyama U., Ishikawa Y., Suzuki N., Saito H., Wu D., Masago-Toda M., Yamawaki-Kataoka Y., Setsu T., Terashima T., Maeda S., Satoh T., and Kataoka T. (2005) Congenital semilunar valvulogenesis defect in mice deficient in phospholipase Cϵ. Mol. Cell. Biol. 25, 2191–2199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cooper H. S., Everley L., Chang W. C., Pfeiffer G., Lee B., Murthy S., and Clapper M. L. (2001) The role of mutant Apc in the development of dysplasia and cancer in the mouse model of dextran sulfate sodium-induced colitis. Gastroenterology 121, 1407–1416 [DOI] [PubMed] [Google Scholar]
- 39.Perše M., and Cerar A. (2012) Dextran sodium sulphate colitis mouse model: traps and tricks. J. Biomed. Biotechnol. 2012, 718617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cicalese L., Caraceni P., Nalesnik M. A., Borle A. B., and Schraut W. H. (1996) Oxygen free radical content and neutrophil infiltration are important determinants in mucosal injury after rat small bowel transplantation. Transplantation 62, 161–166 [DOI] [PubMed] [Google Scholar]
- 41.Boivin G. P., Washington K., Yang K., Ward J. M., Pretlow T. P., Russell R., Besselsen D. G., Godfrey V. L., Doetschman T., Dove W. F., Pitot H. C., Halberg R. B., Itzkowitz S. H., Groden J., and Coffey R. J. (2003) Pathology of mouse models of intestinal cancer: consensus report and recommendations. Gastroenterology 124, 762–777 [DOI] [PubMed] [Google Scholar]
- 42.Farooq S. M., Stillie R., Svensson M., Svanborg C., Strieter R. M., and Stadnyk A. W. (2009) Therapeutic effect of blocking CXCR2 on neutrophil recruitment and dextran sodium sulfate-induced colitis. J. Pharmacol. Exp. Ther. 329, 123–129 [DOI] [PubMed] [Google Scholar]
- 43.Platt A. M., Bain C. C., Bordon Y., Sester D. P., and Mowat A. M. (2010) An independent subset of TLR expressing CCR2-dependent macrophages promotes colonic inflammation. J. Immunol. 184, 6843–6854 [DOI] [PubMed] [Google Scholar]
- 44.Banno T., Gazel A., and Blumenberg M. (2005) Pathway-specific profiling identifies the NF-κB-dependent tumor necrosis factor α-regulated genes in epidermal keratinocytes. J. Biol. Chem. 280, 18973–18980 [DOI] [PubMed] [Google Scholar]
- 45.Wu J. M., Xu Y., Skill N. J., Sheng H., Zhao Z., Yu M., Saxena R., and Maluccio M. A. (2010) Autotaxin expression and its connection with the TNF-α-NF-κB axis in human hepatocellular carcinoma. Mol. Cancer 9, 71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Osawa Y., Banno Y., Nagaki M., Brenner D. A., Naiki T., Nozawa Y., Nakashima S., and Moriwaki H. (2001) TNF-α-induced sphingosine 1-phosphate inhibits apoptosis through a phosphatidylinositol 3-kinase/Akt pathway in human hepatocytes. J. Immunol. 167, 173–180 [DOI] [PubMed] [Google Scholar]
- 47.Ling L., Cao Z., and Goeddel D. V. (1998) NF-κB-inducing kinase activates IKK-α by phosphorylation of Ser-176. Proc. Natl. Acad. Sci. U.S.A. 95, 3792–3807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Delhase M., Hayakawa M., Chen Y., and Karin M. (1999) Positive and negative regulation of IκB kinase activity through IKKβ subunit phosphorylation. Science 284, 309–313 [DOI] [PubMed] [Google Scholar]
- 49.Bain J., Plater L., Elliott M., Shpiro N., Hastie C. J., McLauchlan H., Klevernic I., Arthur J. S., Alessi D. R., and Cohen P. (2007) The selectivity of protein kinase inhibitors: a further update. Biochem. J. 408, 297–315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yamaguchi T., Kakefuda R., Tajima N., Sowa Y., and Sakai T. (2011) Antitumor activities of JTP-74057 (GSK1120212), a novel MEK1/2 inhibitor, on colorectal cancer cell lines in vitro and in vivo. Int. J. Oncol. 39, 23–31 [DOI] [PubMed] [Google Scholar]
- 51.Jamieson T., Clarke M., Steele C. W., Samuel M. S., Neumann J., Jung A., Huels D., Olson M. F., Das S., Nibbs R. J., and Sansom O. J. (2012) Inhibition of CXCR2 profoundly suppresses inflammation-driven and spontaneous tumorigenesis. J. Clin. Invest. 122, 3127–3144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kojouharoff G., Hans W., Obermeier F., Männel D. N., Andus T., Schölmerich J., Gross V., and Falk W. (1997) Neutralization of tumour necrosis factor (TNF) but not of IL-1 reduces inflammation in chronic dextran sulphate sodium-induced colitis in mice. Clin. Exp. Immunol. 107, 353–358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Noguchi M, Hiwatashi N, Liu Z, and Toyota T. (1998) Secretion imbalance between tumour necrosis factor and its inhibitor in inflammatory bowel disease. Gut 43, 203–209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sands B. E., Anderson F. H., Bernstein C. N., Chey W. Y., Feagan B. G., Fedorak R. N., Kamm M. A., Korzenik J. R., Lashner B. A., Onken J. E., Rachmilewitz D., Rutgeerts P., Wild G., Wolf D. C., Marsters P. A., Travers S. B., Blank M. A., and van Deventer S. J. (2004) Infliximab maintenance therapy for fistulizing Crohn's disease. N. Engl. J. Med. 350, 876–885 [DOI] [PubMed] [Google Scholar]
- 55.Rutgeerts P., Sandborn W. J., Feagan B. G., Reinisch W., Olson A., Johanns J., Travers S., Rachmilewitz D., Hanauer S. B., Lichtenstein G. R., de Villiers W.J., Present D., Sands B. E., and Colombel J. F. (2005) Infliximab for induction and maintenance therapy for ulcerative colitis. N. Engl. J. Med. 353, 2462–2476 [DOI] [PubMed] [Google Scholar]
- 56.Ajuebor M. N., and Swain M. G. (2002) Role of chemokines and chemokine receptors in the gastrointestinal tract. Immunology 105, 137–143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ishikawa T. O., Oshima M., and Herschman H. R. (2011) Cox-2 deletion in myeloid and endothelial cells, but not in epithelial cells, exacerbates murine colitis. Caricinogenesis 32, 417–426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Dudhgaonkar S. P., Tandan S. K., Kumar D., Raviprakash V., and Kataria M. (2007) Influence of simultaneous inhibition of cyclooxygenase-2 and inducible nitric oxide synthase in experimental colitis in rats. Inflammopharmacology 15, 188–195 [DOI] [PubMed] [Google Scholar]
- 59.Fu Y., and Rubin C. S. (2011) Protein kinase D: coupling extracellular stimuli to the regulation of cell physiology. EMBO Rep. 12, 785–796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Cacalano G., Lee J., Kikly K., Ryan A. M., Pitts-Meek S., Hultgren B., Wood W. I., and Moore M. W. (1994) Neutrophil and B cell expansion in mice that lack the murine IL-8 receptor homolog. Science 265, 682–684 [DOI] [PubMed] [Google Scholar]
- 61.Griffith J. W., Sokol C. L., and Luster A. D. (2014) Chemokines and chemokine receptors: positioning cells for host defence and immunity. Annu. Rev. Immunol. 32, 659–702 [DOI] [PubMed] [Google Scholar]
- 62.Kucharzik T., Hudson J. T. 3rd, Lügering A., Abbas J. A., Bettini M., Lake J. G., Evans M. E., Ziegler T. R., Merlin D., Madara J. L., and Williams I. R. (2005) Acute induction of human IL-8 production by intestinal epithelium triggers neutrophil infiltration without mucosal injury. Gut 54, 1565–1572 [DOI] [PMC free article] [PubMed] [Google Scholar]