

Abstract
We have recently discovered that the ZZ zinc finger domain represents a novel small ubiquitin-like modifier (SUMO) binding motif. In this study we identify the binding epitopes in the ZZ domain of CBP (CREB-binding protein) and SUMO1 using NMR spectroscopy. The binding site on SUMO1 represents a unique epitope for SUMO interaction spatially opposite to that observed for canonical SUMO interaction motifs (SIMs). HADDOCK docking simulations using chemical shift perturbations and residual dipolar couplings was employed to obtain a structural model for the ZZ domain-SUMO1 complex. Isothermal titration calorimetry experiments support this model by showing that the mutation of key residues in the binding site abolishes binding and that SUMO1 can simultaneously and non-cooperatively bind both the ZZ domain and a canonical SIM motif. The binding dynamics of SUMO1 was further characterized using 15N Carr-Purcell-Meiboom-Gill (CPMG) relaxation dispersions, which define the off rates for the ZZ domain and SIM motif and show that the dynamic binding process has different characteristics for the two cases. Furthermore, in the absence of bound ligands SUMO1 transiently samples a high energy conformation, which might be involved in ligand binding.
Keywords: biophysics, nuclear magnetic resonance (NMR), protein structure, protein-protein interaction, structural biology, structural model, SUMO-interacting motif (SIM), protein dynamics
Introduction
SUMO2 (small ubiquitin-like modifier) is a protein structurally homologous to ubiquitin that functions as a post-translational modifier involved in diverse cellular processes such as chromatin remodeling, ubiquitin E3 ligation, autophagy, cytoskeletal scaffolding, and DNA repair. Higher eukaryotes encode at least three different functional SUMO isoforms, in the human genome designated as SUMO1–3, which can conjugate to a number of target proteins (1–3). SUMO modified target proteins are recognized by SUMO interacting motifs (SIMs), the minimal consensus sequence, which is ψKXE, where ψ consists of a large hydrophobic amino acid and where X can be any amino acid (4, 5). In addition to this canonical type of SIM, closely related variants such as inverted SIMs and phosphorylation-dependent SIMs have also been recently described (6–8). The binding site for the canonical SIM is located in a groove between the α-helix and β-sheet in SUMO, where the SIM motif can bind either in a parallel or anti-parallel fashion (6).
We recently showed that the ubiquitin ligase HERC2 can bind SUMO1 through its zinc finger ZZ domain (9), which therefore represents a new class of SUMO binding motifs. Isothermal titration calorimetry (ITC) showed that the ZZ domain binds to SUMO1 with μm affinity in the same range as the canonical SIMs (10). The ZZ domain consists of two anti-parallel β-sheets and a short α-helix coordinating two zinc ions as revealed by the first published ZZ domain structure originating from CBP (CREB-binding protein/p300) (11). There are ∼20 ZZ domains (11, 12) identified in the human genome, but a general biological function has not been assigned.
Here we present the complex between SUMO1 and the ZZ domain from CBP, where we map the interaction interface using NMR spectroscopy. The structure of this particular ZZ domain has previously been determined in solution by NMR and thus has favorable properties for structural studies (11). Using 15N HSQC spectra, we could identify the interaction surfaces on the ZZ domain and SUMO1, where the residues affected by the binding were broadened beyond detection. On SUMO1 this constituted a unique binding epitope that subsequently could be used to model the protein-protein complex using HADDOCK docking simulations (13).
Furthermore, to address the protein dynamics between different states of SUMO1, we performed CPMG relaxation dispersion experiments to map the micro- to millisecond dynamics of SUMO1, where we discovered that apoSUMO1 experiences intrinsic conformational exchange. The conformational exchange was quenched in SIM-bound SUMO1, whereas the intrinsic conformational exchange in ZZ domain-bound SUMO1 was largely unperturbed.
Experimental Procedures
Molecular Biology
DNA sequences for the ZZ domain from CBP (UniProtKB = Q92793) and SUMO1 (UniProtKB = P63165) were synthesized by Geneart and subcloned into a pNIC28-BsaI vector using ligation independent cloning (14). The resulting expression constructs contained a His-tag capture sequence and a tobacco etch virus protease cleavage site at the N terminus preceding the expressed protein of interest. To obtain SUMO1 protein that behaves as monomers in solution without the need of the addition of an reducing agent, the single cysteine residue in SUMO1, Cys-52, was mutated to serine using QuikChange site-directed mutagenesis (15) and transformed into MACH-1 cells. The SUMO1 mutant was shown to have the same structural and binding properties as the wild type protein (data not shown). The C52S mutant was used in all subsequent experiments and is hereafter referred to as SUMO1. The sequences for the expression constructs were verified by sequencing. Finally, to test the importance of residues in the identified binding epitopes, one mutant was created for the ZZ domain, denoted ZZmut, corresponding to residues N36A, K38A, and S39A, whereas two mutants were designed for SUMO1 corresponding to residues H75A, K78A, and E83A denoted SUMO1mut1 and K16A, H43A, H75A, K78A, and E83A denoted SUMO1mut2, respectively.
Protein Expression and Purification
Plasmids were transformed into Escherichia coli BL21 (DE3) Rosetta cells before protein expression. Protein expression was started from overnight cultures grown in Terrific Broth (TB) medium supplemented with 50 μg/ml kanamycin and 30 μg/ml chloramphenicol. SUMO1 and the SUMO1 mutants were expressed in TB media with 50 μg/ml kanamycin and 30 μg/ml chloramphenicol, grown to A600 ∼ 0.6–0.7 at 37 °C, induced with 0.5 mm isopropyl-β-thiogalactopyranoside (IPTG), and grown overnight at 18 °C. The ZZ domain and the ZZ mutant were expressed in TB media as above with the addition of 150 μm ZnCl2 according to a previously published protocol (11). For the NMR experiments, SUMO1 and the ZZ domain were labeled with 15N and 13C using minimal medium (16). SUMO1 was grown in minimal medium to A600 ∼ 0.6–0.7, induced with 0.5 mm IPTG, and grown overnight at 18 °C. The ZZ domain was grown in minimal media with 150 μm ZnCl2 until reaching an A600 ∼ 1.2–1.5, induced with 1.0 mm IPTG, and grown overnight at 18 °C. Cells were harvested by centrifugation at 6000 rpm for 10 min, resuspended in 100 mm Tris-HCl, pH 8.0, 300 mm NaCl, and 10 mm imidazole with benzonase and Complete-EDTA free tablets (Roche Life Sciences), homogenized using a French press cell disruptor, and centrifuged at 11,000 × g for 30 min. Purification of the proteins was performed using immobilized metal ion affinity chromatography (IMAC) on an ÄKTA express system (GE Healthcare) at 4 °C with the HiTrap chelating columns (17) equilibrated with 50 mm Tris-HCl, pH 8.0, 300 mm NaCl, 10 mm imidazole and eluted using a linear gradient of 10–500 mm imidazole. Protein identity was verified using mass spectrometry (MS). The eluted fractions with protein were collected, dialyzed (molecular mass cutoff 3500 Da) against 50 mm Tris-HCl, pH 7.5, and processed with tobacco etch virus protease to remove the His tag. The tobacco etch virus protease treatment retained the amino acid pair Ser-Met at the N terminus of the expressed proteins. We, therefore, designated the first two residues as Ser-0 and Met-1 for both the ZZ domain and SUMO1. The final amino acid sequences for the two proteins are, therefore: ZZ domain, SM1QDRFVYTCNECKHHVETRWHCTVCEDYDLCINCYNTKSHAHKMVKWGLGLDD53 (the underline indicates Met at position 2); SUMO1, SM1SDQEAKPSTEDLGDKKEGEYIKLKVIGQDSSEIHFKVKMTTHLKKLKESYSQRQGVPMNSLRFLFEGQRIADNHTPKELGMEEEDVIEVYQEQTGGHSTV101.
In the final purification step the proteins were loaded onto an ion exchange Mono Q column (ÄKTA, GE Healthcare) equilibrated with 50 mm Tris, pH 7.5, and eluted with a linear gradient of 0–0.4 mm NaCl. The purity was verified by SDS-PAGE and electrospray ionization mass spectrometry (ESI-MS).
Peptide Synthesis
The SIM-peptide from PIASX (6), hereinafter denoted SIMPX, corresponding to the sequence KVDVIDLTIESSSDEEEDPPAKRQM, was synthesized by Biosyntan Gmbh (Berlin, Germany). Purity and molecular mass was confirmed by MALDI-TOF MS.
Protein Characterization
The secondary structure of the ZZ domains (ZZ wild type and ZZmut) and the SUMO1 variants (SUMO1, SUMO1mut1, and SUMO1mut2) was verified by circular dichroism (CD) spectroscopy using 0.5–1.0 mg/ml protein in 20 mm MOPS buffer at pH 6.8.
ITC Experiments
ITC experiments were performed using a VP-ITC200 instrument (GE Healthcare). The samples were extensively dialyzed against 20 mm MOPS, pH 6.8. For the experiments addressing the binding properties of the ZZ domains (ZZ wild type and ZZmut) and the SUMO variants (SUMO1, SUMO1mut1, and SUMO1mut2), 0.5 mm SUMO1 or SUMO1 mutant (SUMO1mut1 or SUMO1mut2) was titrated into 0.04 mm ZZ wild type or ZZ mutant. The concentrations were determined from the respective absorbance at 280 nm. In the experiments investigating the SUMO binding of the ZZ domain and the SIM peptide SIMPX, 0.5 mm SIMPX was titrated into the ITC sample cell containing 0.05 mm SUMO1. Finally, 0.5 mm SIMPX was titrated into a sample containing the pre-formed ZZ domain-SUMO1 complex. All experiments were performed at 10 °C and run until saturation was achieved. The data were fitted using a model describing one binding site using the software provided by the manufacturer (18).
NMR Experiments and Resonance Assignments
Protein samples were dialyzed against 20 mm MOPS, pH 6.8, and concentrated to 0.3–0.5 mm using Vivaspin concentrators with a molecular mass cut-off of 3500 Da. D2O was added to a concentration of 7% (v/v) for the spectrometer lock, and NaN3 was added to a concentration of 0.02% (w/v) to prevent bacterial growth in the samples. The backbone resonance assignments for SUMO1 and the ZZ domain were based on published assignments (11, 19) in combination with standard three-dimensional triple-resonance experiments for the backbone assignments (HNCA (20), HNCOCA (21), HNCACB (22), and CBCACONH (23)). Spectra were processed with NMRpipe (24), applying zero-filling and linear prediction in the indirect dimensions and solvent filter and polynomial baseline correction in the direct dimension. CCPNMR (25) was used for visualization of spectra and resonance connectivity analysis. Resonance assignment was achieved for 98% of the backbone amides in the ZZ domain and 86% in SUMO1. The SIMPX-bound SUMO1 was assigned by performing a titration of SIMPX into a SUMO1 sample and by following the chemical shift changes upon complex formation. The samples for measuring residual dipolar couplings (RDCs) were prepared by adding the PF1 phage (ASLA Biotech) to a final concentration of 10 mg/ml in the NMR samples.
RDC Measurements and Molecular Modeling
RDCs were measured using interleaved in-phase-anti-phase experiments (26) at 25 °C with spectral widths of 8000 Hz (1H) and 1835 Hz (15N) sampled over 1024 and 256 points, respectively. RDCs were measured for each component of the ZZ domain-SUMO1 complex using two separate samples, in which one protein was labeled with 13C,15N and the other unlabeled. In total, 91 RDCs were collected, 44 for SUMO1 and 47 for the ZZ domain.
Formation of the ZZ-SUMO1 and SIMPX-SUMO1 Complexes
To confirm binding and establish complexes with 1:1 stoichiometry, the NMR samples were prepared by ITC titrations of unlabeled ZZ domain into 13C,15N-labeled SUMO1 and vice versa. The final NMR samples of the ZZ domain-SUMO1 complex contains 4% unbound SUMO1, as calculated from the Kd and protein concentrations. The complex between 13C,15N-labeled SUMO1 and SIMPX was formed in a similar way using ITC titrations resulting in NMR samples of the SIMPX-SUMO1 complex that contains 2% unbound SUMO1. 15N HSQC experiments were subsequently run on the complexes, and the resulting spectra were compared with those for unbound proteins.
Structure Calculations
Structural refinements of SUMO1 and the ZZ domain was done using Xplor-NIH (27). For the ZZ domain, NOE restraints and dihedral and hydrogen bond restraints were adopted from the published structure (Protein Data Bank code 1TOT) (11) and imported from the Biological Magnetic Resonance Bank (BMRB) and converted from Amber (28) to Xplor-NIH format using in-house scripts. Zinc ions were included into the structure calculations by adding distance and angle restraints in a similar way as for the published structure of the ZZ domain of CBP (11). Structure calculations were initiated from an elongated protein structure using a simulated annealing protocol in which NOE, dihedral, hydrogen bond, and RDC restraints were applied as well as restraints for the zinc coordination. Structure calculations were performed in an iterative fashion, where NOE, dihedral angle, and RDC restraints were iteratively pruned from the calculations to obtain a converged molecular structure ensemble. Structure analysis was done using the protein structure validation software suite (PSVS) (29) and PALES (30). Residues in the N and C termini (1–5, 44, 50–53) were considered flexible based on observed peak intensities in the 15N HSQC and were excluded from the structure calculations. In the case of the NMR structure of SUMO1 (Protein Data Bank code 1A5R), the restraints are not available. Synthetic NOE distance restraints were, therefore, created by collecting all 1H-1H distances in the first model of the structural ensemble of SUMO1 excluding all restraints corresponding to distances longer than 6.5Å or shorter than 2.8Å. The structure calculation protocol was otherwise the same as for the ZZ domain, where NOE distance restraints and RDCs were iteratively pruned. The weight of RDC restraints in the structure calculations was increased as compared with the calculations of the structure of the ZZ domain due to the use of synthetic NOE restraints. Residues in the N and C termini were considered flexible (1–20, 95–103) based on R20 values from the CPMG relaxation dispersion experiments and were excluded from the structure calculations. The structure analysis were performed using the PSVS suite (29) and PALES (30).
Docking Simulations
Docking simulations were performed using HADDOCK (13) and CNS (31) following published protocols (32). Chemical shift changes upon binding were used as ambiguous interaction restraints in combination with RDCs measured for the ZZ domain-SUMO1 complex. The docking simulations involved active restraints for those residues experiencing chemical exchange, whereas neighboring residues were treated as passive. Residues with a solvent-accessible surface area (SASA) <20% were filtered out. The active residues in the ZZ domain were Asn-36, Lys-38, and Ala-41, whereas the passive residues were Glu-11, Cys-12, Trp-20, Val-24, Cys-31, Ile-32, Asn-33, Tyr-35, Thr-37, Ser-39, and His-42. The active residues in SUMO1 were His-43, Lys-46, Met-82, Glu-83, and Glu-85, whereas the passive residues were Lys-23, Lys-25, Thr-42, Lys-45, Gly-81, Glu-84, and Val-87. RDCs belonging to residues in the flexible N and C termini were not included as restraints in the docking simulations. The protonation state of the two histidine residues (His-40 and His-42), coordinating the zinc ion in the metal site of the ZZ domain, was set to match the metal coordination pattern (11). Docking simulations were run using the standard HADDOCK protocol with 1000 initial docking simulations followed by 200 refinement simulations and subsequently 200 final refinement simulations including explicit water molecules. RDCs were used as intervector projection angle restraints (33) in the first two simulation steps and as direct restraints in the final water refinement procedure.
NMR Relaxation Dispersion Experiments
NMR 15N CPMG relaxation dispersion was measured using continuous time CPMG relaxation dispersion experiments (34, 35) at static magnetic field strengths of 14.1 and 18.9 tesla at 10 °C. The temperature was calibrated using a methanol sample before each series of experiments. The constant relaxation time was 40 ms. The experiments utilized the phase cycle proposed by Yip and Zuiderweg (36) to suppress artifacts due to off-resonance effects. The effective relaxation rate (R2eff) at a given CPMG refocusing frequency was determined from two data points as described (35). Relaxation dispersions were sampled using 18–24 R2eff values. The spectral widths were 8,000 Hz (1H) and 1,835 Hz (15N) at 14.1 tesla and 10,666 Hz (1H) and 2447 Hz (15N) at 18.9 tesla, with 512 and 64 points collected in direct and indirect dimensions, respectively. Spectra were processed using NMRpipe (24). The processing protocol involved a solvent filter, cosine-squared apodization functions, zero filling to twice the number of increments in all dimensions, and a polynomial baseline correction in the 1H dimension. Peak intensities were measured as the integral over 5 × 3 points (1H/15N) centered on the peak maximum. The signal-to-noise ratio was estimated by calculating the standard deviation of 200 samples of integrated 5 × 3-point windows in empty regions of each spectrum. Errors in the extracted R2eff were estimated from the signal-to-noise ratio using error propagation. The relaxation dispersion data were analyzed using CPMGfit v2.3 (38). Relaxation dispersion curves were fitted to the Carver-Richards (39) two-state exchange model (40),
in which
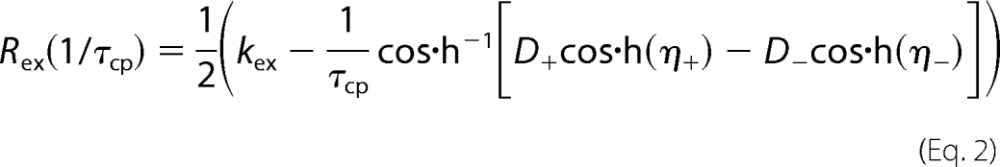 |
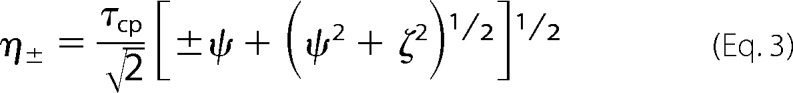 |
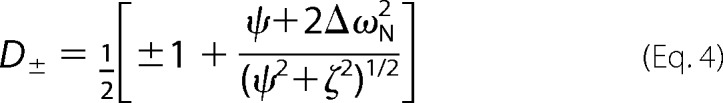 |
and ψ = kex2 − Δω2, ζ = −2Δωkex(1 − 2pm), and kex = k1 + k−1 is the sum of the forward and reverse rate constants, Δω is the chemical shift difference between the exchanging conformations, R20 is the average limiting value of the relaxation rate constant for processes other than chemical exchange, pm is the relative population of the minor (less populated) conformational state, which is related to the major conformational state pM as pm = 1 − pM, and τcp = 1/ncp is the spacing between refocusing pulses in the CPMG train.
The statistical significance of each fit was assessed by also fitting the data to a constant R20 value (i.e. modeling a flat dispersion profile, indicating the absence of exchange), and the F test was used to discriminate between models by rejecting the null hypothesis that the model with more parameters does not provide a significantly better fit than the simpler model at the level p < 0.001. Errors in the fitted parameters were estimated from 1000 synthetic data sets created using Monte-Carlo simulations (41, 42).
Results
To define the molecular details in the interaction between the ZZ domain and SUMO1, we employed a combination of chemical shift mapping, RDCs, docking simulations, and protein engineering. In addition, experiments addressing the dynamic properties of the interactions between SUMO1 and SIMs were performed to advance our understanding of SUMO recognition.
Chemical Shift Mapping of the Binding Interface between SUMO1 and the ZZ Domain
The binding interface on the ZZ domain and SUMO1 was mapped using binding-induced chemical shift differences measured in 15N HSQC spectra, where the binding was verified by ITC. A comparison of these 15N HSQC spectra shows that six residues in the labeled SUMO1 are broadened beyond detection upon the addition of the ZZ domain due to intermediate chemical exchange between the free and bound states (Fig. 1A). In a similar fashion, five residues in the ZZ domain are broadened beyond detection upon adding SUMO1 (Fig. 1B). The affected residues in SUMO1 are located in a contiguous region of the structure: Leu-24 is situated in a β-strand, and Met-82, Glu-83, and Glu-85 are found in an adjacent loop, whereas His-43 and Lys-46 are located in the N-terminal part of the α-helix and preceding loop (Fig. 1C). This binding site is spatially distinct from that observed for classical SIMs, which is located on the opposite side of the protein (4–6). The perturbed residues in the ZZ domain are located in the α-helix and the following loop (Fig. 1D). The side chains of Cys-34 and His-40 are part of the metal coordination for both of the Zn2+ ions present in the ZZ domain, indicating that the presence of bound Zn2+ ions might be important for binding SUMO1. The observed chemical shift changes define a binding site for the interaction between the ZZ domain and SUMO1 not previously described. To further define the binding interface, attempts were made to record intermolecular NOEs using three-dimensional 13C,15N-filtered NOESY experiments and two-dimensional 13C,15N-filtered NOESY experiments, where the three-dimensional experiment did not provide reliable intermolecular NOEs due to low signal-to-noise, whereas the two-dimensional experiments produced highly ambiguous intermolecular NOEs due to extensive spectral overlap. No intermolecular NOEs were, therefore, included in the generation of the structure of the ZZ domain-SUMO1 complex.
FIGURE 1.

Chemical shift differences observed when titrating the unlabeled ZZ domain or SUMO1 into 13C,15N-labeled SUMO1 or ZZ domain, respectively. A, cutout of 15N HSQC showing backbone amides in SUMO1 affected by the binding of the ZZ domain. B, cutout of 15N HSQC showing backbone amides in the ZZ domain affected by the binding of SUMO1. Contours colored in black correspond to the apo form, whereas contours colored in red correspond to ZZ domain-bound SUMO1. C, residues in SUMO1 affected by the binding of ZZ are highlighted in red: Leu-24, His-43, Lys-46, Met-82, Glu-83, and Glu-85 (Protein Data Bank code 1A5R). D, residues in the ZZ domain affected by the binding of SUMO1 are highlighted in red: Cys-34, Asn-36, Lys-38, His-40, and Ala-41. Zinc ions are depicted as blue spheres (Protein Data Bank code 1TOT). Protein images were made using Molmol (37).
RDC Measurements and Molecular Refinements
Backbone H-N bond vector orientations were determined from 44 RDCs for SUMO1 and 47 RDCs for the ZZ domain. The alignment tensors were calculated for each protein using published NMR structures (SUMO1, Protein Data Bank code 1A5R (Ref. 19) and the ZZ domain, Protein Data Bank code 1TOT (Ref. 11)) with the structure software PALES (30) with Q values of 0.93 and 0.90, respectively (43). The initial agreement between the RDCs back-calculated from the published structures and the experimental RDCs is low (Fig. 2, A and B), most likely because the structures have not been refined using RDC restraints. To improve the correlation between experimental and theoretical RDCs, the published protein structures were, therefore, first refined versus the experimental RDCs. Given that the changes in chemical shifts are minor upon forming the protein-protein complex, we do not expect any major conformational changes upon formation of the ZZ domain-SUMO1 complex. Hence, as an initial approximation, we assumed that the structure of the complex could be modeled using the published structures and the RDCs measured for the complex. Structural restraints have been published for the ZZ domain (Protein Data Bank code 1TOT), and these were converted to Xplor-NIH format using a structure refinement protocol from the program suite (27). RDCs measured for the protein-protein complex as well as NOE distance restraints, dihedral angle restraints, and hydrogen bond restraints were used for the refinement. Before refinement, the protein was modeled to correspond to the human ZZ domain by adding a C-terminal Asp and mutating Thr-41 to Ala-41 followed by energy minimization using Xplor-NIH. Structural refinement was done using the first model of the published structure ensemble as the input structure. Residues in the N and C termini (1–5, 49–53) were considered flexible based on the observed peak intensities in the 15N HSQC. Refinement was performed in an iterative manner until the resulting structure showed no NOE violations >0.5 Å. Because there are no published distance restraints available for SUMO1, we employed a different approach for the RDC refinement of this structure. Synthetic NOE distance restraints were created by calculating all 1H-1H distances in the first model of the published protein structure (Protein Data Bank code 1A5R), filtering out distances larger than 6.5 Å and shorter than 2.8 Å. In addition, the protein structure was prepared for refinement by changing Cys-52 to Ser-52. Refinement was done iteratively using the same protocol as for the ZZ domain. Residues in the N and C termini were considered flexible (1–20, 95–103) based on CPMG relaxation dispersion experiments and excluded from the calculations. The refinement was performed iteratively until the resulting structure had no NOE violations >0.5 Å. For each iteration, NOE violations >0.5 Å and distances >6.5 Å were filtered out. Q values after refinement were 0.30 and 0.26 for the ZZ domain and SUMO1, respectively (Fig. 2, C and D) (43). The refined protein structures for the ZZ domain and SUMO1 are highly similar to the original structures after RDC refinement with r.m.s.d. values for Cα of 0.29 Å and 0.68 Å for the ZZ domain (residues 6–48) and SUMO1 (residues 21–94), respectively. The main structural differences in terms of Cα r.m.s.d. for the ZZ domain are observed for residues 10–18 in β-strand 1 and in the SUMO1 binding loop 36–46. The main differences in Cα r.m.s.d. for SUMO1 are seen for residues 33–43 in β-strand 2 and for residues 70–87 in the ZZ domain binding loop.
FIGURE 2.

Residual dipolar couplings for SUMO1 (A and C) and the ZZ domain (B and D) displayed before and after structure refinement using residual dipolar couplings as additional restraints. Experimental RDCs are plotted versus back-calculated RDCs as calculated from the initial (A and B) and the final structures after refinement (C and D).
Structural Model for the ZZ Domain-SUMO1 Complex
After final refinement in water, the docked structures were clustered using the standard HADDOCK algorithm, where 115 structures were clustered into one ensemble (Fig. 3, A and B). Nine additional clusters of structure ensembles were found, each containing 10 structures or fewer. The r.m.s.d. of all residues in the interacting protein interface is 1.6 ± 0.6 Å, whereas the r.m.s.d. between all backbone atoms in the docked structure was 1.6 ± 0.6 Å. The r.m.s.d. between the protein interface and backbone atoms were 1.3 ± 0.4 Å and 2.2 ± 0.6 Å for SUMO1 and the ZZ domain, respectively (Fig. 3, C and D). The HADDOCK score for the final structure ensemble was −43 ± 18, and the Q value calculated for the complex was 0.36, showing that the experimental RDCs demonstrate a good agreement with the calculated RDCs for the modeled protein complex (Fig. 3E) (43). The magnitude and orientation of the RDC alignment tensor for the ZZ domain and SUMO1 and the ZZ domain-SUMO1 complex was calculated using Module (45), where the magnitudes and rhombocities are shown in Table 1. Using Module, the RDC alignment tensor was fitted separately for the ZZ domain and SUMO1 in the protein complex indicating highly similar tensor orientations and magnitudes (Fig. 3F). The observed structures for the ZZ domain and SUMO1 in the protein complex are nearly identical to the individually refined structures of the uncomplexed proteins with Cα r.m.s.d. of 0.001 Å for both the ZZ domain and SUMO1. By extending the comparisons between complexed and uncomplexed proteins to include all atoms in the assessments, also incorporating all surface accessible side chains, the r.m.s.d. were 2.32 Å and 2.29 Å for the ZZ domain and SUMO1 respectively.
FIGURE 3.

A, model of the ZZ domain-SUMO1 complex shown in a surface representation made using PyMOL (44). SUMO1 is colored green, the ZZ domain is colored red, and the peptide corresponding to a SIM motif is colored in blue (Protein Data Bank code 2ASQ; Ref. 6). B, model of the ZZ domain-SUMO1 complex shown in a ribbon representation using the same color scheme as in A with the two zinc ions depicted as blue spheres. The residues in SUMO1 and the ZZ domain affected by the interaction in the NMR epitope mapping experiments are indicated in the model, corresponding to the same residues shown for the individual protein models in Fig. 1, C and D. C, per residue r.m.s.d. between interface residues and backbone Cα atoms in the modeled complex for SUMO1 residues 1–103. The location for the secondary structure elements of SUMO1 are indicated by arrows (red) for β-strands and a cylinder for the single α-helix (yellow). D, per residue r.m.s.d. between interface residues and backbone Cα atoms in the modeled complex for the ZZ domain residues 1–53. The location for the secondary structure elements of the ZZ domain is indicated by arrows (red) for β-strands and a cylinder for the helical segment (yellow). E, experimental RDCs plotted versus calculated RDCs for the modeled protein complex. F, orientations of the RDC alignment tensor for the ZZ domain and SUMO1 in the ZZ domain-SUMO1 complex in which tensor orientations were fitted using Module (45). The ZZ domain (yellow) and SUMO1 (blue) are shown in ribbon representations.
TABLE 1.
Summary of structural statistics for the structural refinement using RDCs of the ZZ domain and SUMO1 as well as structural statistics of the docked ZZ domain-SUMO1 complex
Structural analysis was performed using PSVS (29) and PALES (30). The magnitude and orientation of the RDC alignment tensor for the ZZ domain and SUMO1 and the ZZ domain-SUMO1 complex were calculated using Module (45).
| NMR and refinement statistics | Protein |
ZZ domain-SUMO1 complex | |
|---|---|---|---|
| ZZ domain | SUMO1 | ||
| NMR distance and dihedral constraints | |||
| Distance constraints | |||
| Total NOE | 547 | 3304 | |
| Intra-residue | 135 | 476 | |
| Inter-residue | |||
| Sequential (|i − j| = 1) | 172 | 1073 | |
| Medium range (|i − j| <4) | 78 | 644 | |
| Long range (|i − j| >5) | 162 | 1111 | |
| Intermolecular, hydrogen bonds | 5 | 0 | |
| Total dihedral angle restraints | 80 | 0 | |
| Residual dipolar coupling restraints | 42 | 37 | 79 |
| AIR restraintsa | 7 | ||
| Magnitude (Da) | 6.03 | 5.95 | 5.89 |
| Rhombicity | 0.56 | 0.536 | 0.56 |
| Q-factor | 0.30 | 0.26 | 0.36 |
| Structure statistics violations (RMS) | |||
| Distance constraints (Å) | 0.08 Å | 0.05 Å | |
| Dihedral angle constraints (°) | 1.73° | ||
| Max. dihedral angle violation (°) | 8.60° | ||
| Max. distance constraint violation (Å) | 0.55 Å | 0.77 Å | |
| Structure quality factors; overall statistics | |||
| Procheck G-factor (φ/ψ only) | −1.18 | −0.65 | −0.83 |
| Procheck G-factor (all dihedral angles) | −1.04 | −0.39 | −0.56 |
| Verify three-dimensional | 0.27 | 0.30 | 0.29 |
| ProsaII (−ve) | −0.08 | 0.41 | −0.29 |
| MolProbity clash score | 64.01 | 43.88 | 54.67 |
| Ramachandran plot summary from Procheck | |||
| Most favored regions (%) | 63.3 | 70.0 | 77.6 |
| Additionally allowed regions (%) | 30.6 | 22.2 | 15.1 |
| Generously allowed regions (%) | 2.0 | 4.4 | 7.2 |
| Disallowed regions (%) | 4.1 | 3.3 | 0.1 |
a AIR restraints indicate the ambiguous restraints used in the HADDOCK docking procedure.
As expected, the binding interface for the docked protein-complex corresponds to the surface residues experiencing chemical shift perturbations upon binding. The binding surface on the ZZ domain consists of the residues spanning the C-terminal end of the α-helix to the loop connecting the α-helix to β-strand 1. The binding surface on SUMO1 consists of residues belonging to β-strand 1, the N-terminal part of the α-helix, and the preceding loop as well as the long loop connecting β-strands 3 and 4. The binding surface on SUMO1 consists of a convex groove on the protein surface. This interaction surface represents a completely new binding epitope for SUMO1, previously not observed in the Protein Data Bank, which is clearly separated in space from known interaction motifs. The previously known interaction motifs for SUMO1 are the SUMO-SIM motif (4–6), the Ubc9 motif, and the SENP/RanGAP motif (46, 47). None of these interaction motifs overlapped with the ZZ domain-SUMO1 binding site, suggesting that the ZZ domain can bind to SUMO1 in the presence of other interaction motifs.
To verify the binding mode observed in the ZZ domain-SUMO1 complex, we created one mutant of the ZZ domain and two mutants of SUMO1 where we mutated residues in the binding interface to alanines. The residues selected for alanine substitution where either residues with charged side chains or with side chains with the potential to form hydrogen bonds, thus removing important electrostatic interactions in the binding interface. We find that the ZZ domain mutant still had the ability to bind SUMO1, albeit with significantly lower enthalpy of binding as compared with the wild type (Fig. 4, A and B, and Table 2). The two mutants of SUMO1 did, however, completely abolish binding to the ZZ domains (Fig. 5, A–D), clearly indicating that these residues are important for the complex formation and further supporting the molecular model derived from the NMR data.
FIGURE 4.

ITC experiments show the binding between the ZZ domain and SUMO1, the SIM peptide and SUMO1, and the binding of the SIM peptide to the complex between the ZZ domain and SUMO1. The experiments show the titrations of the binding of the wild type ZZ domain (ZZwt) to the wild type SUMO1 (SUMO1wt) (A), the binding of the ZZ domain mutant (ZZmut) to the wild type SUMO1 (SUMO1wt) (B), the binding of the SIM peptide to SUMO1 (C), and the binding of the SIM peptide to the preformed complex between the ZZ domain and SUMO1 (D). The raw data of the experiments are presented on the top panel. The area underneath each injection peak is equal to the total heat released for that injection.
TABLE 2.
Summary of thermodynamic parameters from ITC experiments of the binding of the ZZ domain wild type (ZZwt), the ZZ domain mutant (ZZmut), and the SIM peptide SIMPX (6) to SUMO1, respectively
The fourth row shows the data for the binding of SIMPX to the preformed complex between the ZZ domain (ZZwt) and SUMO1.
| Protein complex | N (sites) | KD | ΔH | ΔS |
|---|---|---|---|---|
| μm | kcal/mol | cal/mol/deg | ||
| ZZwt/SUMO1 | 1.04 ± 0.06 | 5.4 ± 0.2 | −1.7 ± 0.1 | 18.1 |
| ZZmut/SUMO1 | 0.86 ± 0.07 | 6.5 ± 0.3 | −0.72 ± 0.08 | 21.2 |
| SIMPX/SUMO1 | 0.87 ± 0.01 | 6.2 ± 0.6 | −2.49 ± 0.05 | 15.5 |
| SIMPX/(ZZwt-SUMO1) | 0.88 ± 0.02 | 6.1 ± 0.7 | −2.27 ± 0.06 | 16.2 |
FIGURE 5.

ITC experiments between ZZ domain variants and SUMO1 variants. The experiments show the titrations of wild type ZZ domain (ZZwt) to the SUMO1 mutant 1 (SUMO1mut1) (A), ZZwt to SUMO1 mutant 2 (SUMOmut2) (B), ZZ domain mutant (ZZmut) to SUMO1mut1 (C), and ZZmut to SUMO1mut2 (D). The raw data of the experiments are presented on the top panel. The area underneath each injection peak is equal to the total heat released for that injection.
To compare the SUMO binding characteristics of the ZZ domain with a canonical SIM motif, we performed ITC experiments between SUMO1 and a SIM peptide denoted SIMPX from PIASx (6). As expected, we could show that SIMPX binds to SUMO1 with a similar affinity as the ZZ domain (Fig. 4C and Table 2). To investigate whether SIMPX and the ZZ domain can bind to SUMO simultaneously, ITC experiments between SIMPX and a preformed complex between the ZZ domain and SUMO1 were carried out. These experiments revealed that SIMPX binds with a similar affinity and enthalpy of binding to SUMO1 as it does in the absence of the ZZ domain, indicating that SIMPX can bind SUMO1 independent of the ZZ domain in a non-cooperative manner (Fig. 4, C and D, and Table 2).
CPMG Relaxations Dispersion Experiments
15N CPMG relaxation dispersions were measured for apo-, SIMPX-bound, and ZZ domain-bound SUMO1 at static magnetic field strengths of 14.1 and 18.9 tesla (supplemental Table 1). ApoSUMO1 showed 52 backbone amides experiencing significant conformational exchange (p < 0.01), as exemplified in Fig. 6A. The exchanged residues are highlighted in Fig. 6D, showing that large parts of the core domain (residues 20–95) underwent exchange. Each residue was initially fitted to residue-specific exchange parameters, kex and pm. Residues in the core domain with a kex < 2000 s−1 were fitted to a global exchange process, resulting in kex = 1185 ± 91 s−1 and pM = 0.987 ± 0.002.
FIGURE 6.

Example of 15N CPMG relaxation dispersion curves for apo-, SIMPX-bound, and ZZ domain-bound states of SUMO1 showing transverse relaxation rates R2 plotted versus the effective field νcp. The solid line corresponds to a CPMG relaxation dispersion curve fitted to a two-state chemical exchange process, whereas the dotted line corresponds to a model with no chemical exchange. A, CPMG relaxation dispersion curve for Val-38 in apoSUMO1. B, CPMG relaxation dispersion curve for Glu-20 in SIMPX-bound SUMO1. C, CPMG relaxation dispersion curve for Val-38 in the ZZ domain-bound SUMO1. Residues exhibiting significant (p < 0.01) CPMG relaxation dispersion curves are colored red on the structure of SUMO1. Structures are depicted in schematic representation. D, residues with significant CPMG relaxation dispersions in apoSUMO1: 7, 13, 15–16, 18–26, 28–38, 40, 42, 45–47, 49–50, 54–56, 61, 65, 67, 69–70, 74–76, 78, 81–83, 87, 90, 92, 94, 100–101 (Protein Data Bank code 1A5R). E, residues with significant CPMG relaxation dispersions in SIMPX-bound SUMO1: 7, 10, 15, 17–18, 20–21, 23, 26–28, 32, 38, 42–43, 48, 55, 57, 61–62, 64–65, 70, 74, 80–81, 83, 87, 100–101 (Protein Data Bank code 2ASQ). SIMPX is colored blue. F, residues with significant CPMG relaxation dispersions in ZZ domain-bound SUMO1: 2–5, 7–8, 10, 13–16, 18–22, 24–38, 40, 42, 45, 47, 49–50, 60–61, 67, 70, 74, 81–82, 90, 92, 94, 100. The ZZ domain is colored blue, and the zinc ions are depicted as green spheres. In panels D, E, and F a selected set of residues from different regions of SUMO1 with significant CPMG relaxation dispersions are indicated with their respective residue number.
15N CPMG relaxation dispersions for the SIMPX-bound SUMO1 revealed 30 residues experiencing exchange (Fig. 6, B and E). Residues were selected for a global fit based on the same criteria as for apoSUMO1 and were fitted to a global exchange rate kex = 479 ± 30 s−1 and major population pM = 0.942 ± 0.01 (Table 3).
TABLE 3.
Chemical exchange rates (kex), off-rates (koff) and major populations (pM) for the three different states of SUMO1 extracted from the global fits of significant CPMG relaxation dispersion curves
| State | kex | koff | pM |
|---|---|---|---|
| s−1 | s−1 | ||
| ApoSUMO1 | 1185 ± 91 | 28 ± 2 | 0.987 ± 0.002 |
| SIMPX-SUMO1 | 479 ± 30 | 15 ± 1 | 0.942 ± 0.01 |
| ZZ domain-SUMO1 | 1418 ± 203 | 31 ± 5 | 0.978 ± 0.019 |
15N CPMG relaxation dispersion experiments on ZZ domain-bound SUMO1 showed 48 residues with significant exchange (Fig. 6, C and F). Again, residues in the core domain of SUMO1 were selected for the global fit, giving a global exchange rate kex = 1418 ± 203 s−1 and major population pM = 0.978 ± 0.019 (Table 3). In comparison to apoSUMO1, the ZZ domain bound SUMO1 had a larger number of residues in the flexible N-terminal exhibiting significant dispersions. Residues belonging to the N-terminal part (2–5, 7–8, 10, 13–16, and 18–19) were fitted to a global exchange rate kex = 1924 ± 145 s−1 and a major population pM = 0.990 ± 0.002.
Comparison of Exchanging States in SUMO1
Upon the addition of SIMPX, most of the backbone residues in SUMO1 experienced a significant change in chemical environment as seen from a comparison of the cross-peak positions in the 15N HSQC for SIMPX-bound and apoSUMO1 (Fig. 7A). Residues not affected by the binding of SIMPX are located in the N- and C-terminal parts of SUMO1, whereas most backbone residues in the core region of SUMO1 experience a shift in chemical environment upon the addition of SIMPX (Fig. 7, B and C). However, only a subset of the residues affected by the binding of the SIM peptide is within 5 Å of SIMPX, as gauged from the SIM-SUMO1 structure (Protein Data Bank code 2ASQ). This result indicates that the binding of SIMPX induces a conformational change in SUMO1. A closer look at the residues exhibiting conformational exchange in apoSUMO1 reveals that these are primarily located in or close to the SIM binding site. In summary, all residues within 5 Å of SIMPX, with the exception of residues 39, 43, 48, and 53, exhibit conformational exchange in apoSUMO1.
FIGURE 7.
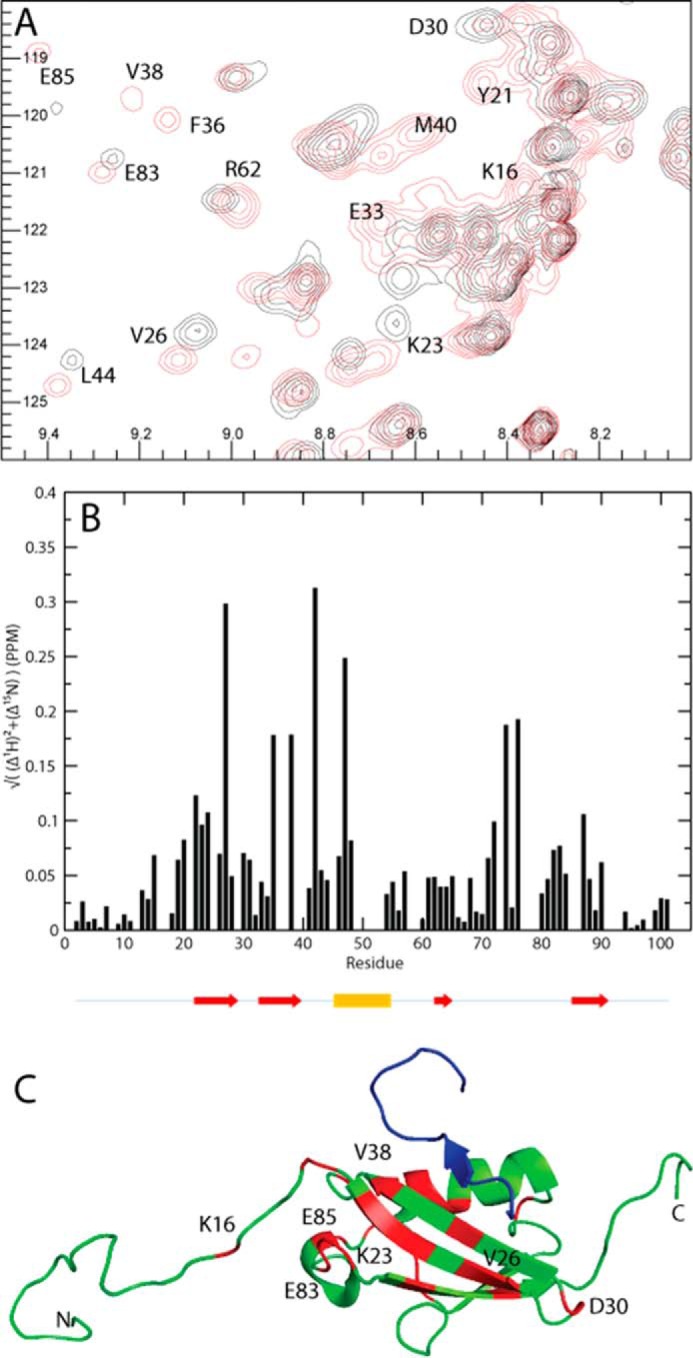
Chemical shift differences observed when titrating SIMPX into 13C,15N SUMO1. A, cutout of 15N HSQC of SUMO1 showing backbone amides in SUMO1 affected by the binding of SIMPX. Black corresponds to apo, and red corresponds to the SIMPX-bound SUMO1. The affected residues are indicated by their respective residue number. B, weighted 1H,15N chemical shift differences between the SIMPX and the apo state of SUMO1 plotted per backbone residue. The location for the secondary structure elements of SUMO1 are indicated by arrows (red) for β-strands and a cylinder for the single α-helix (yellow). C, SUMO1 is colored in green, residues with a significant weighted chemical shift differences (>0.05 PPM) are colored in red, where a subset is indicated by their respective residue number, whereas SIMPX is shown in blue (Protein Data Bank code 2ASQ).
Upon the addition of SIMPX to SUMO1, the exchange is quenched for many residues, but 20 residues show exchange in both apo- and SIMPX-bound SUMO1. Of these, 6 residues are within 5 Å of SIMPX. A small number of residues in the SIMPX-SUMO1 complex experience chemical exchange not detected for apoSUMO1, and of these only 1 residue is within 5 Å of the SIM binding site. The remainders of these residues are located in the flexible N-terminal or in the hydrophobic core of SUMO1, suggesting a change in the hydrophobic packing upon binding.
A comparison of residues that show exchange in the apo- and ZZ domain-bound states of SUMO1 reveals that these groups of residues are similar. In addition, a set of residues shows exchange in the ZZ domain-SUMO1 complex that is not observed in apo-SUMO. Most of these residues are located in the flexible N-terminal region, which is involved in binding the ZZ domain and, therefore, flexible in the apo state. Residues that show exchange for apoSUMO1 but not for the ZZ domain-SUMO1 complex are mainly located in the region indicated to bind the ZZ domain based on the chemical shift perturbations observed for SUMO1.
The 15N chemical shift changes between the major and minor state was determined from global fits of the CPMG relaxation dispersions for the various states of SUMO1. The chemical shift changes extracted from the global fit of the apoSUMO1 CPMG relaxation dispersion experiments does not show a direct one-to-one correspondence with the 15N chemical shift changes observed between the SIMPX-bound and apoSUMO1 (Fig. 8A) with an r.m.s.d. of 0.96 ppm. The data suggest that apoSUMO1 is exchanged between a major ground state and a high energy state that involves other conformations than those of the SIMPX-bound state. In contrast, the chemical shift change between the minor and major state in the SIMPX-SUMO1 complex correlated well with the 15N chemical shift change measured from the 15N HSQCs of the apo- and SIMPX-bound SUMO1 with an r.m.s.d. of 0.08 ppm (Fig. 8B). These data imply that SUMO1 exchanged between free and bound states in the SIMPX-SUMO1 sample. Furthermore, the comparison of the globally fitted chemical shift changes from the CPMG relaxation dispersions between the minor and major state for apo- and ZZ domain-bound SUMO1 yielded a good correlation with an r.m.s.d. of 0.14 ppm (Fig. 8C). Notably, the chemical exchange rates and the populations of ZZ domain-bound SUMO1 and apoSUMO1 are the same within errors and with a similar set of residues showing exchange broadening in both states. These observations might suggest that the binding of the ZZ domain to SUMO1 has no major effect on the intrinsic conformational exchange of SUMO1. A direct comparison of the chemical shift change between the ZZ domain-bound SUMO1 and apoSUMO1 is not feasible due to the lack of 15N cross-peaks caused by exchange broadening upon formation of the ZZ domain-SUMO1 complex. An alternative interpretation of the data is that the CPMG dispersions observed for the ZZ domain-bound SUMO1 sample involves exchange between free and bound states; in this scenario, the good match between the chemical shifts of the minor state of apoSUMO1 and the ZZ domain-bound SUMO1 indicates that SUMO1 binds the ZZ domain by conformational selection. The latter interpretation is further supported by the expectation, based on the binding affinities from ITC and the NMR sample conditions, that both the SIMPX and ZZ domain SUMO1 samples contain a minor population of the peptide-free state, which is in good agreement with the minor state population determined from the CPMG dispersions.
FIGURE 8.

A, 15N chemical shift difference for backbone amides between major and minor state of apoSUMO1 from global fits of CPMG relaxation dispersion curves plotted versus 15N chemical shift difference between apo- and SIMPX-bound states. B, 15N chemical shift difference for backbone amides between the major and minor state of SIMPX-bound SUMO1 from global fits of CPMG relaxation dispersion curves plotted versus 15N chemical shift difference between apo- and SIMPX-bound states. C, 15N chemical shift differences for backbone amides between major and minor state from global fits of CPMG relaxation dispersion curves for apo- and ZZ domain-bound SUMO1 plotted in a covariance graph.
The on and off rates of the various states was calculated based on a global fit of the populations and exchange rates. For the SIMPX-SUMO1 complex, the koff is 28 ± 2 s−1, and kon is 1.6 ± 0.1 × 106 m−1s−1. The calculated value of kon indicates that the binding of SIMPX to SUMO1 is essentially diffusion controlled. In comparison, the off-rate for the ZZ domain-SUMO1 complex is 31 ± 5 s−1, as calculated for the core residues, whereas the results for the N-terminal residues directly involved in binding of the ZZ domain peptide yielded a koff = 19 ± 2 s−1. We speculate that the release of the ZZ domain peptide might involve a two-step process, with slightly different life times of the interactions with the N-terminal segment and the core residues. The overall slower off-rate for the ZZ domain is in keeping with the slightly higher affinity of the ZZ domain-SUMO1 complex than SIMPX-SUMO1. The off-rates observed for the different bound states of SUMO1 are in the same regime as the intrinsic conformational exchange of apoSUMO1, suggesting that these processes might be linked.
Discussion
Protein modifications, such as SUMOylation, play important roles in intricate protein-protein interaction networks involving numerous intracellular processes such as the response to DNA double-strand breaks and chromatin remodeling. In response to DNA damage, the SUMOylated target proteins are recognized by SIMs present in various DNA damage signal and repair proteins. Until recently, one type of SIM was described that is characterized by a signature sequence involving a stretch of charged anionic amino acid residues (10). The newly discovered SUMO binding ZZ domain represents a new SUMO recognition motif distinct from the classical SIMs (9). We now show that the ZZ domain interacts with a unique epitope in SUMO1 and that the complex between the ZZ domain and SUMO1 displays a different binding mode from those previously described for SUMO-mediated interactions. Mutation of residues in the interaction surface for the ZZ domain did not significantly decrease the affinity for SUMO1 even though the enthalpy of binding was reduced. From previous studies we established that the zinc coordination is crucial for the interaction between the HERC2 ZZ domain and SUMO1 (9). By mutating the zinc-coordinating residues in the ZZ domain, the SUMO binding capacity was lost. We, therefore, conclude that the presence of the coordinated zinc in the ZZ domain close to the interaction interface with SUMO1 plays an important role in the ZZ domain-SUMO1 complex formation. Importantly, mutation of two epitopes in SUMO1, representing residues in the ZZ domain-SUMO1 interface, completely eliminated binding to the ZZ domain, clearly indicating the importance of these epitopes in ZZ domain recognition.
The CPMG relaxation dispersion data demonstrate that apoSUMO1 exchanges between the (major) ground state and a (minor) high energy state, which might be linked to ligand binding. However, it is clear that the minor conformation of apoSÚMO1 did not directly match that of the bound state in the case of SIMPX binding, indicating that this peptide did not bind by strict conformational selection but induced additional conformational changes upon binding or that it selected one conformer out of a manifold present in the high energy state. Also in the case of the ZZ domain, the available data can be interpreted in two ways; either the peptide did not affect the intrinsic dynamics of apoSUMO1 or it bound by conformational selection. Thus, the available data clearly showed that the dynamic process of peptide binding to SUMO1 has markedly different signatures for the two cases of SIMPX and the ZZ domain.
Taken together we have shown that the new SUMO-binding ZZ domain binds to SUMO1 through interactions with a unique and spatially distinct site in SUMO1 as compared with the SIMs and that these two kinds of recognition motifs can bind SUMO1 simultaneously in a non-cooperative fashion. Our data, therefore, provide new insights into the interactions between SUMO binding motifs and SUMO and extend the repertoire of proteins recognizing SUMO to also include regulatory proteins harboring ZZ domains.
Author Contributions
C. D. expressed and purified the proteins and conducted the NMR experiments including analysis of results. M. A. contributed with experimental design and interpretation of the CPMG relaxation dispersion experiments. S. B.-J. and N. M. provided biological data assessments and expertise. W. S. and M. W. performed the ITC experiments. M. W. designed the study, served as the project supervisor, and wrote the paper with input from all other authors.
Supplementary Material
Acknowledgment
We thank Havva Koç for excellent technical assistance.
This work was supported by Novo Nordisk Foundation Grant NNF14CC0001, the European Commission under the Seventh Framework Program (FP7) contract Bio-NMR 261863 providing access to the Swedish NMR Centre, Gothenburg, Sweden, and the MAX laboratory, Lund, Sweden, enabling access to the MAX laboratory (ID 20130289). The authors declare that they have no conflicts of interest with the contents of this article.

This article contains supplemental Table 1.
The chemical shifts for the ZZ domain and SUMO1 (code 25553) have been deposited in the Biological Magnetic Resonance Data Bank (www.bmrb.wisc.edu).
The atomic coordinates and structure factors (code 2N1A) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- SUMO
- small ubiquitin-related modifier
- SIM
- SUMO interacting motif
- CBP
- CREB-binding protein
- ZZ domain
- CBP/p300 ZZ domain
- RDC
- residual dipolar coupling
- HSQC
- heteronuclear single quantum coherence
- ITC
- isothermal titration calorimetry
- HADDOCK
- high ambiguity driven protein-protein docking
- r.m.s.d.
- root mean square deviation(s)
- CPMG
- Carr-Purcell-Meiboom-Gill.
References
- 1.Geiss-Friedlander R., and Melchior F. (2007) Concepts in sumoylation: a decade on. Nat. Rev. Mol. Cell Biol. 8, 947–956 [DOI] [PubMed] [Google Scholar]
- 2.Gareau J. R., and Lima C. D. (2010) The SUMO pathway: emerging mechanisms that shape specificity, conjugation, and recognition. Nat. Rev. Mol. Cell Biol. 11, 861–871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Becker J., Barysch S. V., Karaca S., Dittner C., Hsiao H. H., Berriel Diaz M., Herzig S., Urlaub H., and Melchior F. (2013) Detecting endogenous SUMO targets in mammalian cells and tissues. Nat. Struct. Mol. Biol. 20, 525–531 [DOI] [PubMed] [Google Scholar]
- 4.Rodriguez M. S., Dargemont C., and Hay R. T. (2001) SUMO-1 conjugation in vivo requires both a consensus modification motif and nuclear targeting. J. Biol. Chem. 276, 12654–12659 [DOI] [PubMed] [Google Scholar]
- 5.Sampson D. A., Wang M., and Matunis M. J. (2001) The small ubiquitin-like modifier-1 (SUMO-1) consensus sequence mediates Ubc9 binding and is essential for SUMO-1 modification. J. Biol. Chem. 276, 21664–21669 [DOI] [PubMed] [Google Scholar]
- 6.Song J., Zhang Z., Hu W., and Chen Y. (2005) Small ubiquitin-like modifier (SUMO) recognition of a SUMO binding motif: a reversal of the bound orientation. J. Biol. Chem. 280, 40122–40129 [DOI] [PubMed] [Google Scholar]
- 7.Chang C. C., Naik M. T., Huang Y. S., Jeng J. C., Liao P. H., Kuo H. Y., Ho C. C., Hsieh Y. L., Lin C. H., Huang N. J., Naik N. M., Kung C. C., Lin S. Y., Chen R. H., Chang K. S., Huang T. H., and Shih H. M. (2011) Structural and functional roles of Daxx SIM phosphorylation in SUMO paralog-selective binding and apoptosis modulation. Mol. Cell 42, 62–74 [DOI] [PubMed] [Google Scholar]
- 8.Cappadocia L., Mascle X. H., Bourdeau V., Tremblay-Belzile S., Chaker-Margot M., Lussier-Price M., Wada J., Sakaguchi K., Aubry M., Ferbeyre G., and Omichinski J. G. (2015) Structural and functional characterization of the phosphorylation-dependent interaction between PML and SUMO1. Structure 23, 126–138 [DOI] [PubMed] [Google Scholar]
- 9.Danielsen J. R., Povlsen L. K., Villumsen B. H., Streicher W., Nilsson J., Wikström M., Bekker-Jensen S., and Mailand N. (2012) DNA damage-inducible SUMOylation of HERC2 promotes RNF8 binding via a novel SUMO-binding zinc finger. J. Cell Biol. 197, 179–187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hecker C.-M., Rabiller M., Haglund K., Bayer P., and Dikic I. (2006) Specification of SUMO1-and SUMO2-interacting motifs. J. Biol. Chem. 281, 16117–16127 [DOI] [PubMed] [Google Scholar]
- 11.Legge G. B., Martinez-Yamout M. A., Hambly D. M., Trinh T., Lee B. M., Dyson H. J., and Wright P. E. (2004) ZZ domain of CBP: an unusual zinc finger fold in a protein interaction module. J. Mol. Biol. 343, 1081–1093 [DOI] [PubMed] [Google Scholar]
- 12.Ponting C. P., Blake D. J., Davies K. E., Kendrick-Jones J., and Winder S. J. (1996) ZZ and TAZ: new putative zinc fingers in dystrophin and other proteins. Trends Biochem. Sci. 21, 11–13 [PubMed] [Google Scholar]
- 13.Dominguez C., Boelens R., and Bonvin A. M. (2003) HADDOCK: a protein-protein docking approach based on biochemical or biophysical information. J. Am. Chem. Soc. 125, 1731–1737 [DOI] [PubMed] [Google Scholar]
- 14.Aslanidis C., and de Jong P. J. (1990) Ligation-independent cloning of PCR products (LIC-PCR). Nucleic Acids Res. 18, 6069–6074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang W., and Malcolm B. (1999) Two-stage PCR protocol allowing introduction of multiple mutations, deletions and insertions using QuikChange site-directed mutagenesis. Biotechniques 26, 680–682 [DOI] [PubMed] [Google Scholar]
- 16.Neidhardt F. C., Bloch P. L., and Smith D. F. (1974) Culture medium for enterobacteria. J. Bacteriol. 119, 736–747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gileadi O., Burgess-Brown N. A., Colebrook S. M., Berridge G., Savitsky P., Smee C. E., Loppnau P., Johansson C., Salah E., and Pantic N. H. (2008) High throughput production of recombinant human proteins for crystallography. In Structural Proteomics, pp. 221–246, Springer-Verlag New York Inc., New York: [DOI] [PubMed] [Google Scholar]
- 18.Wiseman T., Williston S., Brandts J. F., and Lin L. N. (1989) Rapid measurement of binding constants and heats of binding using a new titration calorimeter. Anal. Biochem. 179, 131–137 [DOI] [PubMed] [Google Scholar]
- 19.Bayer P., Arndt A., Metzger S., Mahajan R., Melchior F., Jaenicke R., and Becker J. (1998) Structure determination of the small ubiquitin-related modifier SUMO-1. J. Mol. Biol. 280, 275–286 [DOI] [PubMed] [Google Scholar]
- 20.Clore G. M., Bax A., Driscoll P. C., Wingfield P. T., and Gronenborn A. M. (1990) Assignment of the side-chain proton and carbon-13 resonances of interleukin-1β using double-and triple-resonance heteronuclear three-dimensional NMR spectroscopy. Biochemistry 29, 8172–8184 [DOI] [PubMed] [Google Scholar]
- 21.Bax A., and Ikura M. (1991) An efficient 3D NMR technique for correlating the proton and 15N backbone amide resonances with the α-carbon of the preceding residue in uniformly 13C,15N-enriched proteins. J. Biomol. NMR 1, 99–104 [DOI] [PubMed] [Google Scholar]
- 22.Wittekind M., and Mueller L. (1993) HNCACB, a high-sensitivity 3D NMR experiment to correlate amide-proton and nitrogen resonances with the α-and β-carbon resonances in proteins. J. Magn. Reson. B 101, 201–205 [Google Scholar]
- 23.Grzesiek S., and Bax A. (1992) Correlating backbone amide and side chain resonances in larger proteins by multiple relayed triple resonance NMR. J. Am. Chem. Soc. 114, 6291–6293 [Google Scholar]
- 24.Delaglio F., Grzesiek S., Vuister G. W., Zhu G., Pfeifer J., and Bax A. (1995) NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR 6, 277–293 [DOI] [PubMed] [Google Scholar]
- 25.Vranken W. F., Boucher W., Stevens T. J., Fogh R. H., Pajon A., Llinas M., Ulrich E. L., Markley J. L., Ionides J., and Laue E. D. (2005) The CCPN data model for NMR spectroscopy: development of a software pipeline. Proteins 59, 687–696 [DOI] [PubMed] [Google Scholar]
- 26.Ottiger M., Delaglio F., and Bax A. (1998) Measurement of J and dipolar couplings from simplified two-dimensional NMR spectra. J. Magn. Reson. 131, 373–378 [DOI] [PubMed] [Google Scholar]
- 27.Schwieters C. D., Kuszewski J. J., Tjandra N., and Clore G. M. (2003) The Xplor-NIH NMR molecular structure determination package. J. Magn. Reson. 160, 65–73 [DOI] [PubMed] [Google Scholar]
- 28.Case D. A., Cheatham T. E. 3rd, Darden T., Gohlke H., Luo R., Merz K. M. Jr., Onufriev A., Simmerling C., Wang B., and Woods R. J. (2005) The Amber biomolecular simulation programs. J. Comput. Chem. 26, 1668–1688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bhattacharya A., Tejero R., and Montelione G. T. (2007) Evaluating protein structures determined by structural genomics consortia. Proteins 66, 778–795 [DOI] [PubMed] [Google Scholar]
- 30.Zweckstetter M., and Bax A. (2000) Prediction of sterically induced alignment in a dilute liquid crystalline phase: aid to protein structure determination by NMR. J. Am. Chem. Soc. 122, 3791–3792 [Google Scholar]
- 31.Brünger A. T., Adams P. D., Clore G. M., DeLano W. L., Gros P., Grosse-Kunstleve R. W., Jiang J.-S., Kuszewski J., Nilges M., and Pannu N. S., Read R. J., Rice L. M., Simonson T., and Warren G. L. (1998) Crystallography NMR system: a new software suite for macromolecular structure determination. Acta Crystallogr. D Biol. Crystallogr. 54, 905–921 [DOI] [PubMed] [Google Scholar]
- 32.de Vries S. J., van Dijk M., and Bonvin A. M. (2010) The HADDOCK web server for data-driven biomolecular docking. Nat. Protoc. 5, 883–897 [DOI] [PubMed] [Google Scholar]
- 33.Meiler J., Blomberg N., Nilges M., and Griesinger C. (2000) A new approach for applying residual dipolar couplings as restraints in structure elucidation. J. Biomol. NMR 16, 245–252 [DOI] [PubMed] [Google Scholar]
- 34.Loria J. P., Rance M., and Palmer A. G. (1999) A relaxation-compensated Carr-Purcell-Meiboom-Gill sequence for characterizing chemical exchange by NMR spectroscopy. J. Am. Chem. Soc. 121, 2331–2332 [Google Scholar]
- 35.Mulder F. A., Mittermaier A., Hon B., Dahlquist F. W., and Kay L. E. (2001) Studying excited states of proteins by NMR spectroscopy. Nat. Struct. Mol. Biol. 8, 932–935 [DOI] [PubMed] [Google Scholar]
- 36.Yip G. N., and Zuiderweg E. R. (2004) A phase cycle scheme that significantly suppresses offset-dependent artifacts in the R2-CPMG 15N relaxation experiment. J. Magn. Reson. 171, 25–36 [DOI] [PubMed] [Google Scholar]
- 37.Koradi R., Billeter M., and Wüthrich K. (1996) MOLMOL: a program for display and analysis of macromolecular structures. J. Mol. Graph. 14, 51–55 [DOI] [PubMed] [Google Scholar]
- 38.Weininger U., Modig K., and Akke M. (2014) Ring Flips revisited: 13C relaxation dispersion measurements of aromatic side chain dynamics and activation barriers in basic pancreatic trypsin inhibitor. Biochemistry 53, 4519–4525 [DOI] [PubMed] [Google Scholar]
- 39.Carver J. P., and Richards R. E. (1972) A general two-site solution for the chemical exchange produced dependence of T2 upon the carr-Purcell pulse separation. J. Magn. Reson. (1969) 6, 89–105 [Google Scholar]
- 40.Davis D. G., Perlman M. E., and London R. E. (1994) Direct measurements of the dissociation-rate constant for inhibitor-enzyme complexes via the T1 [rho] and T2 (CPMG) methods. J. Magn. Reson. B 104, 266–275 [DOI] [PubMed] [Google Scholar]
- 41.Press W. H., Flannery B. P., Teukolsky S. A., and Vetterling W. T. (1986) Numerical Recipes: The Art of Scientific Computing, Cambridge University Press, Cambridge [Google Scholar]
- 42.Mandel A. M., Akke M., and Palmer A. G. (1995) Backbone dynamics of Escherichia coli ribonuclease HI: correlations with structure and function in an active enzyme. J. Mol. Biol. 246, 144–163 [DOI] [PubMed] [Google Scholar]
- 43.Cornilescu G., Delaglio F., and Bax A. (1999) Protein backbone angle restraints from searching a database for chemical shift and sequence homology. J. Biomol. NMR 13, 289–302 [DOI] [PubMed] [Google Scholar]
- 44.DeLano W. L. (2002) The PyMOL Molecular Graphics System, Schrodinger, LLC, New York [Google Scholar]
- 45.Dosset P., Hus J. C., Marion D., and Blackledge M. (2001) A novel interactive tool for rigid-body modeling of multi-domain macromolecules using residual dipolar couplings. J. Biomol. NMR 20, 223–231 [DOI] [PubMed] [Google Scholar]
- 46.Bernier-Villamor V., Sampson D. A., Matunis M. J., and Lima C. D. (2002) Structural basis for E2-mediated SUMO conjugation revealed by a complex between ubiquitin-conjugating enzyme Ubc9 and RanGAP1. Cell 108, 345–356 [DOI] [PubMed] [Google Scholar]
- 47.Reverter D., and Lima C. D. (2006) Structural basis for SENP2 protease interactions with SUMO precursors and conjugated substrates. Nat. Struct. Mol. Biol. 13, 1060–1068 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.