

Abstract
Copper-dependent lytic polysaccharide monooxygenases (LPMOs) are enzymes that oxidatively deconstruct polysaccharides. The active site copper in LPMOs is coordinated by a histidine-brace. This utilizes the amino group and side chain of the N-terminal His residue with the side chain of a second His residue to create a T-shaped arrangement of nitrogen ligands. We report a structural, kinetic, and thermodynamic appraisal of copper binding to the histidine-brace in an auxiliary activity family 10 (AA10) LPMO from Streptomyces lividans (SliLPMO10E). Unexpectedly, we discovered the existence of two apo-SliLPMO10E species in solution that can each bind copper at a single site with distinct kinetic and thermodynamic (exothermic and endothermic) properties. The experimental EPR spectrum of copper-bound SliLPMO10E requires the simulation of two different line shapes, implying two different copper-bound species, indicative of three and two nitrogen ligands coordinating the copper. Amino group coordination was probed through the creation of an N-terminal extension variant (SliLPMO10E-Ext). The kinetics and thermodynamics of copper binding to SliLPMO10E-Ext are in accord with copper binding to one of the apo-forms in the wild-type protein, suggesting that amino group coordination is absent in the two-nitrogen coordinate form of SliLPMO10E. Copper binding to SliLPMO10B was also investigated, and again it revealed the presence of two apo-forms with kinetics and stoichiometry of copper binding identical to that of SliLPMO10E. Our findings highlight that heterogeneity exists in the active site copper coordination sphere of LPMOs that may have implications for the mechanism of loading copper in the cell.
Keywords: copper monooxygenase, electron paramagnetic resonance (EPR), kinetics, thermodynamics, x-ray crystallography
Introduction
Lytic polysaccharide monooxygenases (LPMOs)3 are enzymes that utilize copper as their functional active site metal and act to enhance the depolymerization of recalcitrant polysaccharides in nature such as cellulose and chitin (1, 2). Identification and characterization of members of this cuproenzyme family from fungal and bacterial species have gathered pace in recent years due in part to their promising uses in biorefinery applications of biomass to derive second generation biofuels (2, 3). LPMOs are classed into auxiliary activity (AA) families in the CAZy (carbohydrate active enzyme) database with four AA families, AA9, AA10, AA11, and AA13 identified so far (4). AA9 (formerly GH61) and AA10 (formerly CBM33) families have been the most extensively studied, with AA9 members having activity for cellulose, hemicellulose, and cellodextrin substrates (5–8) and AA10 members having activity with either cellulose or chitin substrates (9, 10). The AA11 and AA13 families are the most recently discovered, and so far have been determined to have activity with chitin and starch substrates, respectively (11, 12). All LPMOs coordinate a single copper ion by three nitrogen ligands to from the active site. Two of these are provided from the amino group (Nt) and side chain of the N-terminal His with the third from a second His side chain to give a T-shaped copper coordination geometry referred to as the His-brace (6). To initiate enzymatic activity, an electron source is utilized to reduce Cu(II) to Cu(I), with the latter required to activate dioxygen (13, 14). Two principal mechanisms of glycosidic bond cleavage have been proposed (15, 16) with the resulting C1 or C4 oxidation of a sugar ring (6, 8, 15, 17–19) leading to chain breaks that are susceptible to hydrolases for further degradation.
Streptomycetes are the dominant bacterial genus responsible for aerobic biomass decomposition in soil environments and considered vital players in the decomposition of cellulose and other biomass polymers (20–22). Streptomycetes genomes encode a relatively high percentage of genes for putative celluloytic and chitinolytic enzymes. For example, the secretome of Streptomyces sp. SirexAA-E (ActE) has been analyzed through growth on a variety of carbon sources, including cellulose and chitin (23). Streptomyces coelicolor and Streptomyces griseus have comparable putative genomic compositions of CAZy proteins to ActE; however, these two streptomycetes are less capable of growing on cellulose (23). Thus, not all streptomycetes, despite their abilities to grow on plant polysaccharides (21), are efficient cellulose decomposers.
In liquid-grown cultures, the growth of many streptomycetes is characterized by the formation of large biofilm-like aggregates, called pellets (24, 25). The competence of streptomycetes to directly secrete enzymes into the culture broth makes them attractive hosts for the heterologous production of commercially valuable enzymes (26, 27). However, the pellets suffer from poor efficiency in oxygen and nutrient uptake and thus compromise the overall efficiency of the fermentation process. The synthesis of extracytoplasmic glycans is important for pellet formation (28–31). In Streptomyces lividans enzymes encoded by the cslA-glxA locus produce a β-(1,4)-glycan at the hyphal tip, of which the chemical composition is unknown (28–30). CslA belongs to the glycosyltransferase family 2, which includes cellulose and chitin synthases, among others (32). Deletion of cslA abolishes pellet formation in liquid-grown cultures and also blocks the formation of reproductive aerial hyphae on solid medium (28, 29). GlxA is a newly characterized member of the AA5 family of mono-copper oxidase possessing a Tyr-Cys cross-linked redox cofactor (30). A glxA null mutant blocks development and abolishes pellet formation coinciding with the loss of glycan deposition at hyphal tips, the same phenotype as observed with the cslA deletion (30, 33). This is consistent with a model whereby CslA and GlxA cooperatively function in glycan deposition at the hyphal tip and are jointly responsible for the formation of reproductive aerial structures and, in liquid environments, for the formation of pellets (30).
Recent studies in S. lividans have identified an extracellular copper-trafficking pathway involving two copper chaperone proteins (Sco and ECuC) (34, 35) and a DyP-type heme peroxidase (DtpA) (36) that are all required to facilitate GlxA maturation (i.e. copper-loading and subsequent Tyr-Cys cross-link formation (30)). Based on these studies, a model has been developed that proposes the presence of a large protein complex at the hyphal tip for synthesis and deposition of the glycan required for morphogenesis (36). Downstream of glxA is the cslZ gene, encoding for an endoglucanase, and upstream of cslA is a gene (SLI3182) predicted to encode for an AA10 LPMO. The S. lividans genome has seven genes encoding putative AA10s (SliLPMO10A-G) (37). RNA-sequence data of S. lividans grown in a mannitol/glucose media reveal two slilpmo10 transcripts to be constitutively expressed (slilpmo10B and slilpmo10E), although the remaining five may be under transcriptional control of a repressor or activator and depend upon induction by a polysaccharide substrate (Table 1).
TABLE 1.
Transcription analysis of the seven lpmo genes in S. lividans
RNA sequence data obtained and reported earlier in liquid minimal media and glucose/mannitol as a carbon source (61) were analyzed with the S. lividans 1326 genome sequence as input (37).
| Gene | Protein | S. lividans 1326a (RPKM)b | S. lividans 1326 + Cua (RPKM)b |
|---|---|---|---|
| SLI0440 | SliLPMO10A | 0 | 0 |
| SLI0614 | SliLPMO10B | 10 | 9 |
| SLI1466 | SliLPMO10C | 0 | 6 |
| SLI2039 | SliLPMO10D | 0 | 2 |
| SLI3182 | SliLPMO10E | 22 | 27 |
| SLI6742 | SliLPMO10F | 0 | 0 |
| SLI7441 | SliLPMO10G | 0 | 3 |
a The transcriptomes of WT S. lividans 1326 were obtained without addition of copper to the growth medium. The copper-induced transcriptome was obtained after a 2-h exposure to 400 μm Cu(II).
b The selected expression measure is the RPKM and is defined as the reads/kb of exon/million mapped reads, i.e. dividing the total number of exon reads (in this case one exon per reference sequence) by the number of mapped reads (in millions) times the exon length (in kb) (62).
In this study, we have set out to establish the biochemical and structural properties of SliLPMO10E along with investigating the kinetic and thermodynamic properties of Cu(II) binding to the His-brace. Interestingly, our data are consistent with the His-brace of apo-SliLPMO10E existing in two non-equilibrating forms that can coordinate Cu(II) by three or two nitrogen ligands. Mixed Cu(II) complexes of equal stoichiometry are also inferred from EPR spectral simulations, with the parameters derived indicating that AA10s may not be as rhombic as originally thought but rather reflect a mixture of Cu(II) species.
Experimental Procedures
Cloning of LPMOs and Site-directed Mutagenesis
A pET26b plasmid (Novagen) was modified by QuikChange mutagenesis to relocate the NcoI site 6 nucleotides downstream. This modification enabled the overexpression of mature LPMOs correctly processed at the N-terminal His residue following cleavage of the pelB-leader sequence. SLI3182 (nucleotides 88–606) encoding for SliLPMO10E and SLI0614 (nucleotides 127–687) encoding for SliLPMO10B were amplified from S. lividans 1326 and cloned into the modified pET26b plasmid using the new NcoI and original HindIII restriction sites. Site-directed mutagenesis of His-187 to an Ala in SliLPMO10E was carried out by PCR using the wild-type (WT) construct with forward and reverse primers containing the desired nucleotide change. The N-terminal SliLPMO10E extension variant (SliLPMO10E-Ext) whereby His-30 is no longer the N-terminal residue was created by introducing a Gly residue prior to His-30 and cloning into an unmodified pET26b plasmid to give a mature protein with an N-terminal sequence of Met-Gly-His. All constructs were DNA sequenced to corroborate the mutations and insertions.
Overexpression and Purification of LPMOs
All proteins were overexpressed in Escherichia coli BL21 (RIL) cells starting from 37 °C overnight pre-cultures that were used to inoculate 500 ml of LB cultures in 2-liter flasks (37 °C, 180 rpm). At an A600 of 0.4, the temperature was lowered to 16 °C and isopropyl β-d-1-thiogalactopyranoside (Melford) was added to a final concentration of 1 mm on reaching an A600 of 0.6. Cells were harvested after 16 h at 3,501 × g for 20 min at 4 °C. Cell pellets were combined, weighed, and resuspended in 1/30th of the total culture volume in ice-cold 50 mm Tris/HCl, pH 8.0, 1 mm EDTA, and 20% w/v sucrose and stirred at 4 °C for 1 h. For every gram of cell pellet, 60 μl of 1 m MgSO4 was added, and stirring was continued for a further 30 min at 4 °C. The smooth cell suspension was then centrifuged at 38,724 × g for 20 min at 4 °C, and the supernatant was removed and stored at 4 °C. The pellet was then resuspended in ice-cold water (1/30th the volume of the culture), stirred at 4 °C for 1 h, followed by centrifugation at 38,724 × g for 20 min, with the supernatant collected and combined with that from the previous sucrose fractionation. The combined supernatants were then dialyzed overnight against 5 mm Tris/HCl, pH 7, 1 mm EDTA at 4 °C. For SliLPMO10E (pI 8.42) and variants, the dialysate was diluted with buffer A (5 mm Tris/HCl, pH 7) and loaded to a 5-ml HiTrap SP-column (GE Healthcare) equilibrated with buffer A and eluted with a linear salt gradient using buffer A containing 1 m NaCl. For SliLPMO10B (pI 4.24), the dialysate was diluted with buffer B (10 mm Tris/HCl, pH 5.5) and loaded to a pre-equilibrated DEAE column (GE Healthcare) and eluted with a linear salt gradient using buffer B containing 500 mm NaCl. Fractions containing LPMO were pooled and concentrated at 4 °C using a 5-kDa cutoff Centricon (Vivaspin). Concentrated protein was then loaded to a HiLoad 26/60 Superdex-75 column (GE Healthcare) equilibrated with buffer C (10 mm sodium acetate, pH 5, 150 mm NaCl). Fractions eluting from the major peak were concentrated and used in subsequent experiments.
LPMO Preparation
Protein concentrations were determined by UV-visible spectroscopy on a Varian Cary50 UV-visible spectrophotometer using an extinction coefficient (ϵ) at 280 nm of 36,690 m−1 cm−1 for SliLPMO10E, H187A, and SliLPMO10E-Ext and 63,160 m−1 cm−1 for SliLPMO10B (38). Prior to experiments requiring the addition of Cu(II) salts, proteins were incubated for prolonged periods with excess EDTA followed by gel filtration. For Cu(II) titrations and preparation of Cu(II)-loaded enzyme, a stock solution of 100 mm Cu(II)SO4 (Sigma) was prepared and diluted as required. Cu(II)-loaded enzymes were prepared by stoichiometric addition of Cu(II)SO4 followed by desalting to ensure no unbound metal remained.
Substrate Binding Assay
SliLPMO10E (∼20 μm in 50 μl of 50 mm BisTris/HCl, pH 6.2) was incubated with ∼5–10 mg of excess substrate, cellulose (Whatman No. 1, 50 μm), or squid pen β-chitin at room temperature for 3–4 h with occasional agitation. The supernatant was then removed by centrifugation and kept as the unbound fraction. Substrate was then washed three times to remove any unspecific enzyme. Bound enzyme was removed by adding 50 μl of SDS-PAGE loading buffer containing DTT and heating at 95 °C for 10 min. Samples were then analyzed for bound and unbound enzyme content using a 15% SDS-polyacrylamide gel (Bio-Rad).
Mass Spectrometry
Oxidative activity was assayed by mass spectrometry (MS) using a range of substrates (squid pen β-chitin, Avicel (cellulose), Konjac glucomannan, Guar gum, Ivory nut mannan, and Locust bean gum). Reaction mixtures (1 ml total volume) contained 0.2% w/v solid substrate in 10 mm ammonium acetate, pH 5, 1 mm ascorbic acid, and 10 μm copper-loaded LPMO. Control samples were also prepared in the absence of ascorbic acid and protein. Reactions were incubated at 30 °C rotating for 5–12 h before the remaining solid substrate was removed by centrifugation at 16,000 × g at 4 °C for 5 min, and the supernatant was used for analysis. 1 μl of sample was mixed with an equal volume of 10 mg/ml 2,5-dihydroxybenzoic acid in 50% acetonitrile, 0.1% trifluoroacetic acid on a SCOUT-MTP 384 target plate (Bruker). The spotted samples were then dried in a vacuum desiccator before being analyzed on an Ultraflex III matrix-assisted laser desorption ionization-time of flight/time of flight (MALDI-TOF/TOF) instrument (Bruker), as described previously (9).
Electron Paramagnetic Resonance (EPR) Spectroscopy and Spectral Simulations
Samples (80 μm) for EPR analysis were prepared in duplicate in 5 mm Tris/HCl, pH 7, 150 mm NaCl with additions of 0.9 eq of Cu(II)SO4. Wilmad SQ EPR tubes (Wilmad Glass, Buena, NJ) were filled with 250 μl of LPMO solutions, frozen in methanol kept on dry ice, and then transferred to liquid nitrogen. All EPR spectra were measured at 10 K on a Bruker EMX EPR spectrometer (X band). A spherical high quality Bruker resonator ER 4122 SP 9703 and an Oxford Instruments liquid helium system were used to measure the low temperature EPR spectra. Blank spectra of frozen water samples were subtracted from the EPR spectra of the protein samples to eliminate the baseline caused by the resonator's walls, quartz insert, or quartz EPR tube. The baseline was corrected by subtracting a polynomial line drawn through a set of points randomly chosen on the baseline using WinEPR (version 2.22, Bruker Analytik, GmbH). Spectral simulations were performed in SimFonia version 1.26 (Bruker Analytik, GmbH). The gz value and its hyperfine splitting constant AzCu were determined directly from the spectra. Rather than varying gx and gy independently, two other values, gav and Δg, were varied, where gav = (gx + gy)/2 and Δg = gy − gav = gav − gx. To maintain consistency in the simulation procedure while varying the minimal number of parameters, a number of constraints were imposed on the choice of the other simulation parameters thus minimizing the number of varied parameters. Thus, the tensor for the hyperfine interaction of the spin density with copper nucleus ACu (ICu = 3/2) was assumed axial, with the AxCu = AyCu = 0.1 AzCu (small variations around empirically picked factor of 0.1 were found to produce a minimal effect on the simulated spectra). Furthermore, all ACu tensors were assumed co-linear with the g-tensor (all Euler angles were assumed zero). Cu(II) coordinating nitrogen atoms (IN = 1) were assumed to produce isotropic hyperfine interactions (AxN = AyN = AzN = 14 G). In accordance with earlier reported relationship between principal g-factor components and individual line width values along those directions (39), the x, y, and z components of the spectra line width ΔH were assumed to have a close to linear dependence on the g values, the higher the gi, the higher the ΔHi value. This dependence was maintained for all simulations.
Fluorescence Spectroscopy
Fluorescence spectroscopy was carried out at 20 °C on an LS 50B fluorimeter (PerkinElmer Life Sciences) with emission spectra collected between 300 and 400 nm following excitation at 295 nm. The excitation emission slit widths were both set at 5 nm. LPMO proteins (2 μm) were prepared in 10 mm sodium acetate, pH 5, 150 mm NaCl and titrated with microliter aliquots of a Cu(II)SO4 solution.
Stopped-flow Kinetics
Kinetic experiments to measure fluorescence emission changes on Cu(II) binding were carried out on a SX20 stopped-flow spectrophotometer (Applied Photophysics) thermostatted at 25 °C. Trp residues were excited at 295 nm and emitted fluorescence sampled at right angles to the exciting beam after passage through a cutoff filter permitting light with wavelengths above 350 nm to pass, thus removing any scattered exciting light. All LPMO samples (2.5 or 5 μm after mixing) were prepared in 10 mm sodium acetate, pH 5, 150 mm NaCl and were rapidly mixed with a range of Cu(II)SO4 concentrations (25–500 μm after mixing). Following excitation at 295 nm, time courses were taken collecting the total fluorescence emission. Stoichiometry of Cu(II) binding to the LPMOs was monitored by fluorescence amplitude changes. All kinetic data were analyzed using the ProKinetist software (Applied Photophysics).
Isothermal Titration Calorimetry (ITC)
Calorimetric titration experiments were carried out at 25 ± 0.1 °C on a MicroCal VP-ITC calorimeter in (10 mm sodium acetate, pH 5, 150 mm NaCl). Before each run, samples were degassed for 15 min at 23 ± 0.1 °C using the ThermoVac accessory. A solution of Cu(II)SO4 (1 mm) was loaded into the injection syringe and titrated into 100 μm WT or variant SliLPMO10E in the sample cell with stirring at 307 rpm for the duration of the experiment. A reference power of 5 μcal/s was used with either an initial 3- or 5-μl injection of Cu(II)SO4 followed by either a 5- or 10-μl injection for all subsequent titration points. A 60-s initial equilibrium delay with 270-s pauses between subsequent injections were applied throughout. Raw data were analyzed using Origin 7.0 software. The integrated data were corrected for the heat of dilution of buffer into buffer and Cu(II)SO4 into buffer, and the binding isotherms were fitted using binding models provided in the software package.
Crystallization and Structure Determination
An ARI-Gryphon 96-well crystallization robot was used to screen crystallization conditions for SliLPMO10E. A crystal hit was found in 0.1 m sodium acetate, pH 4.6, 25% PEG 4,000 (PEG suite, Qiagen). Scaling-up and optimization of SliLPMO10E crystals from the initial hit were carried out in 24-well VDX plates (Molecular Dimensions) using the hanging drop vapor diffusion method at 18 °C. Equal volumes of protein (15 mg ml−1) and reservoir solution were mixed, and crystals suitable for diffraction studies grew within 2–3 weeks. A single crystal grown from 0.05 m sodium acetate, pH 4.6, 25% PEG 4,000 was transferred to a cryoprotectant solution containing the respective reservoir solution and 20% glycerol prior to flash-cooling by plunging into liquid nitrogen. Crystallographic data were measured at beamline I03, Diamond Light Source, using an x-ray wavelength of 1.00 Å and a Pilatus 6 M-F detector (Dectris). Data were processed automatically using XDS (40) in XIA2 (41) and scaled and merged using Pointless/Scala (42) in the CCP4i suite. The structure was solved using automated molecular replacement in BALBES with an initial model built-in Arp-Warp (43, 44). The structure was refined using Refmac5 (45), with model building between refinement cycles in Coot (46). Riding hydrogen atoms were added when refinement of the protein atoms had converged. Structures were validated using the MolProbity server (47), the JCSG Quality Control Server, and tools within Coot (46). Coordinates and structure factors were deposited in the RCSB Protein Data Bank. A summary of data, refinement statistics, and the quality indicators for the structure are given in Table 2. Additional datasets were collected at wavelengths of 1.20 and 1.33 Å to generate anomalous maps for validation of modeled copper atoms in the structure (Table 2).
TABLE 2.
Crystallographic data processing and refinement statistics
Values in parentheses refer to the outermost resolution shells (1.40–1.38, 2.07–2.02, and 2.07–2.01 Å, respectively). The unit cell was a = 69.6, b = 32.4, c = 61.3 Å, β = 97.8° in space group C2. R.M.S.D. is root mean square deviation. ESU is estimated standard uncertainty; ML is maximum likelihood.
| Wavelength (Å) | 1.0 | 1.33 | 1.20 |
| Resolution (Å) | 1.38 | 2.02 | 2.01 |
| Unique reflections | 27360 | 8755 | 8844 |
| Completeness (%) | 97.3 (92.2) | 97.6 (90.6) | 96.7 (87.2) |
| Rmerge | 0.055 (0.680) | 0.060 (0.087) | 0.049 (0.084) |
| Mn(I/S.D.) | 14.8 (1.8) | 20.3 (13.7) | 24.1 (15.9) |
| Redundancy | 4.9 (3.5) | 5.5 (4.7) | 5.5 (4.7) |
| Wilson B-factor (Å2) | 9.3 | 8.3 | 9.8 |
| Rcryst | 0.135 | ||
| Rfree | 0.160 | ||
| ESU based on ML (Å) | 0.038 | ||
| R.M.S.D. bond lengths (Å) | 0.017 | ||
| R.M.S.D. bond angles (°) | 1.72 | ||
| Ramachandran favored (%) | 98.8 | ||
| Protein Data Bank code | 5ftz |
Results
Trp Fluorescence of Apo-SliLPMO10E Is Quenched upon Binding Cu(II)
Size-exclusion chromatography revealed SliLPMO10E to be a monomeric species, which migrated as a single band on an SDS-polyacrylamide gel to a mass of ∼18 kDa (predicted mass 18,422 Da for residues 30–201). Samples of as-purified SliLPMO10E were subjected to ICP-MS analysis, and a copper content of <3% (0.03 μg of atoms/liter) was consistently determined, indicating a predominantly apo-form. Titrating Cu(II) into apo-SliLPMO10E resulted in the quenching of the Trp fluorescence at λ 340 nm (Fig. 1), with a break point reached at ∼0.8 to 0.9 eq of copper, indicating a binding stoichiometry close to 1:1 (Fig. 1).
FIGURE 1.

Cu(II) binding to SliLPMO10E at pH 5 and 20 °C. Changes in the Trp emission spectrum (inset) upon titration of 1 mm Cu(II)SO4 to SliLPMO10E (2 μm) and plotted as a function of [Cu(II)]/[SliLPMO10E] are shown. The arrow indicates the direction of the emission change, and the stoichiometry of the reaction is indicated by the intersection of the two solid lines.
X-band EPR Spectroscopy Infers the Existence of Two Cu(II) Complexes
Apo-SliLPMO10E gave a very weak EPR signal consistent with the presence in the sample of a small amount of Cu(II), in agreement with ICP-MS analysis (Fig. 2A). On preparation of holo-SliLPMO10E, a much stronger EPR signal clearly originating from Cu(II) centers is obtained (Fig. 2B). Attempts to simulate this experimental spectrum as a single Cu(II) species proved unsuccessful. However, a simulation was possible if a linear combination of two different line shapes was assumed (Fig. 2C) with the simulation parameters reported in Table 3. Thus, the experimental EPR spectrum recorded at 10 K for SliLPMO10E is best approximated by a superposition of two different EPR lines, further referred to as Sim1 and Sim2, from two different Cu(II) complexes. Sim1 was simulated for three nitrogen atoms coordinating copper and Sim2 for two nitrogen atoms. One noticeable difference between the line shapes of Sim1 and Sim2 is the presence of a strong “overshoot” line in Sim2. This is a consequence of a combination of two factors as follows: a smaller g-factor anisotropy (resulting in a smaller gz value) and a larger AzCu value in Sim2 than in Sim1 (Table 3). The second integrals of Sim1 and Sim2 are similar within 2% accuracy indicating the two Cu(II) complexes are likely to be present in roughly equal concentrations.
FIGURE 2.
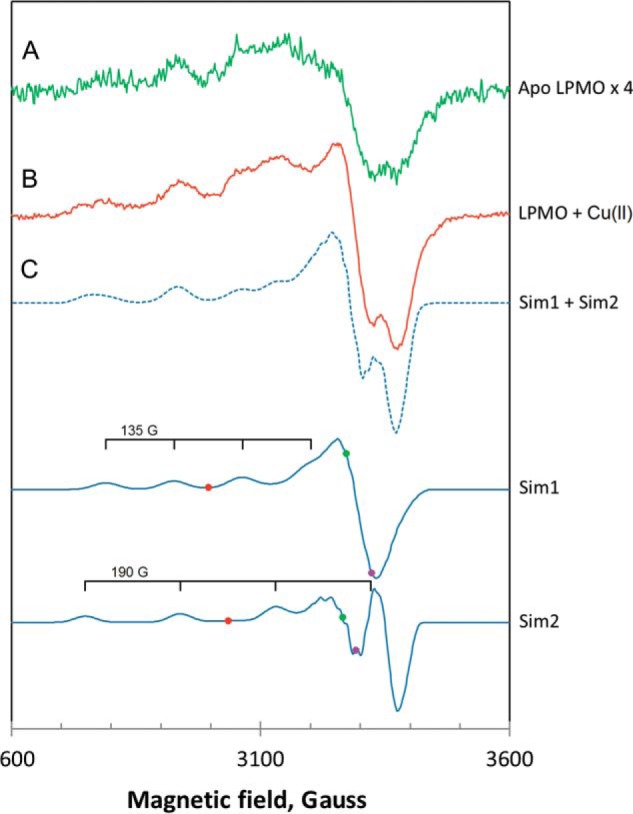
EPR spectra of SliLPMO10E at pH 7 and 10 K. A, as purified magnified ×4, and B, loaded with 0.9 eq of Cu(II). The experimental spectrum in B (solid line) has been approximated by a linear combination (dotted line) of the simulated EPR signals Sim1 and Sim2. C, simulated EPR spectra of the Cu(II) ion coordinated by three nitrogen ligands (Sim1) and by two nitrogen ligands (Sim2). The colored dots on the two simulated spectra indicate the positions of the principal g-values used in the simulations, purple, gx; green, gy; red, gz. Each simulated spectrum is labeled with the equidistant quartet tick marks, centered at gz and showing the positions of lines originated from the hyperfine interaction of the Cu(II) electron spin (S = 1/2) with the copper nuclear spin (I = 3/2). The simulation parameters are given in Table 3.
TABLE 3.
EPR simulation parameters of SliLPMO10E used to simulate spectra Sim1 and Sim2 in Fig. 2
| g | ACu | AN | ΔH | |
|---|---|---|---|---|
| G | G | G | ||
| Sim1a | ||||
| x | 2.033 | 13.5 | 14 | 12 |
| y | 2.067 | 13.5 | 14 | 13 |
| z | 2.260 | 135 | 14 | 26 |
| Sim2b | ||||
| x | 2.055 | 19 | 14 | 12 |
| y | 2.071 | 19 | 14 | 13 |
| z | 2.230 | 190 | 14 | 23 |
a Data were simulated for three coordinating N atoms.
b Data were simulated for two coordinating N atoms.
SliLPMO10E Has Enzymatic Activity with β-Chitin
Initial substrate binding experiments using cellulose and β-chitin were monitored by SDS-PAGE. SliLPMO10E displayed a clear preference for β-chitin based on the proportion of unbound relative to bound fraction observed on the gel (Fig. 3, inset). Substrate specificity and activity were further probed by incubation with diverse glycan substrates (see “Experimental Procedures”) in the presence of ascorbate as the reducing agent. MALDI-TOF MS analysis of the reaction products revealed LPMO activity only for β-chitin (Fig. 3). The observed activity was mainly consistent with previous studies on chitin-active AA10s (9), producing predominantly reducing-end aldonic acid oligosaccharide products (C1 oxidation) with even-numbered degrees of polymerization. However, not consistent was the observation of −2 Da species implicated as possible reducing end lactones prior to ring opening or non-reducing end ketoaldoses resulting from C4 oxidation (Fig. 3). Distinguishing these species by MS is challenging, and so from the present data it is unclear whether SliLPMO10E is capable of oxidizing at the C4 as well as the C1 position.
FIGURE 3.

Substrate binding and activity of SliLPMO10E. Inset SDS-PAGE to monitor the binding of SliLPMO10E to cellulose (filter paper) or squid-pen β-chitin. Fractions shown are untreated SliLPMO10E, the protein content in the supernatant (unbound) or bound to the substrate (bound). MALDI-TOF analysis of SliLPMO10E products from squid-pen β-chitin. DPna1 = aldonic acid; DPn−2 = lactone or C4 ketoaldose (measured molecular weight). DP4−2 + Na+ (851.3), DP4al + Na+ (869.3), DP4al− + 2Na+ (891.3), DP6−2 + Na+ (1257.5), DP6al + Na+ (1275.5), DP6al− + 2Na+ (1297.5), DP8−2 + Na+ (1663.6), DP8al + Na+ (1681.7), DP8al− + 2Na+ (1703.6).
X-ray Crystal Structure of SliLPMO10E Reveals Two Copper-binding Sites
The x-ray structure of holo-SliLPMO10E was determined to a resolution of 1.38 Å and contains one protein molecule in the crystallographic asymmetric unit. Continuous and well defined electron density was found for residues 30–201. The overall structure includes seven anti-parallel β-strands arranged in two β-sheets (Fig. 4A); one contains three antiparallel strands (S1, S3, and S6), and the other contains four antiparallel strands (S4, S5, S7, and S8). This distorted β-sandwich fold is consistent with that of other AA10 structures (48–52). Between strands S1 and S3 is the loop 2 region, which shows the most variability in size between LPMO families. In SliLPMO10E, this region consists of two α-helices (H1 and H2) and a small β-strand (S2). Two disulfide bonds are also present, Cys-43/Cys-51 and Cys-78/Cys-196, with the latter connecting loop 2 to the C-terminal β-strand (S8) (Fig. 4A). Disulfide bonds have been identified in other LPMOs and most likely confer structural stability.
FIGURE 4.
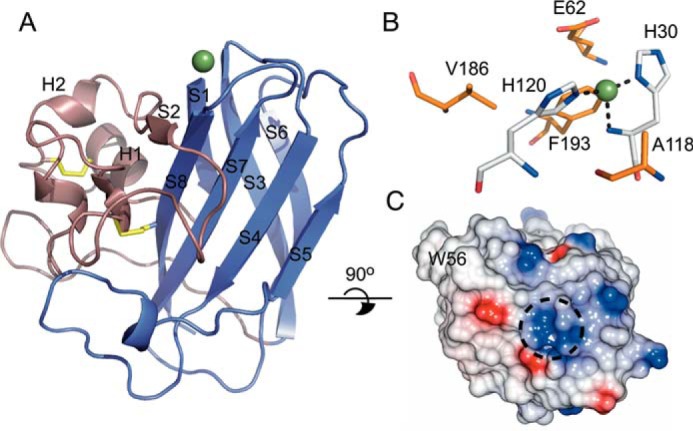
X-ray crystal structure of SliLPMO10E. A, schematic representation with the core β-sheet fold shown in purple and the loop 2 region displayed in violet. Disulfide bonds are shown in yellow, and the copper represented as a sphere. B, stick representation of the active site. Coordinating His residues are colored in silver and residues surrounding the first copper coordination sphere shown in orange. Bonds to the copper ion are indicated by dashed lines. C, electrostatic surface representation of the proposed substrate-binding surface with W56 labeled and the location of the copper indicated by a dashed circle.
A well defined 16-σ-peak is observed in the σA-weighted Fo − Fc difference map into which a copper ion was modeled with coordination by three equatorial nitrogen ligands; the Nt of His-30 and the Nδ1 and Nϵ2 atoms of His-30 and His-120, respectively (Fig. 4B). This results in the T-shaped His-brace coordination geometry, with bond lengths and angles reported in Table 4. A number of residues commonly found in chitin-active AA10s are noted and include Ala-118, suggested to influence the solvent-facing copper site and Phe-193 (Fig. 4B) (52). The latter lies with its Cζ atom only 3.6 Å from the active site copper, a feature consistently found in AA10s but replaced by Tyr in the cellulose-active S. coelicolor ScLPMO10B (a homologue to SliLPMO10B) (52). The side chain orientations of Glu-62 and Val-186 in the vicinity of the copper site make a cavity, which is a feature found in other chitin-active AA10s and has been suggested to accommodate oxygen or the acetyl group of an N-acetylglucosamine moiety (52).
TABLE 4.
Bond lengths and angles of active site and symmetry-related metal-mediated dimer site (MMD)
| Bond | Length/angle (Å, °) |
|---|---|
| Active site | |
| His30 Nδ1 | 1.94 |
| His30 N | 2.24 |
| His120 Nϵ2 | 1.91 |
| His30 Nδ1 -Cu- His120 N | 95 |
| His30 Nδ1 -Cu- His120 Nϵ2 | 160 |
| His30 N -Cu- His120 Nϵ2 | 103 |
| MMD site | |
| His187 Nϵ2 | 1.95 |
| Asp188 Oδ2 | 2.40 |
| His187 Nϵ2a | 1.94 |
| Asp188 Oδ2a | 2.42 |
a Symmetry-related molecule is shown.
The putative substrate-binding surface of SliLPMO10E is flat with two small stretches of opposite surface charge, running either side of the positively charged copper ion (Fig. 4C). A prominent surface aromatic residue, Trp-56, is located in the loop 2 region and is considered important in AA10s for substrate specificity.
A second well defined σ-peak in the σA-weighted Fo − Fc difference map consistent with the presence of a metal ion is also observed. Modeling a copper ion into this peak results in coordination with His-187 and Asp-188 from the monomer in the asymmetric unit and the same residues from a symmetry-related molecule, thus creating a metal-mediated dimer (Fig. 5A). Crystallization conditions contained no metal salts, and the excess of Cu(II) used to form the holo-SliLPMO10E was removed prior to crystallization. Although every care was taken to ensure no serendipitous binding of rogue metals, there is the possibility that a divalent metal such as Zn(II) could out-compete Cu(II) for this site. Therefore, to confirm the identity of the metals in both sites (copper or zinc), anomalous difference maps were generated by measuring a single crystal at two different x-ray wavelengths (1.2 and 1.33 Å). At 1.2 Å the energy is above that of both copper and zinc K-edges, whereas at 1.33 Å only the copper K-edge contributes to the anomalous scattering. Anomalous maps generated at these different wavelengths for both the active and dimer sites are consistent with the presence of a copper ion in both sites (Fig. 5B). The copper coordination geometry in the symmetry-generated dimer is a distorted square planar, with the Nϵ2 atoms of the His residues coordinating in a linear fashion and the Oδ2 atoms of the aspartates bent (Table 4). The existence of such a metal-mediated dimer is not readily supported from solution measurements (Fig. 1).
FIGURE 5.

Second metal-binding site creates a metal-mediated SliLPMO10E dimer. A, surface representation of the symmetry-generated metal-mediated dimer of SliLPMO10E together with a 2Fo − Fc electron density map contoured at 1 σ of the copper coordination site at the dimer interface. Copper ions are shown in spheres. B, anomalous electron density maps contoured at 5 σ at x-ray wavelengths of 1.2 Å (above the copper and zinc K-edges) and 1.33 Å (below the zinc but above the copper K-edges) for the active and MMD sites.
Kinetics of Cu(II) Binding to Apo-SliLPMO10E Reveals Two Binding Forms
Apo-SliLPMO10E was mixed with Cu(II) solutions of known concentrations and the quenching of Trp fluorescence monitored as a function of time in a stopped-flow spectrophotometer. Upon mixing, two phases of fluorescence quenching were observed, the rates of which were [Cu(II)]-dependent (Fig. 6A, inset). Pseudo first-order rate constants (kobs) were linearly dependent on the [Cu(II)], with the slopes yielding second-order rate constants, k1 and k2 (Fig. 6A, inset, and Table 5). The linear plots run through/close to the origin, indicating that the Cu(II) affinity of the binding sites is high (i.e. the dissociation rate constants for Cu(II) are small compared with the association rates), and the relative amplitudes of the two phases are independent of the [Cu(II)]. Binding stoichiometries were measured by first loading apo-SliLPMO10E with sub- to super-stoichiometric amounts of Cu(II) followed by mixing with excess Cu(II). Upon mixing, the two phases were retained, but as expected, the total amplitude of the fluorescence quench decreased as the concentration of the Cu(II) preloaded into SliLPMO10E increased (i.e. Cu(II) can now only bind to the empty sites). The relative amplitudes of the two phases changed as a result of pre-mixing with Cu(II), the faster phase being abolished at lower preloaded Cu(II) additions and the slow phase at higher preloaded [Cu(II)]. This enables the stoichiometry of each phase to be determined as illustrated in Fig. 6B. The faster phase titrated with a stoichiometry of ∼0.5, and the slower phase, which titrates once the site responsible for the faster binding is filled, also with a stoichiometry of ∼0.5. Thus, the overall stoichiometry is 1:1, consistent with the interpretation of the static fluorescence titration (Fig. 1). Furthermore, the proportions of the two kinetic phases are unaffected at low [NaCl]. Therefore the observed [Cu(II)] dependence is not an ionic strength effect, and the data are consistent with Cu(II) binding to two forms of a single site.
FIGURE 6.

Fluorescence stopped-flow kinetics of Cu(II) binding to SliLPMO10E at pH 5 and 25 °C. A, biphasic time courses and fits to a double exponential function on reacting 2.5 μm (after mixing) SliLPMO10E with various [Cu(II)] as indicated. Inset, first-order rate constants for the fast (kobs, 1, squares) and the slow (kobs,2, triangles) phase of Cu(II) binding as a function of [Cu(II)]. B, stoichiometry of Cu(II) binding monitored by fluorescence amplitude changes to SliLPMO10E (5 μm after mixing) pre-loaded with sub- to super-stoichiometric amounts of Cu(II), following rapid mixing with excess Cu(II). The fast phase (kobs, 1, squares) titrates with an ∼0.5 stoichiometry with the slow phase (kobs, 2, triangles) being observed following completion of the fast phase and with an ∼0.5 stoichiometry.
TABLE 5.
Kinetic and thermodynamic parameters determined from stopped-flow fluorescence, fluorescence titrations, and ITC at 25 °C for Cu(II) binding to the various forms of SliLPMO10E
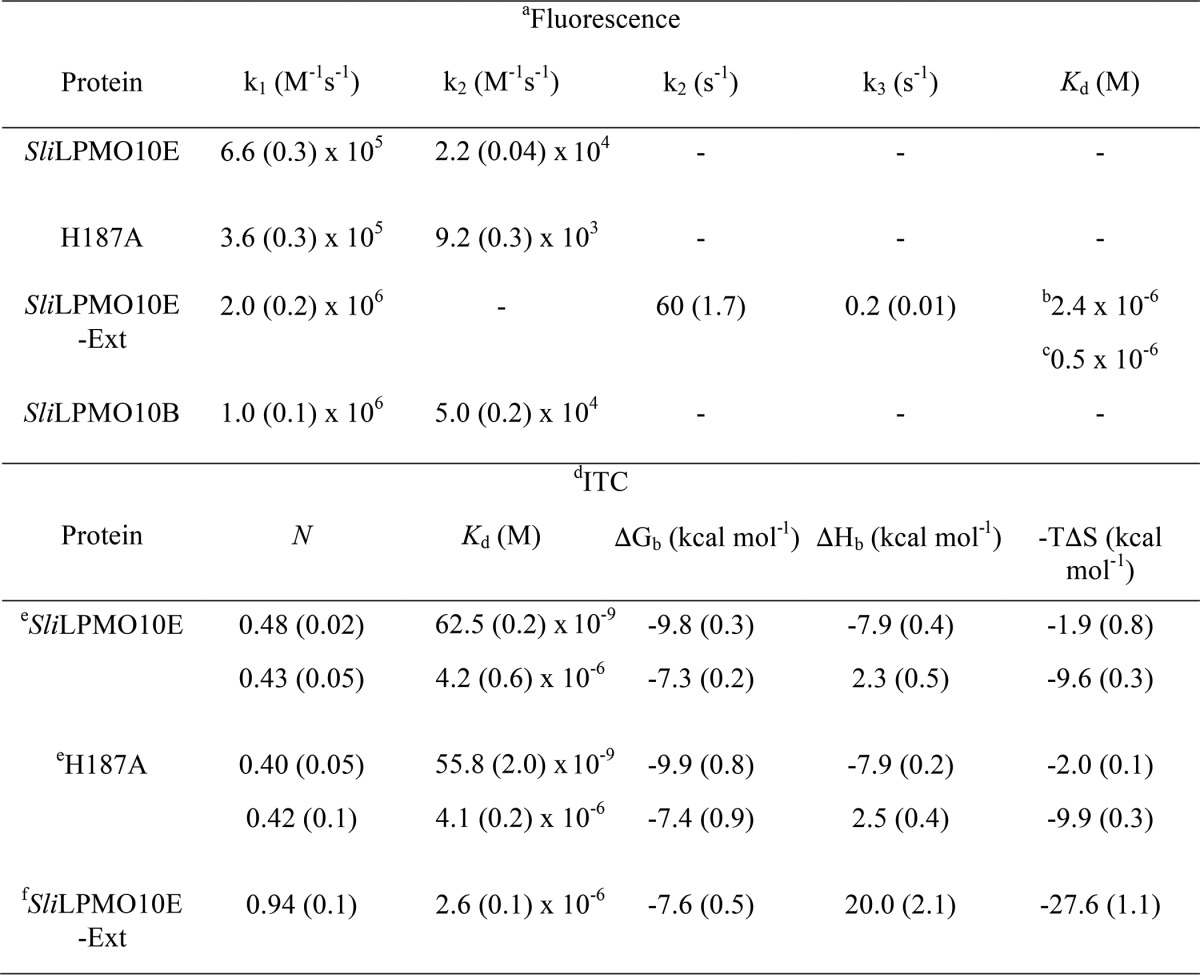
a Experimental errors are reported in parentheses and are the standard errors from triplicate experiments.
b Data were determined from static fluorescence titration.
c Data were determined from stopped-flow fluorescence.
d Experimental errors are reported in parentheses and are the standard errors from triplicate experiments for SliLPMO10E and duplicates for H187A and SliLPMO10E-EXT.
e Parameters were derived from a sequential binding model.
f Parameters were derived from a one set of sites binding model.
The two [Cu(II)]-dependent phases observed in the kinetic measurements prompted us to consider whether the slow phase of Cu(II) binding was due to the presence of a second copper-binding site. Kinetic experiments, with the H187A variant, gave essentially identical results to that of the WT SliLPMO10E, i.e. two clear phases both [Cu(II)]-dependent with k1 and k2 values reported in Table 5. Thus, the contribution of this dimer site observed in the crystal structure to the quenching of fluorescence in the WT protein may be discounted.
ITC Identifies Two Thermodynamically Distinct Cu(II)-binding Sites
A typical ITC profile obtained by titrating Cu(II) into apo-SliLPMO10E is shown in Fig. 7, where two binding phases are detected. The first of these is an exothermic process with the second endothermic in nature. A two-site model was used to fit the data with the parameters obtained reported in Table 5. Notably, the stoichiometry (N) for each binding phase is ∼0.5 giving a total stoichiometry of 1 and thus in keeping with the kinetic data. The first binding phase has a nanomolar Kd value for Cu(II) and exhibits a favorable enthalpic binding (ΔHb) contribution, with a small but favorable entropic (−TΔS) contribution leading to a favorable free energy of binding (ΔGb) (Table 5). The second phase displays a weaker affinity for Cu(II) with a micromolar Kd value (Table 5). Moreover, this binding event is characterized by a positive ΔHb (unfavorable) that is compensated for by an increased −TΔS, thus maintaining a favorable ΔGb (Table 5). The H187A mutant gave the same ITC profile with the parameters obtained from fitting to a two-site model comparable with WT SliLPMO10E (Table 5), reconfirming that the second metal site observed in the crystal is not that detected by either kinetics or ITC.
FIGURE 7.

Thermodynamics of Cu(II) binding to SliLPMO10E at pH 5 and 25 °C. An ITC thermogram and fit to a sequential binding model (solid line) upon titrating SliLPMO10E with Cu(II). Thermodynamic parameters obtained are reported in Table 5. Inset reports the heat of dilutions from a buffer to buffer and a Cu(II) to buffer titration.
Kinetics of Cu(II) Binding to SliLPMO10B Are Identical to Those of SliLPMO10E
To investigate whether the complex kinetics of Cu(II) binding is unique to SliLPMO10E or may be typical for AA10s, a second AA10 was tested. SliLPMO10B is a homologue of ScLPMO10B from S. coelicolor whose structure has been reported and is cellulose-active (52). Size-exclusion chromatography of purified SliLPMO10B revealed a monomer species that migrated as a single band on an SDS-polyacrylamide gel to a mass of ∼20 kDa (predicted mass 20,723.4 Da for residues 43–229). Trp fluorescence at λ ≥350 nm was quenched upon addition of Cu(II) aliquots, until a break point at ∼0.8 to 0.9 eq was reached, indicating a binding stoichiometry close to 1:1. On rapid mixing in the stopped-flow with excess Cu(II), the fluorescence was quenched as seen for SliLPMO10E with time courses again found to compose two phases, the rate constants of which were linearly dependent on [Cu(II)] (Table 5).
Cu(II) Binding to the SliLPMO10E-Ext Variant Reveals a Single Cu(II) Form
The mature SliLPMO10E-Ext variant has a Met-Gly-His N-terminal sequence and was produced with the aim to abolish/perturb the Nt coordination of the His-brace. SliLPMO10E-Ext remains capable of binding Cu(II) as determined from the quenching of the Trp fluorescence. However, in contrast to SliLPMO10E where the tight binding of Cu(II) precluded the determination of a Kd value (Fig. 1), SliLPMO10E-Ext possesses a weaker affinity for Cu(II) enabling a Kd value to be determined (Table 5). Stopped-flow fluorescence measurements yielded time courses now comprising of three phases after mixing with excess Cu(II). The fastest phase was linearly dependent on [Cu(II)] (Fig. 8A) with a second order rate constant (k1) reported in Table 5. Although binding to this site is rapid, it is with low affinity as seen by the intercept on the kobs, 1 axis, which is interpreted as the dissociation rate constant (koff) of Cu(II) from this site (koff = 230 s−1) (Fig. 8A) enabling a Kd of 1.2 × 10−4 m to be determined. The two slower phases display little [Cu(II)] dependence and are assigned to first-order reorganization steps (k2 and k3, Table 5) in the Cu(II)-bound SliLPMO10E-Ext that each lead to enhanced quenching of the Trp fluorescence. Titration experiments performed by mixing Cu(II) solutions with SliLPMO10E-Ext preloaded with sub- to super-stoichiometric [Cu(II)] were also carried out. Unlike for SliLPMO10E, the three distinct phases all titrate together indicating binding to a single site that undergoes rearrangement. A striking result is that the Kd value determined following preloading with Cu(II) (Fig. 8B and Table 5), is 2 orders of magnitude lower (tighter) than the initial (fast) binding site (Kd 1.2 × 10−4 m) and in line with the Kd value determined from static fluorescence titration experiments (Table 5). Together, these indicate that following initial Cu(II) binding, subsequent rearrangements detected in the slower kinetic phases lock Cu(II) into the site. This is supported by the observed decrease in the amplitude of the slower phase as the [Cu(II)] with which it is mixed increases. This is expected when the [Cu(II)] spans the value of the Kd for the initial binding site. Thus, at 100 μm Cu(II), the amplitude of the fast phase reflects half-occupancy of this site, and full binding then occurs at the rate of the slower phase. At 250 μm Cu(II), ∼70% of the initial site is rapidly filled and reflected in the amplitude of the initial binding process, and the remaining sites are filled at the rate of the reorganization.
FIGURE 8.

Kinetics and thermodynamics of Cu(II) binding to SliLPMO10E-Ext at pH 5 and 25 °C. A, first-order rate constants for the fast (kobs, 1) phase of Cu(II) binding to SliLPMO10E-Ext as a function of [Cu(II)]. B, titration of Cu(II) to SliLPMO10E-Ext plotted as a function of fractional saturation defined as Y = 1 − ΔF/ΔFT, where ΔF is the fluorescence change and ΔFT is the fluorescent change from zero to fully saturated SliLPMO10E with Cu(II). C, ITC binding profile and fit to a single-site binding model (solid line) upon titrating SliLPMO10E-Ext with Cu(II). Thermodynamic parameters obtained from the fit are reported in Table 5.
The ITC profile in Fig. 8C shows an endothermic binding event, fitted to a single-site binding model to give the parameters reported in Table 5 for Cu(II) binding to SliLPMO10E-Ext. A stoichiometry of ∼1 Cu(II) is determined with a Kd value in the micromolar range correlating well to that determined from the static fluorescence titration and stopped-flow kinetics (Table 5). The unfavorable ΔHb is compensated for by a large favorable −TΔS contribution, maintaining a favorable ΔGb (Table 5). Thus, the thermodynamic profile for the SliLPMO10E-Ext variant has a strong resemblance to the second endothermic binding event observed with the WT SliLPMO10E.
Discussion
Two cellulose-degrading AA10s from S. coelicolor (ScLPMO10B and ScLPMO10C) and a chitin-degrading AA10 from S. griseus (SgLPMO10F) have so far been characterized from streptomycetes (52–54). ScLPMO10C and SgLPMO10F have been shown to oxidize strictly at the C1 carbon of the sugar (52, 54), whereas ScLPMO10B can oxidize at the C1 and C4 positions (52). SliLPMO10E is active only on chitin, with the predominant oxidation product revealed from MS indicative of C1 cleavage (Fig. 3). As described for other AA10s, a Phe and a conserved Ala residue are located in the vicinity of the copper site, with their hydrophobic side chains oriented so as to impede access/binding of exogenous ligands to the axial positions of the copper ion (Fig. 4B) (51, 55). This has led to the suggestion that dioxygen activation is most likely at the fourth equatorial position in AA10s resulting in strict C1 substrate oxidation. Conversely, access to the solvent-facing axial position has been suggested to be responsible for C1/C4 oxidation in AA9s lacking an Ala and in ScLPMO10B where the Ala is present but the side chain is positioned so as not to obstruct ligand access (16, 52). In SliLPMO10E, the Ala-118 side chain superimposes with other strict C1-oxidizing AA10s. This structural feature is consistent therefore with the MS interpretation of C1-oxidized products of the chitin substrate and furthermore predicts that the −2 Da species identified in the MS likely arises not from C4 oxidation products but reducing end lactones.
Although a major focus of LPMOs has been on their roles and activities with biomass-derived polysaccharides substrates, the constitutive expression and proximity of slilpmo10E to the cslA-glxA operon could imply a role in assisting with the attachment or further processing/modification of the cslA-glxA glycan produced during the development cycle. The chemical composition of this glycan essential for morphological development in S. lividans has not yet been characterized. Typically, genes involved in bacterial cellulose synthesis are organized in an operon, containing the bcsABCD genes (56). However, Streptomyces lacks a c-di-GMP-binding protein (BcsB), conserved in cellulose-producing organisms and essential for the synthesis of cellulose (57). Therefore, coupled with the activity of SliLPMO10E only for chitin hints at the possibility that the glycan produced by CslA has N-acetylglucosamine functionalities.
The x-ray crystal structure of SliLPMO10E reveals the active site copper to have the characteristic His-brace coordination associated with all other LPMOs (Fig. 4). A second coordinating copper ion was also identified in the x-ray structure, with subsequent solution studies providing no evidence to indicate that this second copper site is occupied or that the metal-mediated dimer that can be generated in the crystal is formed. Despite this, the orientation of the two monomers is noteworthy (Fig. 5), as dimerization does not occlude the substrate interaction surface.
Spectral simulation (Sim1 and Sim2) of the EPR spectrum of SliLPMO10E indicates the presence of two Cu(II) species. The gz and AzCu values for these are 2.26/2.23 and 135/190 G, respectively, with the 2.26 and 135 G pair representing the “typical” distorted AA10 coordination where the three nitrogen atoms of the His-brace are all coordinating to the Cu(II). The gz and AzCu values determined directly from the spectral parallel region are the most reliable parameters for comparison with EPR data obtained from other LPMOs. In contrast, the perpendicular g values (gx and gy) are estimated through simulation. From this study it is clear that the simulation of spectra for a mixed species gives a significantly lower degree of rhombicity (difference between gx and gy) compared with other AA10s (51, 53). Thus previous AA10 values, although recognizant of the difficulty of simulating gx and gy values, may reflect a mixture of species rather than true rhombicity (51). For chitin-active AA10s Az values between 109 and 118 G have been reported (53), placing them between the normal Peisach-Blumberg classification of type 1 and type 2 copper centers (58). In contrast cellulose-active AA10s and AA9s have higher Az values of ∼147 G and are firmly within the type 2 classification (53). Thus SliLPMO10E lies between cellulose-active AA9s/AA10s and chitin-active AA10s, suggesting a more axial geometry compared with other chitin-active AA10s. The second species has a lower gz value of 2.23 and is in a clearly axial geometry (Az = 190 G). This could arise from a chloride ion binding to the Cu(II), as a consequence of 150 mm NaCl being present in the buffer, in accord with the gz value of 2.23 recently determined for an AA9 LPMO where chloride acts as the exogenous ligand on Cu(II) (59). The axial geometry may arise from two nitrogen atoms coordinating to the Cu(II), with the decomplexation of one of the His residues or the Nt a possibility, with the coordinated mono-atomic chloride ion able to move around until it overlaps well with the d(x2-y2) orbital giving rise to the lower gz of 2.23 because of enhanced covalency of the singularly occupied molecular orbital across the chloride ligand (59).
In vitro Cu(II) binding studies to SliLPMO10E and SliLPMO10B are most consistent with the mechanism depicted in Fig. 9. Our data suggest that the as-isolated apo-form of these AA10s exist as two non-equilibrating species (I and I*, Fig. 9). Cu(II) first binds to species I in an exothermic reaction with a nanomolar Kd value, which we attribute to Cu(II) coordinating all three nitrogen ligands of the His-brace. Once the Cu(II)-binding site in species I is filled, the second apo-species (I*) binds Cu(II) in an endothermic manner with a micromolar Kd value and is attributed to Cu(II) coordinating two nitrogen ligands of the His brace, with H2O (low salt conditions) or chloride ions (high salt conditions) completing the equatorial coordination sphere (X in Fig. 9). Thus, under the conditions investigated (low and high salt), Cu(II) binds to a single site in two binding forms, each with distinct kinetic and thermodynamic parameters (Fig. 9 and Table 5). Comparison of the kinetic and thermodynamic data for the SliLPMO10E-Ext variant with that of the WT gives a strong indication that the two coordinate nitrogen species arises from the absence of Nt coordination (Fig. 9 and Table 5). This interpretation arises from the unfavorable ΔHb and Kd values that mirror the second binding form of the WT protein (overall Kd 10−6 m) and the presence of a single Cu(II)-dependent binding step (Table 5). The proposition that the kinetics and EPR results arise from two forms of a single site must be set against the structural data that show a single unique copper-binding configuration. One way to account for this apparent discrepancy is by proposing that one of the forms discerned in the kinetic experiments resides at lower energy than the other form. Once Cu(II) is bound, the whole of the protein slowly converts to the lower energy form with the Nt moving inward and completing the three coordinate His-brace geometry as seen in the x-ray crystal structure (dashed arrow, Fig. 9). This final conversion would be difficult to detect in fluorescence experiments, as it would occur in the Cu(II)-bound form in which fluorescence is largely quenched.
FIGURE 9.

Mechanism of Cu(II) binding to SliLPMO10E and the N-terminal extension variant SliLPMO10E-Ext. Dissociation constants (Kd) and rates (k) for Cu(II) binding are reported in blue (ITC measurement) and red (stopped-flow kinetics), with parameters obtained from both experimental approaches given in orange, including the stoichiometry of Cu(II) binding (N). The gz and Az values determined by EPR for the two Cu(II) species in WT SliLPMO10E are reported. The minimum number of ligands for Cu(II) is four. X represents either H2O or chloride ligand depending on the experimental conditions (i.e. high or low NaCl) with chloride more likely under the high NaCl conditions based on the gz value obtained by EPR.
The fact that we have observed heterogeneity in the His-brace coordination geometry in all batches of SliLPMO10E and SliLPMO10B prepared lends credence to the view that coordination flexibility is inherent in the His-brace motif. The question therefore arises as to what biological function this may be assigned. It is possible that the two forms have different enzymic functions or the flexibility of the copper site is important for loading copper from an in vivo donor (i.e. chaperone or low molecular weight ligand). In this respect any putative donor will have a very high affinity for copper with the metal only being transferred through docking and orienting the donor and acceptor ligands to enable facile metal transfer through a ligand exchange mechanism (60). Here, we are loading copper in a way that would never happen in vivo, namely copper is added in large excess in a non-coordinating form. Therefore, what we may be observing is a heterogeneity of the metal-binding site that in some way reflects different states of the protein, which maybe populated transiently in the true biological transfer. Finally, it is worth considering that these AA10s are Sec substrates and fold following translocation across the membrane. As the protein passes into the extracellular space, copper may become quickly bound (via a chaperone or low molecular weight ligand) prior to cleavage of the signal sequence that then enables for the final nitrogen coordination by the Nt. This is reminiscent of copper binding to the Ext mutant that incorporates the metal into a single site, which once having undergone rearrangement binds copper tightly (Fig. 9). If this happens with the signal sequence in place, then cleavage and coordination of the final nitrogen ligand occur when the site is already full. The as-purified WT LPMO proteins lack the signal sequence and in its absence result in two species. These can adopt two folded forms of the His-brace in the apo-state (Nt “out” or “in”) that upon in vitro copper loading result in the heterogeneity observed, whereas the Ext mutant may replicate the true mechanism of copper binding in vivo.
Author Contributions
A. K. C. conducted all experiments, prepared all samples, and analyzed all data. M. T. W. assisted with stopped-flow experiments and analyzed the data. M. A. H. supervised the crystallography and structure determination. D. A. S. carried out EPR experiments and spectral simulations. P. H. W. and G. R. H. contributed reagents, mass spectrometry, and EPR discussion. E. V. carried out RNA-sequence analysis. J. A. R. W. designed the study and wrote the manuscript with contributions from all co-authors.
Acknowledgments
We thank Professor R. Hider for access to ICP-MS measurements at the Institute of Pharmaceutical Science, King's College London. We also thank Dr. Dennis Claessen for critical reading of the manuscript. We acknowledge the Diamond Light Source for access to beamline I03 (East of England Macromolecular Crystallography BAG, MX7461) and use of the JCSG Quality Control Server.
The authors declare that they have no conflicts of interest with the contents of this article.
The atomic coordinates and structure factors (code 5ftz) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- LPMO
- lytic polysaccharide monooxygenase
- AA
- auxiliary activity
- BisTris
- 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol
- ITC
- isothermal titration calorimetry
- Nt
- N-terminal amino group.
References
- 1.Beeson W. T., Vu V. V., Span E. A., Phillips C. M., and Marletta M. A. (2015) Cellulose degradation by polysaccharide monooxygenases. Annu. Rev. Biochem. 84, 923–946 [DOI] [PubMed] [Google Scholar]
- 2.Hemsworth G. R., Johnston E. M., Davies G. J., and Walton P. H. (2015) Lytic polysaccharide monooxygenases in biomass conversion. Trends Biotechnol. 33, 747–761 [DOI] [PubMed] [Google Scholar]
- 3.Müller G., Várnai A., Johansen K. S., Eijsink V. G., and Horn S. J. (2015) Harnessing the potential of LPMO-containing cellulase cocktails poses new demands on processing conditions. Biotechnol. Biofuels 8, 187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lombard V., Golaconda Ramulu H., Drula E., Coutinho P. M., and Henrissat B. (2014) The carbohydrate-active enzymes database (CAZy) in 2013. Nucleic Acids Res. 42, D490–D495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Harris P. V., Welner D., McFarland K. C., Re E., Navarro Poulsen J. C., Brown K., Salbo R., Ding H., Vlasenko E., Merino S., Xu F., Cherry J., Larsen S., and Lo Leggio L. (2010) Stimulation of lignocellulosic biomass hydrolysis by proteins of glycoside hydrolase family 61: structure and function of a large, enigmatic family. Biochemistry 49, 3305–3316 [DOI] [PubMed] [Google Scholar]
- 6.Quinlan R. J., Sweeney M. D., Lo Leggio L., Otten H., Poulsen J. C., Johansen K. S., Krogh K. B., Jørgensen C. I., Tovborg M., Anthonsen A., Tryfona T., Walter C. P., Dupree P., Xu F., Davies G. J., and Walton P. H. (2011) Insights into the oxidative degradation of cellulose by a copper metalloenzyme that exploits biomass components. Proc. Natl. Acad. Sci. U.S.A. 108, 15079–15084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Agger J. W., Isaksen T., Várnai A., Vidal-Melgosa S., Willats W. G., Ludwig R., Horn S. J., Eijsink V. G., and Westereng B. (2014) Discovery of LPMO activity on hemicelluloses shows the importance of oxidative processes in plant cell wall degradation. Proc. Natl. Acad. Sci. U.S.A. 111, 6287–6292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Isaksen T., Westereng B., Aachmann F. L., Agger J. W., Kracher D., Kittl R., Ludwig R., Haltrich D., Eijsink V. G., and Horn S. J. (2014) A C4-oxidizing lytic polysaccharide monooxygenase cleaving both cellulose and cello-oligosaccharides. J. Biol. Chem. 289, 2632–2642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vaaje-Kolstad G., Westereng B., Horn S. J., Liu Z., Zhai H., Sørlie M., and Eijsink V. G. (2010) An oxidative enzyme boosting the enzymatic conversion of recalcitrant polysaccharides. Science 330, 219–222 [DOI] [PubMed] [Google Scholar]
- 10.Forsberg Z., Vaaje-Kolstad G., Westereng B., Bunæs A. C., Stenstrøm Y., MacKenzie A., Sørlie M., Horn S. J., and Eijsink V. G. (2011) Cleavage of cellulose by a CBM33 protein. Protein Sci. 20, 1479–1483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hemsworth G. R., Henrissat B., Davies G. J., and Walton P. H. (2014) Discovery and characterization of a new family of lytic polysaccharide monooxygenases. Nat. Chem. Biol. 10, 122–126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lo Leggio L., Simmons T. J., Poulsen J. C., Frandsen K. E., Hemsworth G. R., Stringer M. A., von Freiesleben P., Tovborg M., Johansen K. S., De Maria L., Harris P. V., Soong C. L., Dupree P., Tryfona T., Lenfant N., et al. (2015) Structure and boosting activity of a starch-degrading lytic polysaccharide monooxygenase. Nat. Commun. 6, 5961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tan T. C., Kracher D., Gandini R., Sygmund C., Kittl R., Haltrich D., Hällberg B. M., Ludwig R., and Divne C. (2015) Structural basis for cellobiose dehydrogenase action during oxidative cellulose degradation. Nat. Commun. 6, 7542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Westereng B., Cannella D., Wittrup Agger J., Jørgensen H., Larsen Andersen M., Eijsink V. G., and Felby C. (2015) Enzymatic cellulose oxidation is linked to lignin by long-range electron transfer. Sci. Rep. 5, 18561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Beeson W. T., Phillips C. M., Cate J. H., and Marletta M. A. (2012) Oxidative cleavage of cellulose by fungal copper-dependent polysaccharide monooxygenases. J. Am. Chem. Soc. 134, 890–892 [DOI] [PubMed] [Google Scholar]
- 16.Kim S., Ståhlberg J., Sandgren M., Paton R. S., and Beckham G. T. (2014) Quantum mechanical calculations suggest that lytic polysaccharide monooxygenases use a copper-oxyl, oxygen-rebound mechanism. Proc. Natl. Acad. Sci. U.S.A. 111, 149–154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Phillips C. M., Beeson W. T., Cate J. H., and Marletta M. A. (2011) Cellobiose dehydrogenase and a copper-dependent polysaccharide monooxygenase potentiate cellulose degradation by Neurospora crassa. ACS Chem. Biol. 6, 1399–1406 [DOI] [PubMed] [Google Scholar]
- 18.Li X., Beeson W. T. 4th, Phillips C. M., Marletta M. A., and Cate J. H. (2012) Structural basis for substrate targeting and catalysis by fungal polysaccharide monooxygenases. Structure 20, 1051–1061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Vu V. V., Beeson W. T., Phillips C. M., Cate J. H., and Marletta M. A. (2014) Determinants of regioselective hydroxylation in the fungal polysaccharide monooxygenases. J. Am. Chem. Soc. 136, 562–565 [DOI] [PubMed] [Google Scholar]
- 20.Crawford D. L. (1978) Lignocellulose decomposition by selected Streptomyces strains. Appl. Environ. Microbiol. 35, 1041–1045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Goodfellow M., and Williams S. T. (1983) Ecology of actinomycetes. Annu. Rev. Microbiol. 37, 189–216 [DOI] [PubMed] [Google Scholar]
- 22.McCarthy A. J., and Williams S. T. (1992) Actinomycetes as agents of biodegradation in the environment. Gene 115, 189–192 [DOI] [PubMed] [Google Scholar]
- 23.Takasuka T. E., Book A. J., Lewin G. R., Currie C. R., and Fox B. G. (2013) Aerobic deconstruction of cellulosic biomass by an insect-associated Streptomyces. Sci. Rep. 3, 1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nielsen J. (1996) Modelling the morphology of filamentous microorganisms. Trends Biotechnol. 14, 438–443 [Google Scholar]
- 25.van Dissel D., Claessen D., and van Wezel G. P. (2014) Morphogenesis of Streptomyces in submerged cultures. Adv. Appl. Microbiol. 89, 1–45 [DOI] [PubMed] [Google Scholar]
- 26.Vrancken K., and Anné J. (2009) Secretory production of recombinant proteins by Streptomyces. Future Microbiol. 4, 181–188 [DOI] [PubMed] [Google Scholar]
- 27.Anné J., Maldonado B., Van Impe J., Van Mellaert L., and Bernaerts K. (2012) Recombinant protein production and streptomycetes. J. Biotechnol. 158, 159–167 [DOI] [PubMed] [Google Scholar]
- 28.Xu H., Chater K. F., Deng Z., and Tao M. (2008) A cellulose synthase-like protein involved in hyphal tip growth and morphological differentiation in Streptomyces. J. Bacteriol. 190, 4971–4978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.van Veluw G. J., Petrus M. L., Gubbens J., de Graaf R., de Jong I. P., van Wezel G. P., Wösten H. A., and Claessen D. (2012) Analysis of two distinct mycelial populations in liquid-grown Streptomyces cultures using a flow cytometry-based proteomics approach. Appl. Microbiol. Biotechnol. 96, 1301–1312 [DOI] [PubMed] [Google Scholar]
- 30.Chaplin A. K., Petrus M. L., Mangiameli G., Hough M. A., Svistunenko D. A., Nicholls P., Claessen D., Vijgenboom E., and Worrall J. A. (2015) GlxA is a new structural member of the radical copper oxidase family and is required for glycan deposition at hyphal tips and morphogenesis of Streptomyces lividans. Biochem. J. 469, 433–444 [DOI] [PubMed] [Google Scholar]
- 31.van Dissel D., Claessen D., Roth M., and van Wezel G. P. (2015) A novel locus for mycelial aggregation forms a gateway to improved Streptomyces cell factories. Microb. Cell Fact. 14, 44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cantarel B. L., Coutinho P. M., Rancurel C., Bernard T., Lombard V., and Henrissat B. (2009) The Carbohydrate-Active EnZymes database (CAZy): an expert resource for glycogenomics. Nucleic Acids Res. 37, D233–D238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liman R., Facey P. D., van Keulen G., Dyson P. J., and Del Sol R. (2013) A laterally acquired galactose oxidase-like gene is required for aerial development during osmotic stress in Streptomyces coelicolor. PLoS ONE 8, e54112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Blundell K. L., Wilson M. T., Svistunenko D. A., Vijgenboom E., and Worrall J. A. (2013) Morphological development and cytochrome c oxidase activity in Streptomyces lividans are dependent on the action of a copper bound Sco protein. Open Biol. 3, 20163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Blundell K. L., Hough M. A., Vijgenboom E., and Worrall J. A. (2014) Structural and mechanistic insights into an extracytoplasmic copper trafficking pathway in Streptomyces lividans. Biochem. J. 459, 525–538 [DOI] [PubMed] [Google Scholar]
- 36.Petrus M. L., Vijgenboom E., Chaplin A. K., Worrall J. A., van Wezel G. P., and Claessen D. (2016) The DyP-type peroxidase DtpA is a Tat-substrate required for GlxA maturation and morphogenesis in Streptomyces. Open Biol. 6, 150149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cruz-Morales P., Vijgenboom E., Iruegas-Bocardo F., Girard G., Yáñez-Guerra L. A., Ramos-Aboites H. E., Péronnet J. L., Anné J., van Wezel G. P., and Barone-Gómez F. (2013) The genome sequence of Streptomyces lividans 66 reveals a novel tRNA-dependent peptide biosynthetic system within a metal-related genomic island. Genome Biol. Evol. 5, 1165–1175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gasteiger E., Gattiker A., Hoogland C., Ivanyi I., Appel R. D., and Bairoch A. (2003) ExPASy: The proteomics server for in-depth protein knowledge and analysis. Nucleic Acids Res. 31, 3784–3788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Svistunenko D. A., and Cooper C. E. (2004) A new method of identifying the site of tyrosyl radicals in proteins. Biophys. J. 87, 582–595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kabsch W. (2010) XDS. Acta Crystallogr. D Biol. Crystallogr. 66, 125–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Winter G. (2010) xia2: an expert system for macromolecular crystallography data reduction. J. Appl. Crystallogr. 43, 186–190 [Google Scholar]
- 42.Evans P. R., and Murshudov G. N. (2013) How good are my data and what is the resolution? Acta Crystallogr. D Biol. Crystallogr. 69, 1204–1214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Langer G., Cohen S. X., Lamzin V. S., and Perrakis A. (2008) Automated macromolecular model building for x-ray crystallography using ARP/wARP version 7. Nat. Protoc. 3, 1171–1179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Long F., Vagin A. A., Young P., and Murshudov G. N. (2008) BALBES: a molecular-replacement pipeline. Acta Crystallogr. D Biol. Crystallogr. 64, 125–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Murshudov G. N., Vagin A. A., and Dodson E. J. (1997) Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D Biol. Crystallogr. 53, 240–255 [DOI] [PubMed] [Google Scholar]
- 46.Emsley P., and Cowtan K. (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
- 47.Davis I. W., Leaver-Fay A., Chen V. B., Block J. N., Kapral G. J., Wang X., Murray L. W., Arendall W. B. 3rd, Snoeyink J., Richardson J. S., and Richardson D. C. (2007) MolProbity: all-atom contacts and structure validation for proteins and nucleic acids. Nucleic Acids Res. 35, W375–W383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Vaaje-Kolstad G., Houston D. R., Riemen A. H., Eijsink V. G., and van Aalten D. M. (2005) Crystal structure and binding properties of the Serratia marcescens chitin-binding protein CBP21. J. Biol. Chem. 280, 11313–11319 [DOI] [PubMed] [Google Scholar]
- 49.Vaaje-Kolstad G., Bøhle L. A., Gåseidnes S., Dalhus B., Bjørås M., Mathiesen G., and Eijsink V. G. (2012) Characterization of the chitinolytic machinery of Enterococcus faecalis V583 and high-resolution structure of its oxidative CBM33 enzyme. J. Mol. Biol. 416, 239–254 [DOI] [PubMed] [Google Scholar]
- 50.Wong E., Vaaje-Kolstad G., Ghosh A., Hurtado-Guerrero R., Konarev P. V., Ibrahim A. F., Svergun D. I., Eijsink V. G., Chatterjee N. S., and van Aalten D. M. (2012) The Vibrio cholerae colonization factor GbpA possesses a modular structure that governs binding to different host surfaces. PLoS Pathog. 8, e1002373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hemsworth G. R., Taylor E. J., Kim R. Q., Gregory R. C., Lewis S. J., Turkenburg J. P., Parkin A., Davies G. J., and Walton P. H. (2013) The copper active site of CBM33 polysaccharide oxygenases. J. Am. Chem. Soc. 135, 6069–6077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Forsberg Z., Mackenzie A. K., Sørlie M., Røhr Å. K., Helland R., Arvai A. S., Vaaje-Kolstad G., and Eijsink V. G. (2014) Structural and functional characterization of a conserved pair of bacterial cellulose-oxidizing lytic polysaccharide monooxygenases. Proc. Natl. Acad. Sci. U.S.A. 111, 8446–8451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Forsberg Z., Røhr A. K., Mekasha S., Andersson K. K., Eijsink V. G., Vaaje-Kolstad G., and Sørlie M. (2014) Comparative study of two chitin-active and two cellulose-active AA10-type lytic polysaccharide monooxygenases. Biochemistry 53, 1647–1656 [DOI] [PubMed] [Google Scholar]
- 54.Nakagawa Y. S., Kudo M., Loose J. S., Ishikawa T., Totani K., Eijsink V. G., and Vaaje-Kolstad G. (2015) A small lytic polysaccharide monooxygenase from Streptomyces griseus targeting α- and β-chitin. FEBS J. 282, 1065–1079 [DOI] [PubMed] [Google Scholar]
- 55.Hemsworth G. R., Davies G. J., and Walton P. H. (2013) Recent insights into copper-containing lytic polysaccharide mono-oxygenases. Curr. Opin. Struct. Biol. 23, 660–668 [DOI] [PubMed] [Google Scholar]
- 56.Römling U. (2002) Molecular biology of cellulose production in bacteria. Res. Microbiol. 153, 205–212 [DOI] [PubMed] [Google Scholar]
- 57.Ross P., Mayer R., and Benziman M. (1991) Cellulose biosynthesis and function in bacteria. Microbiol. Rev. 55, 35–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Peisach J., and Blumberg W. E. (1974) Structural implications derived from the analysis of electron paramagnetic resonance spectra of natural and artificial copper proteins. Arch. Biochem. Biophys. 165, 691–708 [DOI] [PubMed] [Google Scholar]
- 59.Frandsen K. E., Simmons T. J., Dupree P., Poulsen J. C., Hemsworth G. R., Ciano L., Johnston E. M., Tovborg M., Johansen K. S., von Freiesleben P., Mar muse L., Fort S., Cottaz S., Driguez H., Henrissat B., et al. (2016) The molecular basis of polysaccharide cleavage by lytic polysaccharide. Nat. Chem. Biol. 12, 298–303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Banci L., Bertini I., Cantini F., Felli I. C., Gonnelli L., Hadjiliadis N., Pierattelli R., Rosato A., and Voulgaris P. (2006) The Atx1-Ccc2 complex is a metal-mediated protein-protein interaction. Nat. Chem. Biol. 2, 367–368 [DOI] [PubMed] [Google Scholar]
- 61.Dwarakanath S., Chaplin A. K., Hough M. A., Rigali S., Vijgenboom E., and Worrall J. A. (2012) Response to copper stress in Streptomyces lividans extends beyond genes under direct control of a copper-sensitive operon repressor protein (CsoR). J. Biol. Chem. 287, 17833–17847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Mortazavi A., Williams B. A., McCue K., Schaeffer L., and Wold B. (2008) Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 5, 621–628 [DOI] [PubMed] [Google Scholar]