

Abstract
One intriguing feature of prion diseases is their strain variation. Prion strains are differentiated by the clinical consequences they generate in the host, their biochemical properties, and their potential to infect other animal species. The selective targeting of these agents to specific brain structures have been extensively used to characterize prion strains. However, the molecular basis dictating strain-specific neurotropism are still elusive. In this study, isolated brain structures from animals infected with four hamster prion strains (HY, DY, 139H, and SSLOW) were analyzed for their content of protease-resistant PrPSc. Our data show that these strains have different profiles of PrP deposition along the brain. These patterns of accumulation, which were independent of regional PrPC production, were not reproduced by in vitro replication when different brain regions were used as substrate for the misfolding-amplification reaction. On the contrary, our results show that in vitro replication efficiency depended exclusively on the amount of PrPC present in each part of the brain. Our results suggest that the variable regional distribution of PrPSc in distinct strains is not determined by differences on prion formation, but on other factors or cellular pathways. Our findings may contribute to understand the molecular mechanisms of prion pathogenesis and strain diversity.
Keywords: neurodegenerative disease, prion, prion disease, protein aggregation, protein misfolding
Introduction
Prions, the causative agents in transmissible spongiform encephalopathies (TSEs),2 are composed mostly or exclusively by misfolded forms of the prion protein (PrPSc). The unconventional mechanism of infection in TSEs is dependent on the PrPSc-directed misfolding of compatible PrPC (normal prion protein) expressed in the host (1, 2).
One of the most intriguing features of infectious prions is the existence of a wide diversity of strains (reviewed in Ref. 3). Although strain variation is sometimes dependent on changes in the amino acid sequence of the prion protein, many phenotypically different variants have been identified within a single PrP primary structure (4–6). Compelling evidence suggests that the main differences between prion strains lie in the differential conformation that PrPSc acquires. Prion strains can be discriminated by the different properties they exert in the host, such as incubation periods to disease, clinical signs, and distribution of brain lesions (5, 7–11). In vitro tests can also be used to identify strain-specific biochemical features (5, 12–14).
Perhaps the most important features used to discriminate prion strains are the time each agent takes to produce disease in a specific host (15) and the brain areas mostly damaged (7, 11, 16). Importantly, the selective accumulation and replication of prions according to their strain type has been reproduced ex vivo (17) and in cell cultures (18). The differential tropism of the agent to specific brain structures likely dictate specific damage responsible for particular clinical signs (7, 11, 16). Nevertheless, the molecular mechanism responsible for brain targeting of different prion strains is unknown. It is possible that neural connectivity, expression of strain-specific cofactors/receptors or differences in biological clearance by certain cell types might be responsible for the specific signature of accumulation each agent generates in affected brains.
Although prion strains have been identified in almost all naturally or experimentally susceptible animal species, the ones observed in Syrian hamsters are especially intriguing due to the wide variety of clinical signs they can generate (19–21). In this work, we dissected and isolated different brain areas of Syrian hamsters independently infected with four prion strains, and the relative accumulation of disease-associated PrP present in each structure was measured. In addition, protein misfolding cyclic amplification (PMCA) reactions using PrPC substrate from different brain regions were used to assess substrate specific replication of the agent in a cell-free environment. Our results show that strain-specific brain distribution of PrPSc in infected animals was not reproduced by in vitro prion replication. Actually, the efficiency of in vitro prion replication depended mostly on the amount of PrPC substrate available. These results suggest that the brain tropism of each prion strain is determined by factors or cellular processes not present in a simplified in vitro replication reaction.
Experimental Procedures
Prion-infected Animals
∼30-day-old female Syrian golden hamsters received intra-hippocampal injections (5–10 μl) of brain homogenates from clinically ill HY (hyper-TME), DY (drowsy-TME) (19), 139H (21), or SSLOW (synthetic strain leading to overweight, (20)) prion strains (n = 9–10/strain) using a stereotaxic apparatus. Animals were periodically observed for the appearance of clinical signs according to the following scales: HY: 1) normal animal; 2) mild behavioral abnormalities including hyperactivity and hypersensitivity to noise; 3) moderate behavioral problems including head tremors, ataxia, wobbling gait, head bobbing, irritability, and aggressiveness; 4) severe behavioral abnormalities including all of the above plus head and body jerks and spontaneous backrolls; and 5) terminal stage of the disease in which the animal lies in the cage and is no longer able to stand up. DY: 1) normal animal; 2) mild behavioral abnormalities including mild lethargy; 3) increased lethargy and curl up sleep posture; 4) same as 3 but decrease in body weight and difficulty to open the eyes; and 5) terminal stage of the disease in which the animal lies in the cage and is no longer able to stand up. 139H: 1) normal animal; 2) mild behavioral abnormalities including minor signs of ataxia; 3) increased food uptake and body weight; 4) same as 3 but slight reduction in body weight and increased ataxia; and 5) terminal stage of the disease in which the animal lies in the cage and is no longer able to stand up. SSLOW: 1) normal animal; 2) mild behavioral abnormalities including slight sensitivity to noise and touch; 3) same as 2 but increase in body weight and apparent skin dryness; 4) progressive increase in body weight, dry skin (sometimes including matted hair, especially in the thoracic area), and mild lethargy; and 5) terminal stage of the disease in which the animal reduces its body weight, lies in the cage and is no longer able to stand up. Animals scoring stage 4 for more than 1 week in HY, DY, and 139H, and longer than 3 weeks in SSLOW, were sacrificed by CO2 inhalation. Injected brain hemispheres were snap frozen and kept at −80 °C. Contra-lateral hemispheres were fixed and used for neuropathological assessments. All manipulations involving living subjects were performed following NIH and institutional guidelines for animal research.
Assessment of Brain Spongiform Degeneration
Brains from animals sacrificed at stage 4 of clinical signs were fixed in Carnoy, dehydrated and embedded in paraplast. 10-μm brain slices were stained with hematoxilin-eosin. Vacuolation scores were assigned as previously described (8).
Isolation of Brain Structures and Homogenate Preparation
Frozen brains from prion infected or uninfected animals (n = 3–5) were placed in ice-cold Petri dishes and quickly dissected using a surgical blade. Dissection resulted in the following isolated structures: olfactory bulb (OB), frontal cortex (FC), parietal cortex (PC), occipital cortex (OC), caudate/putamen (CPu), hippocampus (Hipp), thalamus (Thal), hypothalamus (Hypo), midbrain (Mb), cerebellum (Cb), and medulla/pons (MP). Resulting tissues were immediately homogenized at 10% w/v in phosphate-buffered saline (PBS) supplemented with a mixture of protease inhibitors (Roche, Basel, Switzerland). Homogenates were spun down at 805 × g for 45 s at 4 °C to remove debris. Supernatants were collected and total protein concentration was measured using the bicinchoninic acid (BCA) assay following manufacturer's recommendations (Pierce, Waltham, MA). Samples were diluted in homogenization buffer to match the concentration of homogenates containing the lowest amount of protein in each brain.
Western Blotting and Densitometric Analysis of Protease-resistant PrP (PrP27–30)
Aliquots containing the same amount of total protein by BCA were proteinase K (PK, Sigma) treated (50 μg/ml for 1 h at 37 °C with shaking) and PrP27–30 (the protease-resistant fragment of PrPSc) content assessed by Western blotting (WB). WB procedure was performed as previously described, using the 6D11 anti-PrP antibody (Covance, Princeton, NJ) (22). Levels of PrPC in brain regions from healthy animals were measured as above but without PK treatment. Densitometric signals obtained in each independent membrane were quantified using the Quantify One (4.6.7) software (Bio-Rad). Values obtained for each isolated brain structure were expressed as a percentage of the added signals obtained for all structures within the same brain. Brains from five animals per strain were measured at least three independent times to assess reproducibility. Results were averaged and plotted using the Prism GraphPad software (GraphPad, La Jolla, CA). To additionally control total protein load, all samples were independently probed against the glyceraldehyde-3-phosphate dehydrogenase (GAPDH) using the GAPDH-71.1 antibody (Sigma). GAPDH signals were assessed in separated blots loaded with the same volume of material without PK treatment.
Protein Misfolding Cyclic Amplification (PMCA)
PMCA was performed as previously reported (22) with minimum changes (source of substrate for the reaction). Briefly, ∼30-day-old hamsters were sacrificed and perfused as described (22). Some brains were dissected in the following structures before substrate preparation: cortex (Ctx), caudate/putamen (CPu), hippocampus (Hipp), thalamus/hypothalamus (Thal+Hypo), midbrain (Mb), cerebellum (Cb), and medulla/pons (MP). Levels of in vitro replication for each strain were measured in a single PMCA round using 2-fold serial PrPSc-containing brain homogenate dilutions and whole brain extracts as substrate. For brain region-specific in vitro replication, 5-fold serial dilutions of whole brain extract containing HY, DY, 139H, and SSLOW prions were made on PMCA substrates prepared from isolated brain structures and submitted to a single PMCA round. 5-fold dilutions were chosen for this experiment to cover a wider range of PrPSc dilutions and thus increase chances to detect region specific differences for replication-efficient strains. PMCA efficiency was assessed by comparing to aliquots of the same reaction not submitted to incubation/sonication cycles (non-amplified). Because of the dissimilar behavior of prion strains in PMCA, data were analyzed as a percentage of the maximum folds of amplification obtained for each PrPSc strain. For the calculation of amplification folds, we estimated the last dilution in which PrP27–30 signal was detectable by WB (signal >10% of the background) divided by the last dilution observed without amplification. Percentages were calculated using the log5 PMCA amplification folds for each reaction since tested samples were serially diluted using a 5-fold dilution factor. Correlations between “% of log5 (PMCA amplification folds)” and “% of PrPC” were made using the averaged data obtained from these two data sets. Data obtained from three different sources of infected brains per prion strain were averaged and plotted. The Pearson correlation coefficient was calculated using the Prism GraphPad software (GraphPad, La Jolla, CA).
Results
Pathological and Biochemical Characterization of Hamster Prion Strains
For our experiments, we used four clinically and pathologically different hamster prion strains: HY, DY, 139H, and SSLOW. The specific characteristics of these prion variants have been extensively described elsewhere (5, 7, 16, 19, 20, 23, 24). As depicted in Fig. 1A, the prion strains used for this study showed different incubation periods, being HY the most aggressive (76.6 ± 3.8, average days post injection ± standard error) and SSLOW the one producing disease at longer times (423.5 ± 11.8). Although DY and 139H prions showed similar incubation periods (210.3 ± 9.6 and 203.4 ± 6.6, respectively) they were easily differentiated by their clinical signs (detailed description under “Experimental Procedures”). Histopathological analyses by measuring the degree of spongiform degeneration in three areas of the brain confirmed the different pathology expected for these strains (Fig. 1B). An additional method to differentiate prion strains utilizes their electrophoretic profile after PK digestion. In these analyses, the mobility of the unglycosylated band as well as the preference for specific glycoforms are measured (3). We observed that all these strains have preference for the diglycosylated form of PrP as commonly observed for hamster prions (Fig. 1C). However, only DY prions showed a faster electrophoretic mobility (∼19 kDa) compared with all other strains (∼21 kDa). The results depicted in this figure confirm the differential identity of the prion material used in our experiments.
FIGURE 1.

Pathological and biochemical characterization of hamster prion strains. HY, DY, 139H, and SSLOW prions were intra-cerebrally administered into hamsters and incubation periods were measured from the time of injection until animals reached advanced stages of clinical disease as described under “Experimental Procedures” (A). Collected brains were analyzed for spongiform degeneration in three different brain regions (B) and electrophoretic mobility of PK-resistant PrP (C). Samples in panel C correspond to the frontal cortex of prion-infected animals. Horizontal line at the right side of panel C represents ∼30 kDa molecular weight. Hypo, hypothalamus; Hipp, hippocampus; Cing Ctx, cingulate cortex.
Brain PrP27–30 Accumulation Profile for Different Hamster Prion Strains
Brains of symptomatic animals infected with the four prion strains previously described were dissected out, and content of PrP27–30 was analyzed by WB. As expected, we found different profiles of PrP27–30 accumulation among the different prion strains analyzed (Fig. 2). The distribution of PrP27–30 among the structures examined showed that HY prions distribute similarly in all brain regions with the sole exception of the thalamus and hypothalamus were accumulation of disease associated prion protein was highest (Fig. 2A). This observation is in agreement with a previous report showing a strong presence of diffuse aggregates for this strain in the medial geniculate nucleus (25). Interestingly, DY prions, a strain sharing the same origin with HY prions, showed a considerably distinct pattern of deposition (Fig. 2B). This strain presented a higher burden of prion accumulation along the cerebral cortex and hippocampus. On the contrary, the olfactory bulb presented the lowest relative signal among all strains studied. 139H, a hamster adapted mouse prion strain (7, 16), presented the highest accumulation of PrP27–30 in the cortex, being even in the three cortical regions analyzed (Figs. 2C). The preference of 139H prions for cortical areas is in agreement with previous reports (16). 139H's pattern of prion accumulation differed from DY and HY mostly in the strong signals found in olfactory bulb and lower contribution of thalamus and hypothalamus, respectively. The lowest levels of PrP27–30 for this strain were found in the cerebellum and medulla/pons (Fig. 2C). SSLOW prions have been previously shown to generate large thioflavin S positive deposits in the brain of affected animals, especially in the hippocampus (23). Our results confirmed the important contribution of hippocampus in PrP accumulation for this strain (Fig. 2D). We also observed a strong presence of SSLOW-PrP aggregates in the occipital cortex. The lowest levels of PrP27–30 for this strain were found in the caudate/putamen and cerebellum.
FIGURE 2.

Brain distribution of PrP27–30 in different hamster prion strains. Brain structures isolated from terminally ill Syrian hamster infected with HY (A), DY (B), 139H (C), and SSLOW (D) prions were tested for PrP27–30 levels by WB. Left panels show representative WBs for each strain. GAPDH was used to control protein load among the samples. Blots for GAPDH were independently run using the same volume as the experimental samples, but without PK digestion. Numbers on the right represent molecular weight standards. Graphs in the middle show the average distribution profile obtained from 3–5 different brains (each one tested at least in triplicates). All samples were PK treated before WB. Data are expressed as averages ± S.E. Continuous lines are used to highlight the different profiles of PrP27–30 accumulation for different strains. OB, olfactory bulb; FC, frontal cortex; PC, parietal cortex; OC, occipital cortex; CPu, caudate/putamen; Hipp, hippocampus; Thal, thalamus; Hypo, hypothalamus; Mb, midbrain; Cb, cerebellum; MP, medulla/pons; MW, molecular weight standard. The schematic drawing in the right side represent a heat map of PrP27–30 distribution throughout different brain regions. Data are represented from >12% (darker signal) to <5% (lighter signal). Color scale is graded in 0.5% intervals.
PMCA Amplification of Hamster Prion Strains Using Different Brain Regions as Substrates
To study whether the different profiles of brain PrP27–30 accumulation observed in the four strains analyzed was due to differential replication of prions in distinct brain areas, we performed experiments of in vitro prion replication using PMCA. We have previously shown that PMCA faithfully reproduces the generation and propagation of different prion strains in vitro (12). These results, which have been corroborated for prions from different species (26, 27), suggest that PMCA is suitable to replicate the strain features of prions. To analyze whether differences in strain-specific accumulation of PrPSc in vivo is due to differential replication of prion strains in distinct brain regions, brains from healthy animals designated for PMCA substrate were carefully dissected and different brain structures independently homogenized. The level of PrPC expression in each of these regions was determined by WB (Fig. 3A) followed by densitometric analysis (Fig. 3B). Results show that similar levels of PrPC expression are present throughout the brain with the sole exception of caudate/putamen showing a slight increase, and cerebellum and medulla/pons where levels were considerably lower (approximately less than half of all other regions averaged).
FIGURE 3.

PrPC distribution in different brain regions. Brains from healthy Syrian hamsters were dissected in the same brain regions depicted in Fig. 2 and PrPC content assessed by WB. Left panel (A) shows a representative WB of three independent assays. GAPDH was used to control protein load. Numbers on the right represent molecular weight standards. Graph on the right (B) shows the distribution profile obtained by densitometric analysis of the blots from three different brains. Data are expressed as average ± S.E. A continuous line is used to highlight the profile of PrPC level throughout the brain.
Previous reports have shown that PMCA efficiencies are different for distinct hamsters' prion strains (28). For this reason, we investigated the PMCA efficiency for the four strains used in this study (Fig. 4). Our results showed that under the PMCA conditions used, HY prions showed the highest efficiency of amplification comparable to the yield previously reported for the 263K strain (29). In vitro replication efficacy was followed by SSLOW, 139H and finally DY prions (Fig. 4). The differential in vitro amplification efficiencies found for HY and DY prions were similar to previously published results (28). It is important to mention that PMCA efficiencies were evaluated using similar amounts of the input PrP27–30 as shown in the non-amplified samples (Fig. 4, top panels). The dissimilar efficiencies of amplification suggested that in vitro prion replication by PMCA in isolated brain regions cannot be directly compared among distinct strains, but rather must be analyzed in a relative manner as a function of the maximum amplification for each strain. Therefore, to compare our data among the different groups tested, we calculated the PMCA amplification folds in each condition by comparing the last dilution in which PrP27–30 signal was detectable over the background before and after PMCA. Amplification folds were then expressed as a percentage of the maximum amplification to facilitate the comparison. Fig. 5A illustrates the results of the percentages of amplification folds in different brain regions for the 4 strains studied. The differences were not significantly different (except for some few specific points) and, indeed can be put together in a single curve with relatively small standard errors (Fig. 5B, purple line). The average amplification score for all strains combined was highest using substrate from cortex, caudate/putamen, hippocampus, thalamus/hypothalamus, and midbrain. The efficiency decreased substantially in cerebellum and medulla/pons (Fig. 5B). The profile of amplification score in distinct brain regions mirrored closely the pattern of PrPC expression in the same structures (Fig. 5B, black dotted line). Indeed, there was a good correlation between the % of PMCA amplification folds and the % of PrPC in distinct brain regions (Fig. 5C). These results suggest that the main determinant of PrPSc replication in vitro is the amount of substrate available for the reaction.
FIGURE 4.

PMCA amplification of hamster-adapted prion strains. Brain extracts from terminally ill HY (A), DY (B), 139H (C), and SSLOW (D) hamsters were serially diluted in brain homogenates from healthy animals and submitted to one PMCA round. Previous to PMCA, an aliquot of the PrPSc-substrate mixture was separated and kept frozen (non-amplified control). All samples were treated with PK before WB, with the sole exception of NBH (normal brain homogenate) representing PrPC and used as a control of electrophoretic mobility. First well to the left of each membrane represents a 0.2% of PrPSc-containing brain homogenate. Following wells correspond to 2-fold serially diluted aliquots. Horizontal lines at the right of each blot represent ∼36-kDa molecular weight markers.
FIGURE 5.
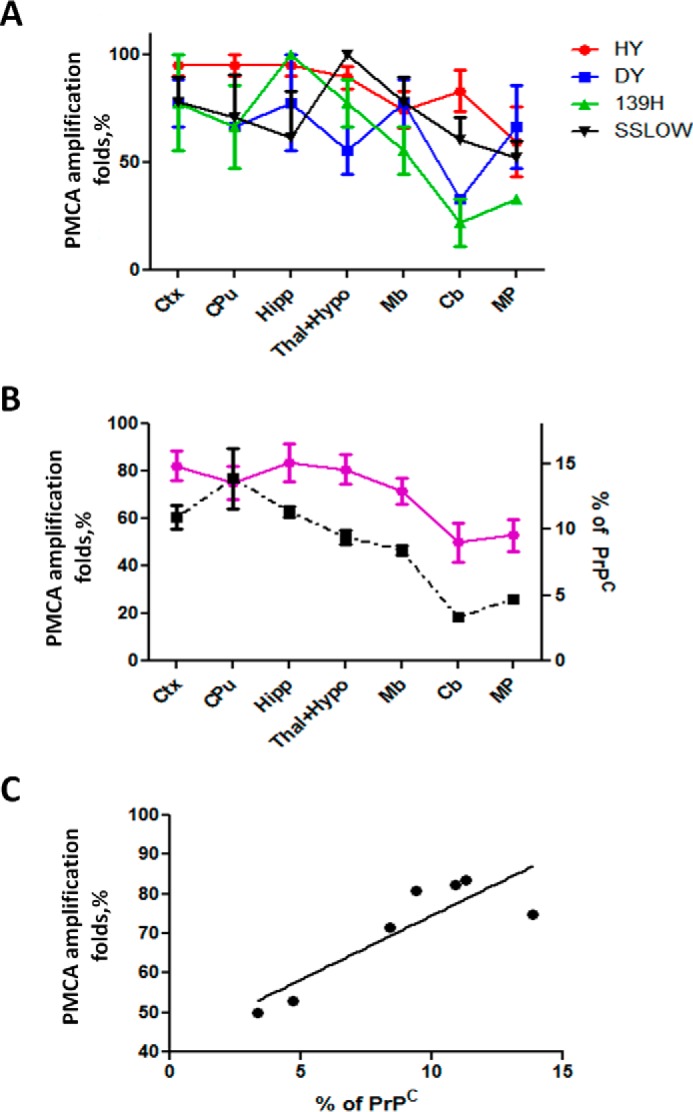
PMCA efficiency for different prion strains using different brain regions as substrate. A, small aliquots of PrPSc-containing brain homogenates from terminally ill prion infected hamsters were mixed with PMCA substrates prepared from isolated healthy brain structures. The amplification folds were estimated as explained under “Experimental Procedures” by comparing the last dilutions in which PrP27–30 signal can be observed over the background before and after PMCA. To compare distinct strains which differed in the amplification efficiency, data are presented as a percentage of the maximum amplification for each strain. Results are displayed as averages ± S.E. of three independent PMCA reactions for each condition. B, percentage of PMCA amplification fold for each strain showed in panel A were averaged for each region and compared with the relative levels of PrPC expression. Purple line corresponds to the average amplification folds in each brain region and dotted black line represents the levels of PrPC, using the right axis. Densitometric data for Ctx were averaged from Frontal, Parietal, and Occipital cortices values. The same procedure was done for Thal+Hypo. C, relationship between the % of PMCA amplification and the % of PrPC in different regions. The data of the PMCA amplification folds correspond to the average of the 4 strains studied in each brain region. Statistical analysis showed a significant relationship (p < 0.05), with a Pearson correlation coefficient of 0.85.
Discussion
One of the most intriguing issues in the prion field is the mechanism determining and controlling strain variation. A surprisingly large number of phenotypically diverse strains can be associated to PrPSc containing the same amino acid sequence (15, 19). The lack of sensitive techniques able to analyze the presumably subtle conformational differences in PrPSc coming from different strains has clouded this area of investigation for years. The differences in clinical manifestation of the disease produced by infection with distinct prion strains are thought to be dependent on the specific targeting of different brain regions. However, the molecular mechanism responsible for the differential neurotropism of prion strains is completely unknown. Accumulation of infectious prions in defined brain structures has been described to be highly specific, reaching the same target tissues independently of the route of administration. The selective effect of prion strains to certain cell types has also been observed in cultured neuroblastoma cells (18, 30) and organotypic brain slice cultures (17). Actually, the preference of certain prion strains to replicate in specific cell types has not been observed only in brain, but also in peripheral tissues (31). Although the selectivity of certain prion variants to recruit specific PrP glycoforms has been proposed as responsible for specific neuronal targeting (7), recent results have discarded this hypothesis (32). Other studies have shown that variation in RNA levels can impact prion replication (33) suggesting that differential prion accumulation in distinct brain areas could be mediated by these biomolecules. However, a limited presence of misfolding-facilitating cofactors has been shown not to be sufficient for maintenance of strain features when used to complement in vitro prion conversion reactions (34).
In agreement with previously published data (7, 11, 25), our results show that each hamster prion strain tested has a particular preference for certain anatomical structures in the brain. For our studies, we analyzed 3–5 different brains per strain, and experiments were done by triplicate per sample. Obtained results showed little variability between independent replicates and high reproducibility in the profiles obtained for distinct brains infected with the same infectious agent. Our data showed that prion accumulation is massively present in cortical areas in all four strains tested. Nevertheless, cortical accumulation was more prevalent in 139H and DY infected animals. HY prions, the most aggressive of all the strain used in this study, displayed a marked thalamic preference. In agreement with previous results reported by Bessen and Marsh, differences were found for PrP accumulation in the occipital cortex when DY and HY were compared, being the accumulation in the former greater than in the latter (25). Although that report based its analysis on scoring immunostained brain slices, the results obtained were comparable to ours.
PMCA has been shown as a useful tool to understand several aspects related to the complex dynamic of prion replication (reviewed in (22)). This technique faithfully replicates strain features in vitro (12) and reproduces the species barrier phenomena (13). As expected from in vivo studies, dissimilar efficiency of in vitro replication has been described for different prion strains (27, 28). Previous studies suggested that the efficiency of in vitro prion replication correlates with the incubation periods to disease generated by different prion agents (28). Although the fastest strain used in our study (HY) showed the greatest PMCA efficiency, SSLOW prions were clearly out of this tendency. These results suggest that other processes besides PrPSc replication are responsible for the length of incubation periods observed in prion diseases. Differential toxicity and cell targeting of each strain to more “sensitive” brain areas could control the extent of incubation periods.
PrPC → PrPSc conversion by PMCA has been shown to depend on the presence of other biomolecules such as RNA and lipids (35, 36). The factors implicated in modulating prion conversion appear to be ubiquitously present in mammalian tissues regardless of the source in terms of cells or species (36). The “Unified Theory” of PrPSc replication proposes that although cofactors are not completely needed for templated misfolding or necessary for infectivity, they could be important accessories to maintain strain properties (37). In support of this view, a previous report showed that in vitro prion conversion reactions supplemented with minimum components converge in a single strain, regardless of the original source of seeds (34). Additionally, the differential expression of cofactors by certain cell types has been proposed to determine the distinct abilities of prion strains to infect diverse cells (18). In this line, we expected that PMCA replication might be susceptible to cofactors differentially expressed by distinct brain regions and perhaps reproduce, at least partially, the natural deposition profile observed for each strain after infection. Unfortunately, our results show that in vitro conversion did not reproduce the pattern of deposition observed in vivo, but rather was dependent on the availability of PrPC substrate present in each preparation. Although negative, these results shed important clues on the mechanism dictating the neurotropism of different prion strain to specific brain structures. Our data strongly suggest that the accumulation of prions in different regions is a dynamic phenomenon involving other cellular pathways in the living brain. Although unlikely, we cannot discard that some rough procedures included in the PMCA methodology (elimination of tissue debris and sonication) could be responsible for eliminating or disrupting some of the components responsible for region-specific protein misfolding.
The net accumulation of misfolded prion protein aggregates depends on the rates of prion replication and biological clearance as well as the transport of prion particles through anatomical connections within the central nervous system. These processes likely operate differently in diverse areas of the brain. Cell-free prion replication systems, such as PMCA, mimic only the process of prion formation, but do not contain the sophisticated biological pathways responsible for prion clearance nor the specific neuronal circuits that may determine the signature deposition of each agent in the brain.
In summary, our results suggest that the neurotropism of prion agents is an intricate process which does not depend on strain-specific differences in prion replication among distinct brain regions, but rather requires the complexity of biological pathways present in the living tissue. The data presented in this report provide additional clues to understand the mechanism dictating prion strain diversity.
Author Contributions
P. P. H. carried out the majority of the experiments, analyzed the results, and prepared the figures. R.M. designed the experiments, coordinated the study, analyzed the data, prepared the final version of the figures, and wrote the draft of the manuscript. C. D. A., I. M. G., and U. K. participated in some of the experiments. C. S. is the principal investigator on the project and was responsible for coordinating research activity, analyzing the data, funding, writing the manuscript, and producing the final version of the article.
Acknowledgments
We thank Dr. Akihiko Urayama for help in isolating brain regions, Dr. Ilia Baskakov for providing original SSLOW material, Javiera Bravo-Alegria for providing blots showing the electrophoretic profiles of prion strains, and Andrea Flores-Ramirez for animal care.
This work was supported in part by National Institutes of Health Grants R01NS049173, P01AI077774, and P01AI106705 (to C. S.). C. S. is the inventor on several patents related to the PMCA technology and is currently Founder, Chief Scientific Officer and Vice-President of Amprion Inc, a biotech company focusing on the commercial utilization of PMCA for prion diagnosis. R. M. is the inventor in some patent applications related to the PMCA technique. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- TSEs
- transmissible spongiform encephalopathies
- PrPSc
- disease-associated/infectious prion protein
- PrPC
- cellular/normal prion protein
- PMCA
- protein misfolding cyclic amplification
- HY
- hyper prion strain
- DY
- drowsy prion strain
- SSLOW
- synthetic prion strain leading to overweight
- GAPDH
- glyceraldehyde-3-phosphate dehydrogenase
- PK
- proteinase K
- PrP27–30
- disease-associated PK-resistant fragment of PrPSc.
References
- 1.Prusiner S. B. (1982) Novel proteinaceous infectious particles cause scrapie. Science 216, 136–144 [DOI] [PubMed] [Google Scholar]
- 2.Prusiner S. B. (1998) Prions. Proc. Natl. Acad. Sci. U.S.A. 95, 13363–13383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Morales R., Abid K., and Soto C. (2007) The prion strain phenomenon: molecular basis and unprecedented features. Biochim. Biophys. Acta 1772, 681–691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bruce M. E., and Fraser H. (1991) Scrapie strain variation and its implications. Curr. Top. Microbiol. Immunol. 172, 125–138 [DOI] [PubMed] [Google Scholar]
- 5.Bessen R. A., and Marsh R. F. (1992) Biochemical and physical properties of the prion protein from two strains of the transmissible mink encephalopathy agent. J. Virol. 66, 2096–2101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gambetti P., Kong Q., Zou W., Parchi P., and Chen S. G. (2003) Sporadic and familial CJD: classification and characterisation. Br. Med. Bull. 66, 213–239 [DOI] [PubMed] [Google Scholar]
- 7.DeArmond S. J., Sánchez H., Yehiely F., Qiu Y., Ninchak-Casey A., Daggett V., Camerino A. P., Cayetano J., Rogers M., Groth D., Torchia M., Tremblay P., Scott M. R., Cohen F. E., and Prusiner S. B. (1997) Selective neuronal targeting in prion disease. Neuron 19, 1337–1348 [DOI] [PubMed] [Google Scholar]
- 8.Fraser H., and Dickinson A. G. (1968) The sequential development of the brain lesion of scrapie in three strains of mice. J. Comp. Pathol. 78, 301–311 [DOI] [PubMed] [Google Scholar]
- 9.Pattison I. H., and Millson G. C. (1961) Scrapie produced experimentally in goats with special reference to the clinical syndrome. J. Comp. Pathol. 71, 101–109 [DOI] [PubMed] [Google Scholar]
- 10.Bruce M. E., McConnell I., Fraser H., and Dickinson A. G. (1991) The disease characteristics of different strains of scrapie in Sinc congenic mouse lines: implications for the nature of the agent and host control of pathogenesis. J. Gen. Virol. 72, 595–603 [DOI] [PubMed] [Google Scholar]
- 11.Taraboulos A., Jendroska K., Serban D., Yang S. L., DeArmond S. J., and Prusiner S. B. (1992) Regional mapping of prion proteins in brain. Proc. Natl. Acad. Sci. U.S.A. 89, 7620–7624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Castilla J., Morales R., Saá P., Barria M., Gambetti P., and Soto C. (2008) Cell-free propagation of prion strains. EMBO J. 27, 2557–2566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Castilla J., Gonzalez-Romero D., Saá P., Morales R., De Castro J., and Soto C. (2008) Crossing the species barrier by PrP(Sc) replication in vitro generates unique infectious prions. Cell 134, 757–768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Safar J., Wille H., Itri V., Groth D., Serban H., Torchia M., Cohen F. E., and Prusiner S. B. (1998) Eight prion strains have PrP(Sc) molecules with different conformations. Nat. Med. 4, 1157–1165 [DOI] [PubMed] [Google Scholar]
- 15.Bruce M. E. (1993) Scrapie strain variation and mutation. Br. Med. Bull. 49, 822–838 [DOI] [PubMed] [Google Scholar]
- 16.Hecker R., Taraboulos A., Scott M., Pan K. M., Yang S. L., Torchia M., Jendroska K., DeArmond S. J., and Prusiner S. B. (1992) Replication of distinct scrapie prion isolates is region specific in brains of transgenic mice and hamsters. Genes Dev. 6, 1213–1228 [DOI] [PubMed] [Google Scholar]
- 17.Falsig J., Sonati T., Herrmann U. S., Saban D., Li B., Arroyo K., Ballmer B., Liberski P. P., and Aguzzi A. (2012) Prion pathogenesis is faithfully reproduced in cerebellar organotypic slice cultures. PLoS. Pathog. 8, e1002985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mahal S. P., Baker C. A., Demczyk C. A., Smith E. W., Julius C., and Weissmann C. (2007) Prion strain discrimination in cell culture: the cell panel assay. Proc. Natl. Acad. Sci. U.S.A. 104, 20908–20913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bessen R. A., and Marsh R. F. (1992) Identification of two biologically distinct strains of transmissible mink encephalopathy in hamsters. J. Gen. Virol. 73, 329–334 [DOI] [PubMed] [Google Scholar]
- 20.Makarava N., Kovacs G. G., Bocharova O., Savtchenko R., Alexeeva I., Budka H., Rohwer R. G., and Baskakov I. V. (2010) Recombinant prion protein induces a new transmissible prion disease in wild-type animals. Acta Neuropathol. 119, 177–187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kimberlin R. H., Cole S., and Walker C. A. (1987) Temporary and permanent modifications to a single strain of mouse scrapie on transmission to rats and hamsters. J. Gen. Virol. 68, 1875–1881 [DOI] [PubMed] [Google Scholar]
- 22.Morales R., Duran-Aniotz C., Diaz-Espinoza R., Camacho M. V., and Soto C. (2012) Protein misfolding cyclic amplification of infectious prions. Nat. Protoc. 7, 1397–1409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jeffrey M., McGovern G., Makarava N., González L., Kim Y. S., Rohwer R. G., and Baskakov I. V. (2014) Pathology of SSLOW, a transmissible and fatal synthetic prion protein disorder, and comparison with naturally occurring classical transmissible spongiform encephalopathies. Neuropathol. Appl. Neurobiol. 40, 296–310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bartz J. C., Bessen R. A., McKenzie D., Marsh R. F., and Aiken J. M. (2000) Adaptation and selection of prion protein strain conformations following interspecies transmission of transmissible mink encephalopathy. J. Virol. 74, 5542–5547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bessen R. A., and Marsh R. F. (1994) Distinct PrP properties suggest the molecular basis of strain variation in transmissible mink encephalopathy. J. Virol. 68, 7859–7868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Green K. M., Castilla J., Seward T. S., Napier D. L., Jewell J. E., Soto C., and Telling G. C. (2008) Accelerated high fidelity prion amplification within and across prion species barriers. PLoS. Pathog. 4, e1000139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Moudjou M., Sibille P., Fichet G., Reine F., Chapuis J., Herzog L., Jaumain E., Laferriere F., Richard C. A., Laude H., Andréoletti O., Rezaei H., and Béringue V. (2014) Highly infectious prions generated by a single round of microplate-based protein misfolding cyclic amplification. MBio. 5, e00829–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ayers J. I., Schutt C. R., Shikiya R. A., Aguzzi A., Kincaid A. E., and Bartz J. C. (2011) The strain-encoded relationship between PrP replication, stability and processing in neurons is predictive of the incubation period of disease. PLoS. Pathog. 7, e1001317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Saá P., Castilla J., and Soto C. (2006) Ultra-efficient replication of infectious prions by automated protein misfolding cyclic amplification. J. Biol. Chem. 281, 35245–35252 [DOI] [PubMed] [Google Scholar]
- 30.Karapetyan Y. E., Saá P., Mahal S. P., Sferrazza G. F., Sherman A., Salès N., Weissmann C., and Lasmézas C. I. (2009) Prion strain discrimination based on rapid in vivo amplification and analysis by the cell panel assay. PLoS ONE 4, e5730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bartz J. C., Dejoia C., Tucker T., Kincaid A. E., and Bessen R. A. (2005) Extraneural prion neuroinvasion without lymphoreticular system infection. J. Virol. 79, 11858–11863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Piro J. R., Harris B. T., Nishina K., Soto C., Morales R., Rees J. R., and Supattapone S. (2009) Prion protein glycosylation is not required for strain-specific neurotropism. J. Virol. 83, 5321–5328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Saá P., Sferrazza G. F., Ottenberg G., Oelschlegel A. M., Dorsey K., and Lasmézas C. I. (2012) Strain-specific role of RNAs in prion replication. J. Virol. 86, 10494–10504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Deleault N. R., Walsh D. J., Piro J. R., Wang F., Wang X., Ma J., Rees J. R., and Supattapone S. (2012) Cofactor molecules maintain infectious conformation and restrict strain properties in purified prions. Proc. Natl. Acad. Sci. U.S.A. 109, E1938-E1946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Deleault N. R., Harris B. T., Rees J. R., and Supattapone S. (2007) Formation of native prions from minimal components in vitro. Proc. Natl. Acad. Sci. U.S.A. 104, 9741–9746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Abid K., Morales R., and Soto C. (2010) Cellular factors implicated in prion replication. FEBS Lett. 584, 2409–2414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Weissmann C. (1991) A 'unified theory' of prion propagation. Nature 352, 679–683 [DOI] [PubMed] [Google Scholar]