

Abstract
Microbial biofilms demonstrate a decreased susceptibility to antimicrobial agents. Various mechanisms have been proposed to be involved in this recalcitrance. We focus on two of these factors. Firstly, the ability of sessile cells to actively mediate efflux of antimicrobial compounds has a profound impact on resistance and tolerance, and several studies point to the existence of biofilm-specific efflux systems. Secondly, biofilm-specific stress responses have a marked influence on cellular physiology, and contribute to the occurrence of persister cells. We provide an overview of the data that demonstrate that both processes are important for survival following exposure to antimicrobial agents.
Keywords: antibiotic resistance, bacteria, biofilm, microbiology, tolerance, efflux
Introduction
Microbial biofilms are surface-attached communities, consisting of cells embedded in an extracellular polymeric matrix that is at least partially composed of polymers produced by the microorganism themselves (1). Biofilms are omnipresent in natural and man-made environments (1, 2), and biofilm-associated bacteria are involved in a wide range of infections, including respiratory tract infections in cystic fibrosis (CF)2 patients, chronically infected wounds, and device-related infections (3, 4). One of the hallmarks of these biofilm-associated infections is the frequent failure of antimicrobial chemotherapy. Although it is often postulated that sessile cells are more resistant to antimicrobial agents, these cells typically do not grow better than planktonic cells in the presence of antibiotics; for example, biofilm-associated and stationary-phase planktonic Burkholderia cepacia complex bacteria showed similar susceptibilities to antibiotics (5). However, it is much more difficult to kill biofilm-associated cells than planktonic cells (6), and various mechanisms that potentially could be involved in this have been described in the literature (see Refs. 7 and 8 for recent reviews as well as Refs. 9 and 10 in this minireview series). Avoiding exposure to (sufficiently high concentrations of) antibiotics, and the presence of a small population of specialized survivor cells that are tolerant toward particular antimicrobial agents (i.e. they are not killed upon exposure to the product) are two important mechanisms that will be discussed in this review.
The Role of Efflux in Biofilm Resistance
Bacteria use specialized membrane-associated proteins to expel a wide range of compounds from the cytoplasm (11). Combined with reduced influx of these compounds and/or enzymatic degradation, efflux pumps are responsible for keeping the cytoplasmic concentrations of certain antimicrobial compounds below a critical threshold (11, 12). Bacterial efflux pumps can be divided into several superfamilies, with members of the RND family being the most studied when it comes to their involvement in bacterial biofilm resistance and/or biofilm formation. They are composed of an inner membrane protein, a periplasmic membrane fusion protein, and an outer membrane protein (13, 14). This complex spans the Gram-negative cell envelope and allows the efficient translocation of a wide range of molecules (supplemental Fig. S1).
Enterobacteriaceae: Escherichia coli, Klebsiella pneumoniae, and Salmonella
In biofilms formed by two E. coli strains, a considerable fraction of genes up-regulated in biofilms as compared with planktonic cells was shown to be involved in efflux and transport (128 out of 600 up-regulated genes) (15). It was claimed that this up-regulation was a direct consequence of the “waste management problem” occurring in the “cramped conditions” encountered in the biofilm. When cells of various E. coli strains and a strain of K. pneumoniae were exposed to the efflux inhibitors thioridazine, phenyl-arginine-β-naphthylamide (PAβN), or 1-(1-naphthylmethyl)-piperazine (NMP), biofilm formation was significantly repressed, suggesting that functional efflux systems are required for full biofilm formation. In addition, these efflux pump inhibitors increased the activity of tetracycline against biofilms (15). In enteroaggregative E. coli, the TolC efflux pump is required for adherence to HEp-2 cells and biofilm formation, probably because it plays an important role in secreting a yet unidentified factor (16).
Salmonella enterica serovar Typhimurium mutants lacking any of the known efflux systems also showed a marked reduction in biofilm formation (17). It was observed that in these mutants, the expression of several curli genes was down-regulated, and these mutants effectively failed to produce curli. As curli fimbriae are an important component of the Salmonella biofilm matrix, and have previously been shown to be involved in adhesion, cell aggregation, and biofilm formation (18), this presents a functional connection between efflux and biofilm formation. Also, in this study, the anti-biofilm effects of inactivating particular efflux systems could be mimicked by adding efflux pump inhibitors, including PAβN, carbonyl cyanide m-chlorophenylhydrazone (CCCP), and chlorpromazine. In a subsequent study, the same authors focused on the S. enterica serovar Typhimurium AcrAB-TolC system and showed that AcrA and TolC are involved in biofilm formation, whereas AcrB is not, and in both the acrA and the tolC mutant, the expression of structural curli genes and regulatory genes involved in curli biosynthesis was significantly down-regulated as compared with the wild type strain (19).
Pseudomonas aeruginosa
The genome of the gamma-proteobacterium P. aeruginosa contains at least 12 RND pump-encoding operons (20, 21). Although initial studies on the mexAB-oprM, mexCD-oprJ, mexEF-oprN, and mexXY efflux systems in P. aeruginosa did not point to a major role in biofilm resistance, expression of these efflux systems in biofilms was found to be heterogeneous, with cells closest to the substrate showing the highest expression levels (22). This should not come as a surprise, as it is currently well established that different populations can be found in microbial biofilms, and these subpopulations of sessile cells are exposed to different chemical and physical environments, and hence show differences in physiology (for reviews on this topic, see Refs. 23 and 24). Subsequent investigations revealed that mexAB-oprM and mexCD-oprJ are essential for P. aeruginosa biofilm formation in the presence of azithromycin, as a mutant in which both systems were knocked out was not capable of forming biofilms in the presence of this macrolide (25). Remarkable in this regard is that mexC was only expressed in azithromycin-exposed sessile cells, whereas mexA expression was observed in both exposed and unexposed planktonic and sessile cells, suggesting that the P. aeruginosa MexCD-OprJ pump is a biofilm-specific defense mechanism against azithromycin (25). Similarly, it was suggested that the P. aeruginosa MexAB-OprM pump provides a biofilm-specific defense mechanism against colistin (26). These initial studies targeting known RND-type efflux pumps already pointed to a role for efflux in biofilm resistance, and this was further confirmed in a large-scale screening study in which ∼4000 P. aeruginosa transposon mutants were investigated (27). In this study, the PA1874–1877 operon was identified as a novel efflux system potentially involved in biofilm-specific resistance to antibiotics: the expression of PA1874 was found to be 10-fold higher in sessile than in planktonic cells, deletion of the operon only affected susceptibility to tobramycin in biofilms, and overexpression of the operon decreased the susceptibility toward selected aminoglycosides and fluoroquinolones in planktonic cells (27). In addition, in Pseudomonas fluorescens, expression of two efflux systems was up-regulated in glutaraldehyde-treated biofilms, and efflux pump inhibitors increased sensitivity of P. aeruginosa and P. fluorescens biofilms to this disinfectant, confirming that not only antibiotic resistance is mediated by efflux (28). Finally, it was observed that planktonic P. aeruginosa cells grown under hypoxic conditions showed an increased expression of MexEF-OprN (29). Although biofilm cells were not investigated in this particular study, the authors speculate that the hypoxia encountered by P. aeruginosa biofilms in the lungs of CF patients could contribute to their antibiotic resistance.
So far, little is known about the regulation of efflux pump expression in biofilms. However, the work of Karin Sauer and co-workers on the P. aeruginosa biofilm-specific MerR-type transcriptional regulator BrlR points to a possible molecular link between efflux pump expression and the biofilm phenotype. BrlR is a transcription factor that responds to changes in the concentration of the secondary messenger c-di-GMP (see Ref. 30 for a recent review as well as Ref. 31 in this minireview series) and regulates the expression of several genes involved in biofilm tolerance, including ndvB (32, 33). Sauer and co-workers (34) also demonstrated that brlR is required for maximal expression of the MexAB-OprM and MexEF-OprN efflux pumps in P. aeruginosa biofilms, and demonstrated a direct regulation of these pumps by BrlR, with BrlR binding to the promotor regions of the mexAB-oprM and mexEF-oprN operons.
In contrast to the work cited above, in two studies carried out by Stewart and co-workers (35, 36), no evidence was found for a role of efflux pumps in tolerance of P. aeruginosa biofilms. These at first sight contradictory findings suggest that the role of efflux systems in protecting sessile cells against antibiotics may depend on the global biofilm physiology (and thus on the experimental conditions) and/or may point to the presence of specific subpopulations in biofilms that benefit from these efflux pumps, whereas others do not. In addition, the regulatory mechanisms linking efflux with the biofilm-phenotype may be strain- and condition-dependent.
Most of the above mentioned studies used antibiotic-exposed P. aeruginosa mutant strains to confirm the role of efflux systems in biofilm tolerance, but the role of these systems in P. aeruginosa biofilm formation as such (in the absence of antibiotics) has not been investigated in great detail. However, using efflux pump inhibitors, including carbonyl cyanide m-chlorophenylhydrazone, chlorpromazine, and PAβN, it was shown that in static as well as in flow conditions, efflux pump inhibitors decreased biofilm formation, leading to the suggestion that efflux pump inhibitors could be used as anti-biofilm agents (19).
Burkholderia cenocepacia
The genome of another CF pathogen, B. cenocepacia, contains genes coding for 22 RND efflux systems (37–40), and high levels of expression of these efflux pumps are frequently observed in B. cepacia complex clinical isolates (41). Exposure of B. cenocepacia biofilms to the disinfectant chlorhexidine results in the up-regulation of eight RND family efflux pumps (42). Using mutants in which single RND efflux pumps were inactivated, it was shown that for some mutants (e.g. the ΔRND-9 mutant), biofilms were less tolerant than wild type biofilms (whereas planktonic susceptibility remaining unaltered). However, in other mutants (e.g. the ΔRND-4 mutant), planktonic cells were more susceptible, whereas the susceptibility of sessile cells remained unchanged, confirming that at least some of the RND efflux pumps in this organism are lifestyle-specific (42).
Expanding on this work, the role of 16 B. cenocepacia efflux systems in resistance to various antibiotics, including tobramycin and ciprofloxacin, was subsequently investigated (43). By measuring susceptibility in planktonic and sessile cultures, it was demonstrated that the RND-3 and RND-4 efflux pumps are important for resistance to various antimicrobial drugs (including tobramycin and ciprofloxacin) in planktonic B. cenocepacia populations, whereas the RND-3, RND-8, and RND-9 efflux pumps are important for protecting sessile cells against tobramycin, again pointing toward the existence of life style-specific RND-type efflux pumps. Interestingly, in the wild type strain, little regulation of expression of these efflux pumps at the mRNA level was observed using RT-quantitative PCR, suggesting that the regulation occurs mainly at the posttranscriptional level and/or at the level of activation of particular efflux systems (43).
Physiological Adaptation in Microbial Biofilms Leads to Reduced Activity of Antimicrobial Agents: Dormancy and the Persister Phenomenon
Bacterial populations are known to contain a subpopulation of cells tolerant to antimicrobial treatment (6). These so-called persister cells are present in both sessile and planktonic cultures, but are especially problematic in a biofilm environment, where they are shielded from the immune system (44, 45). Persisters are not mutants, but phenotypic variants of the wild type that upon re-inoculation produce a culture that again contains both persister and non-persister bacteria like the original population (44). Unlike antibiotic-resistant bacteria, they do not grow in the presence of bactericidal agents, but resume growth after the antibiotics have been removed (46). Because antibiotics kill cells by corrupting specific targets, dormant persisters, in which the antibiotic targets are inactive, escape killing (46). Non-growing or slowly growing bacteria are generally less sensitive to antibiotics, a phenomenon called “drug indifference” (45). However, persistence and drug indifference are different phenomena: the latter reflects the overall reduced sensitivity of dormant/slow-growing microbial populations without a specific mechanistic basis (e.g. stationary phase bacterial cultures surviving treatment with β-lactams), whereas persistence gives rise to a subpopulation with a different phenotype (45). The mechanisms leading to persistence are still largely unknown. Screening knock-out libraries has not led to the identification of mutants completely lacking the ability to form persisters, indicating that the mechanisms involved are redundant (46). However, various studies have identified putative persister genes (47, 48), and a general picture starts to emerge in which multiple (often connected) mechanisms play a role (Fig. 1). Below we will discuss the role of stress responses and metabolism, and focus on the similarities and differences between biofilms and planktonic cultures.
FIGURE 1.

Schematic overview of physiological adaptations in biofilm tolerance.
Oxidative Stress Response
Because bactericidal antibiotics are known to induce oxidative stress (49), lowering cellular hydroxyl radical levels by decreasing their production or by increased detoxification could counteract bactericidal activity. Shatalin et al. (50) showed in both biofilms and stationary phase cultures of various bacteria that H2S induced tolerance by reducing oxidative stress, through sequestration of ferrous iron and stimulation of catalases and superoxide dismutases. Similarly, it was observed that the active lowering of cellular hydroxyl levels played a role in planktonic and sessile P. aeruginosa persistence during starvation (51). Activation of the stringent response increased catalase and superoxide dismutase levels and repressed the production of hydroxy-alkylquinolines, intercellular signaling molecules with pro-oxidant properties (51). Data obtained for tobramycin-treated B. cenocepacia biofilms confirmed that avoiding exposure to antibiotic-induced reactive oxygen species (ROS) is a key factor in survival of persisters in these biofilms (52). Several genes encoding proteins involved in the generation of ROS, including a ferredoxin reductase (involved in recycling Fe3+ to Fe2+ and thus driving the Fenton reaction), were found to be down-regulated in persisters, whereas genes encoding proteins involved in ROS detoxification were up-regulated. These results suggest that persisters are to some extent protected against the detrimental effects of ROS produced upon antibiotic treatment.
The Stringent Response
Several studies have specified a role for guanosine tetra- or pentaphosphate, (p)ppGpp (known as the “alarmone”), the central mediator of the stringent response, in persistence (53–56). The relA-spoT gene pair, involved in the synthesis of (p)ppGpp, was linked to persistence in various studies (57–59). For example, reduced levels of persisters were reported in P. aeruginosa biofilms deficient in (p)ppGpp synthesis (36). Gerdes et al. (53) proposed a model in which (p)ppGpp induces persistence by activating toxin antitoxin (TA) modules via polyphosphate and Lon proteases. It was previously demonstrated that deletion of lon dramatically reduces persistence, whereas a moderate overproduction stimulates persistence by degrading antitoxins and hereby activating their cognate toxins (60). A link between (p)ppGpp and the transcriptional activation of the toxin HokB by Obg, a universally conserved GTPase, was recently identified (61). Obg was shown to control persistence, in both planktonic cultures and P. aeruginosa biofilms, by inducing the expression of HokB, whereas elevated levels of HokB resulted in membrane depolarization and dormancy.
TA Modules
TA modules are thought to be a major player in the induction of persistence (53) (for a review on TA modules, see Ref. 62). Type II TA modules usually consist of two proteins: a toxin that can inhibit an important cellular function and an antitoxin that can form a complex with the toxin and hence inactivates it (63) (Fig. 2) (for a schematic overview of the different types of TA modules, see supplemental Fig. S2). The antitoxin is typically proteolytically degraded during stress, and as a result, the free toxin can impede cellular processes such as DNA replication, translation, ATP, or cell wall synthesis (53, 64). TA modules, although initially thought to be involved only in cell death, have been shown to play a role in various essential cellular processes, including biofilm formation and persistence (53). Because bactericidal antibiotics kill cells by corrupting cellular functions, which are inhibited by toxins, the role of TA modules in persistence has been documented in various studies (53). Toxin inhibition of cell wall synthesis, translation, or replication would prevent antibiotics from killing and give rise to persister cells. HipAB was the first TA module linked to persistence (65), and many key insights on persistence were obtained while studying this particular module. Rotem et al. (66) noticed that persistence occurred once the toxin HipA reached a certain threshold level and that the amount by which the threshold was exceeded determined the duration of dormancy. Since the identification of HipAB, various studies have reported an up-regulation of TA modules in persister cells (65, 67). For example, transcriptome analysis of dormant E. coli cells identified mqsR as the most highly induced gene in persister cells as compared with non-persisters (67). In B. cenocepacia, several toxins were found to be up-regulated in biofilms as compared with planktonic cells, and overexpression of these toxins contributed to persistence in biofilms after treatment with tobramycin or ciprofloxacin (68). Although overproduction of almost any toxin may increase persistence, only two TA pairs have been shown to decrease persistence upon deletion (69, 70). Deletion of mqsRA significantly reduced persistence in E. coli biofilms (69), whereas deletion of tisAB/istr1 led to a decrease in survival in planktonic cultures (70). Simultaneous deletion of multiple other TA systems also decreased the number of persisters in E. coli (60). The latter confirms the role of TA modules in persistence, but again suggests redundancy.
FIGURE 2.
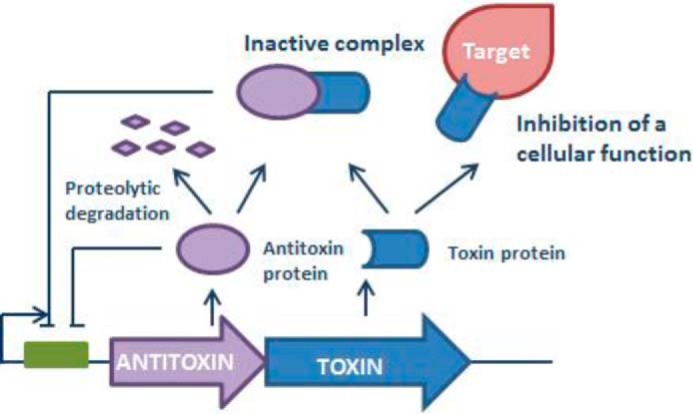
Schematic overview of type II TA modules.
Additionally, recent studies have indicated a role for TA systems in the switch from the planktonic to the sessile lifestyle. MqsRA was the first TA module linked to biofilm formation: in a transcriptome study comparing sessile and planktonic cells, mqsR was found to be induced in biofilms (71), and biofilm formation in E. coli was found to be mediated by MqsRA (72). In support of these observations, the antitoxin MqsA was more recently found to repress rpoS, thereby reducing the c-di-GMP concentration, which leads to increased motility and decreased biofilm formation (69). Degradation of MqsA by Lon proteases in turn resulted in rpoS induction and a switch from the planktonic to the biofilm state. A model was proposed in which there is a spectrum of MqsR activities, and depending on the activity of MqsR, a cell would respond to stress by biofilm formation and the production of proteins to withstand stress, or alternatively, become dormant (69). In this regard, persister cell formation can be seen as an extreme example of the general stress response mediated by MqsR. Further evidence for the involvement of TA modules in E. coli biofilm formation was obtained by studying Δ5, a strain in which five different TA modules were deleted. Deletion of these systems reduced biofilm formation after 8 h but increased biofilm formation after 24 h (73). Transcriptome profiling revealed that deletion of these TA modules induced the expression of an uncharacterized gene, yjgK, which repressed fimbriae at 8 h (73). Although this repression of fimbriae may explain the decrease in biofilm formation at 8 h, a reduction in biofilm dispersal may explain the increase in biofilm formation at 24 h.
The SOS Response
The SOS response is triggered by DNA damage (74). This damage is recognized by RecA, which causes self-cleavage of the LexA repressor, hereby activating SOS genes (74). Based on transcriptome analyses and screening of mutant libraries, lexA and recA were found to be involved in E. coli persistence (58, 65). In line with this observation, deletion of the tisAB/istr1 TA module, which contains a Lex box, dramatically reduced the number of surviving persisters in exponentially growing cultures of E. coli treated with fluoroquinolones, but not after treatment with antibiotics that do not cause DNA damage (70). Moreover, a functional RecA protein was needed for persistence, confirming dependence on the SOS pathway. The induction of persisters by the SOS-induced TisB toxin links two survival strategies, i.e. active repair on the one hand and shutdown of cellular metabolism on the other. This suggests that in the presence of DNA-damaging agents, the optimal strategy is to induce repair and at the same time increase the number of dormant cells that will survive when repair would fail (46). Interestingly, although inactivation of genes involved in the SOS response also increased susceptibility to fluoroquinolones in stationary phase cultures, TisB was not found to be involved, suggesting that persisters form through other mechanisms in non-growing cultures (70). Tolerance to ofloxacin significantly increased in E. coli biofilms upon starvation, whereas starvation did not have a significant effect on tolerance to ofloxacin in planktonic cultures (75). Similarly, tolerance was dependent on the SOS response but independent of known SOS-induced TA modules. Although starvation also induces the stringent response, deletion of relA only partially increased sensitivity to ofloxacin and only upon leucine starvation, suggesting that the stringent response plays a minor role in tolerance to ofloxacin.
Metabolism
A regulator of phosphate metabolism, phoU, was identified as a persister gene as its deletion reduced persistence in planktonic cultures of E. coli and inactivation of phoU was shown to lead to a hyperactive metabolic state (76). However, mutations in sucB or ubiF, leading to reduced ATP synthesis, also negatively affected survival of persisters (77). SucB is a key enzyme of the TCA cycle, whereas UbiF is involved in the biosynthesis of ubiquinone, an acceptor of electrons in the respiratory electron transport chain. These at first sight contradictory results illustrate the critical role regulation of metabolism may play in persistence. Based on persistence assays and FACS sorting of E. coli cells fluorescently labeled according to their metabolic activity, Orman and Brynildsen (78) suggested that a low metabolic activity prior to antibiotic exposure only increases the likelihood of a cell to become a persister. Additionally, they observed that inhibition of respiration decreased the number of persisters surviving treatment with ampicillin (79). Inhibition of respiration during stationary phase may prevent digestion of endogenous proteins and mRNA and thus allow translation and replication to proceed and render bacteria susceptible to antibiotics. In B. cenocepacia biofilms, a metabolic shift bypassing the TCA cycle was observed in persister cells (52). In cells surviving treatment with tobramycin, genes encoding proteins involved in the TCA cycle were down-regulated, and at the same time, the glyoxylate shunt was activated, most likely to sustain ATP production. Mok et al. (80) studied the metabolic aspects of persisters in an E. coli strain in which the antitoxin MazE and the toxin MazF were artificially and independently induced. Upon accumulation of MazF, an endoribonuclease, reversible stasis was achieved and populations were almost entirely tolerant to fluoroquinolones and β-lactams. Although these induced persisters were found to be non-replicative, they maintained oxygen and glucose consumption. Further analysis also indicated accumulation of all four ribonucleotide monophosphates, confirming futile cycling in these persisters. Energy derived from catabolism was used to continue transcription, but at the same time, the transcripts were degraded by MazF.
An obvious question is whether or not persister cells can be considered as dormant, given the mechanisms involved. Based on studies indicating the role of different stress responses in persister cells, it was suggested that persistence is an actively maintained state (81). However, according to Wood et al. (82), most evidence points to a role for dormancy, toxin induction, down-regulation of metabolic pathways, and shutdown of protein synthesis in persistence. Although both viewpoints seem contradictory, they could be reconciled as active responses to stress may play a role in inducing dormancy because most stress responses lead to slowing down or inhibition of cell growth (81, 82). Additionally, although shutdown of metabolic processes may be involved in the entry into a dormant antibiotic tolerant state, residual metabolic activity may be required to maintain viability, and reactivation is necessary to resume growth after removal of the antibiotics (for recent reviews on targeting metabolism, see Refs. 83 and 84). For example, in stationary phase cultures of E. coli, there is no difference in DNA damage between persisters and non-persisters (85). Moreover, neither the level of SOS machinery before nor during treatment impacted the level of persisters, but the cell's ability to repair ofloxacin-induced DNA damage during recovery when the antibiotic was removed was critical to maintain persistence.
Concluding Remarks
Many different mechanisms leading to biofilm resistance and tolerance have been described, including efflux-mediated removal of antimicrobial agents from the cytoplasm and the occurrence of tolerant and persistent subpopulations of cells. Although there is currently no evidence linking increased efflux in bacterial populations with cell density, it is clear that at least in some bacterial species particular efflux systems are involved in biofilm formation and/or resistance. The currently available evidence suggests that life style-specific efflux pumps (i.e. efflux systems that are preferentially used in either planktonic or sessile populations) exist, as well as general systems that are used by bacteria to get rid of unwanted compounds irrespective of the mode of growth. Little is known about the regulation of the expression of these efflux pumps and how that would be affected by growth in a biofilm, although work with the regulator BrlR in P. aeruginosa points to a role for c-di-GMP.
Persister cells have been described in all bacteria examined so far, and persisters present in biofilms are thought to be an important reason for treatment failure. Persisters are generally considered as dormant cells in which the antibiotic targets are inactive. Recent research, however, suggest that persisters are not necessarily inactive but rather have a different metabolism. Several studies points to the importance of residual metabolic activity to maintain viability and the ability to resuscitate after removal of the antibiotics. Additionally, active responses to stress are thought to induce persistence, and different stress responses are likely involved depending on the conditions.
Supplementary Material
This work was supported by FWO-Vlaanderen and the Interuniversity Attraction Poles Programme initiated by the Belgian Science Policy Office. This is the fifth article in the Thematic Minireview series “Biofilms.” The authors declare that they have no conflicts of interest with the contents of this article.

This article contains supplemental Figs. S1 and S2.
- CF
- cystic fibrosis
- RND
- resistance-nodulation-division
- PAβN
- phenyl-arginine-β-naphthylamide
- c-di-GMP
- cyclic-di-guanosine monophosphate
- (p)ppGpp
- guanosine tetra- or pentaphosphate
- ROS
- reactive oxygen species
- TA
- toxin antitoxin
- TCA
- tricarboxylic acid.
References
- 1.Costerton J. W. (2007) The Biofilm Primer, Springer, Berlin [Google Scholar]
- 2.Hall-Stoodley L., Costerton J. W., and Stoodley P. (2004) Bacterial biofilms: from the natural environment to infectious diseases. Nat. Rev. Microbiol. 2, 95–108 [DOI] [PubMed] [Google Scholar]
- 3.Wolcott R. D., Rhoads D. D., Bennett M. E., Wolcott B. M., Gogokhia L., Costerton J. W., and Dowd S. E. (2010) Chronic wounds and the medical biofilm paradigm. J. Wound Care 19, 45–53 [DOI] [PubMed] [Google Scholar]
- 4.Bjarnsholt T. (2013) The role of bacterial biofilms in chronic infections. APMIS. Suppl., 1–51 [DOI] [PubMed] [Google Scholar]
- 5.Peeters E., Nelis H. J., and Coenye T. (2009) In vitro activity of ceftazidime, ciprofloxacin, meropenem, minocycline, tobramycin and trimethoprim/sulfamethoxazole against planktonic and sessile Burkholderia cepacia complex bacteria. J. Antimicrob. Chemother. 64, 801–809 [DOI] [PubMed] [Google Scholar]
- 6.Lewis K. (2008) Multidrug tolerance of biofilms and persister cells. Curr. Top. Microbiol. Immunol. 322, 107–131 [DOI] [PubMed] [Google Scholar]
- 7.Lebeaux D., Ghigo J. M., and Beloin C. (2014) Biofilm-related infections: bridging the gap between clinical management and fundamental aspects of recalcitrance toward antibiotics. Microbiol. Mol. Biol. Rev. 78, 510–543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Van Acker H., Van Dijck P., and Coenye T. (2014) Molecular mechanisms of antimicrobial tolerance and resistance in bacterial and fungal biofilms. Trends Microbiol. 22, 326–333 [DOI] [PubMed] [Google Scholar]
- 9.Sheppard D. C., and Howell P. L. (2016) Biofilm exopolysaccharides of pathogenic fungi: lessons from bacteria. J. Biol. Chem. 291, 12529–12537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gunn J. S., Bakaletz L. O., and Wozniak D. J. (2016) What's on the outside matters: The role of the extracellular polymeric substance of Gram-negative biofilms in evading host immunity and as a target for therapeutic intervention. J. Biol. Chem. 291, 12538–12546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Routh M. D., Zalucki Y., Su C. C., Zhang Q., Shafer W. M., and Yu E. W. (2011) Efflux pumps of the resistance-nodulation-division family: a perspective of their structure, function, and regulation in Gram-negative bacteria. Adv. Enzymol. Relat. Areas Mol. Biol. 77, 109–146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Soto S. M. (2013) Role of efflux pumps in the antibiotic resistance of bacteria embedded in a biofilm. Virulence 4, 223–229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tseng T. T., Gratwick K. S., Kollman J., Park D., Nies D. H., Goffeau A., and Saier M. H. Jr. (1999) The RND permease superfamily: an ancient, ubiquitous and diverse family that includes human disease and development proteins. J. Mol. Microbiol. Biotechnol. 1, 107–125 [PubMed] [Google Scholar]
- 14.Nikaido H. (2011) Structure and mechanism of RND-type multidrug efflux pumps. Adv. Enzymol. Relat. Areas Mol. Biol. 77, 1–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kvist M., Hancock V., and Klemm P. (2008) Inactivation of efflux pumps abolishes bacterial biofilm formation. Appl. Environ. Microbiol. 74, 7376–7382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Imuta N., Nishi J., Tokuda K., Fujiyama R., Manago K., Iwashita M., Sarantuya J., and Kawano Y. (2008) The Escherichia coli efflux pump TolC promotes aggregation of enteroaggregative E. coli 042. Infect. Immun. 76, 1247–1256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Baugh S., Ekanayaka A. S., Piddock L. J., and Webber M. A. (2012) Loss of or inhibition of all multidrug resistance efflux pumps of Salmonella enterica serovar Typhimurium results in impaired ability to form a biofilm. J. Antimicrob. Chemother. 67, 2409–2417 [DOI] [PubMed] [Google Scholar]
- 18.Barnhart M. M., and Chapman M. R. (2006) Curli biogenesis and function. Annu. Rev. Microbiol. 60, 131–147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Baugh S., Phillips C. R., Ekanayaka A. S., Piddock L. J., and Webber M. A. (2014) Inhibition of multidrug efflux as a strategy to prevent biofilm formation. J. Antimicrob. Chemother. 69, 673–681 [DOI] [PubMed] [Google Scholar]
- 20.Kumar A., and Schweizer H. P. (2005) Bacterial resistance to antibiotics: active efflux and reduced uptake. Adv. Drug Deliv. Rev. 57, 1486–1513 [DOI] [PubMed] [Google Scholar]
- 21.Fernández L., and Hancock R. E. (2012) Adaptive and mutational resistance: role of porins and efflux pumps in drug resistance. Clin. Microbiol. Rev. 25, 661–681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.De Kievit T. R., Parkins M. D., Gillis R. J., Srikumar R., Ceri H., Poole K., Iglewski B. H., and Storey D. G. (2001) Multidrug efflux pumps: expression patterns and contribution to antibiotic resistance in Pseudomonas aeruginosa biofilms. Antimicrob. Agents Chemother. 45, 1761–1770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Stewart P. S., and Franklin M. J. (2008) Physiological heterogeneity in biofilms. Nat. Rev. Microbiol. 6, 199–210 [DOI] [PubMed] [Google Scholar]
- 24.Coenye T. (2010) Response of sessile cells to stress: from changes in gene expression to phenotypic adaptation. FEMS Immunol. Med. Microbiol. 59, 239–252 [DOI] [PubMed] [Google Scholar]
- 25.Gillis R. J., White K. G., Choi K. H., Wagner V. E., Schweizer H. P., and Iglewski B. H. (2005) Molecular basis of azithromycin-resistant Pseudomonas aeruginosa biofilms. Antimicrob. Agents Chemother. 49, 3858–3867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pamp S. J., Gjermansen M., Johansen H. K., and Tolker-Nielsen T. (2008) Tolerance to the antimicrobial peptide colistin in Pseudomonas aeruginosa biofilms is linked to metabolically active cells, and depends on the pmr and mexAB-oprM genes. Mol. Microbiol. 68, 223–240 [DOI] [PubMed] [Google Scholar]
- 27.Zhang L., and Mah T. F. (2008) Involvement of a novel efflux system in biofilm-specific resistance to antibiotics. J. Bacteriol. 190, 4447–4452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Vikram A., Bomberger J. M., and Bibby K. J. (2015) Efflux as a glutaraldehyde resistance mechanism in Pseudomonas fluorescens and Pseudomonas aeruginosa biofilms. Antimicrob. Agents Chemother. 59, 3433–3440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schaible B., Taylor C. T., and Schaffer K. (2012) Hypoxia increases antibiotic resistance in Pseudomonas aeruginosa through altering the composition of multidrug efflux pumps. Antimicrob. Agents Chemother. 56, 2114–2118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Römling U., Galperin M. Y., and Gomelsky M. (2013) Cyclic di-GMP: the first 25 years of a universal bacterial second messenger. Microbiol. Mol. Biol. Rev.: MMBR 77, 1–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Valentini M., and Filloux A. (2016) Biofilms and c-di-GMP signaling: lessons from Pseudomonas aeruginosa and other bacteria. J. Biol. Chem. 291, 12547–12555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liao J., and Sauer K. (2012) The MerR-like transcriptional regulator BrlR contributes to Pseudomonas aeruginosa biofilm tolerance. J. Bacteriol. 194, 4823–4836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chambers J. R., Liao J., Schurr M. J., and Sauer K. (2014) BrlR from Pseudomonas aeruginosa is a c-di-GMP-responsive transcription factor. Mol. Microbiol. 92, 471–487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liao J., Schurr M. J., and Sauer K. (2013) The MerR-like regulator BrlR confers biofilm tolerance by activating multidrug efflux pumps in Pseudomonas aeruginosa biofilms. J. Bacteriol. 195, 3352–3363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Folsom J. P., Richards L., Pitts B., Roe F., Ehrlich G. D., Parker A., Mazurie A., and Stewart P. S. (2010) Physiology of Pseudomonas aeruginosa in biofilms as revealed by transcriptome analysis. BMC Microbiol. 10, 294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Stewart P. S., Franklin M. J., Williamson K. S., Folsom J. P., Boegli L., and James G. A. (2015) Contribution of stress responses to antibiotic tolerance in Pseudomonas aeruginosa biofilms. Antimicrob. Agents Chemother. 59, 3838–3847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Guglierame P., Pasca M. R., De Rossi E., Buroni S., Arrigo P., Manina G., and Riccardi G. (2006) Efflux pump genes of the resistance-nodulation-division family in Burkholderia cenocepacia genome. BMC Microbiol. 6, 66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Buroni S., Pasca M. R., Flannagan R. S., Bazzini S., Milano A., Bertani I., Venturi V., Valvano M. A., and Riccardi G. (2009) Assessment of three Resistance-Nodulation-Cell Division drug efflux transporters of Burkholderia cenocepacia in intrinsic antibiotic resistance. BMC Microbiol. 9, 200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bazzini S., Udine C., Sass A., Pasca M. R., Longo F., Emiliani G., Fondi M., Perrin E., Decorosi F., Viti C., Giovannetti L., Leoni L., Fani R., Riccardi G., Mahenthiralingam E., and Buroni S. (2011) Deciphering the role of RND efflux transporters in Burkholderia cenocepacia. PLoS ONE 6, e18902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rushton L., Sass A., Baldwin A., Dowson C. G., Donoghue D., and Mahenthiralingam E. (2013) Key role for efflux in the preservative susceptibility and adaptive resistance of Burkholderia cepacia complex bacteria. Antimicrob. Agents Chemother. 57, 2972–2980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tseng S. P., Tsai W. C., Liang C. Y., Lin Y. S., Huang J. W., Chang C. Y., Tyan Y. C., and Lu P. L. (2014). The contribution of antibiotic resistance mechanisms in clinical Burkholderia cepacia complex isolates: an emphasis on efflux pump activity. PLoS ONE 9, e104986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Coenye T., Van Acker H., Peeters E., Sass A., Buroni S., Riccardi G., and Mahenthiralingam E. (2011) Molecular mechanisms of chlorhexidine tolerance in Burkholderia cenocepacia biofilms. Antimicrob. Agents Chemother. 55, 1912–1919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Buroni S., Matthijs N., Spadaro F., Van Acker H., Scoffone V. C., Pasca M. R., Riccardi G., and Coenye T. (2014) Differential roles of RND efflux pumps in antimicrobial drug resistance of sessile and planktonic Burkholderia cenocepacia cells. Antimicrob. Agents Chemother. 58, 7424–7429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lewis K. (2007) Persister cells, dormancy and infectious disease. Nat. Rev. Microbiol. 5, 48–56 [DOI] [PubMed] [Google Scholar]
- 45.Jayaraman R. (2008) Bacterial persistence: some new insights into an old phenomenon. J. Biosci. 33, 795–805 [DOI] [PubMed] [Google Scholar]
- 46.Lewis K. (2010) Persister cells. Annu. Rev. Microbiol. 64, 357–372 [DOI] [PubMed] [Google Scholar]
- 47.Hansen S., Lewis K., and Vulić M. (2008) Role of global regulators and nucleotide metabolism in antibiotic tolerance in Escherichia coli. Antimicrob. Agents Chemother. 52, 2718–2726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.De Groote V. N., Verstraeten N., Fauvart M., Kint C. I., Verbeeck A. M., Beullens S., Cornelis P., and Michiels J. (2009) Novel persistence genes in Pseudomonas aeruginosa identified by high-throughput screening. FEMS Microbiol. Lett. 297, 73–79 [DOI] [PubMed] [Google Scholar]
- 49.Dwyer D. J., Collins J. J., and Walker G. C. (2015) Unraveling the physiological complexities of antibiotic lethality. Annu. Rev. Pharmacol. Toxicol. 55, 313–332 [DOI] [PubMed] [Google Scholar]
- 50.Shatalin K., Shatalina E., Mironov A., and Nudler E. (2011) H2S: a universal defense against antibiotics in bacteria. Science 334, 986–990 [DOI] [PubMed] [Google Scholar]
- 51.Nguyen D., Joshi-Datar A., Lepine F., Bauerle E., Olakanmi O., Beer K., McKay G., Siehnel R., Schafhauser J., Wang Y., Britigan B. E., and Singh P. K. (2011) Active starvation responses mediate antibiotic tolerance in biofilms and nutrient-limited bacteria. Science 334, 982–986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Van Acker H., Sass A., Bazzini S., De Roy K., Udine C., Messiaen T., Riccardi G., Boon N., Nelis H. J., Mahenthiralingam E., and Coenye T. (2013) Biofilm-grown Burkholderia cepacia complex cells survive antibiotic treatment by avoiding production of reactive oxygen species. PLoS ONE 8, e58943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gerdes K., and Maisonneuve E. (2012) Bacterial persistence and toxin-antitoxin loci. Annu. Rev. Microbiol. 66, 103–123 [DOI] [PubMed] [Google Scholar]
- 54.Germain E., Castro-Roa D., Zenkin N., and Gerdes K. (2013) Molecular mechanism of bacterial persistence by HipA. Mol. Cell 52, 248–254 [DOI] [PubMed] [Google Scholar]
- 55.Maisonneuve E., Castro-Camargo M., and Gerdes K. (2013). (p)ppGpp controls bacterial persistence by stochastic induction of toxin-antitoxin activity. Cell 154, 1140–1150 [DOI] [PubMed] [Google Scholar]
- 56.Ramisetty B. C., Natarajan B., and Santhosh R. S. (2015) mazEF-mediated programmed cell death in bacteria: “What is this?” Crit. Rev. Microbiol. 41, 89–100 [DOI] [PubMed] [Google Scholar]
- 57.Viducic D., Ono T., Murakami K., Susilowati H., Kayama S., Hirota K., and Miyake Y. (2006) Functional analysis of spoT, relA and dksA genes on quinolone tolerance in Pseudomonas aeruginosa under nongrowing condition. Microbiol. Immunol. 50, 349–357 [DOI] [PubMed] [Google Scholar]
- 58.Fauvart M., De Groote V. N., and Michiels J. (2011) Role of persister cells in chronic infections: clinical relevance and perspectives on anti-persister therapies. J. Med. Microbiol. 60, 699–709 [DOI] [PubMed] [Google Scholar]
- 59.Korch S. B., Henderson T. A., and Hill T. M. (2003) Characterization of the hipA7 allele of Escherichia coli and evidence that high persistence is governed by (p)ppGpp synthesis. Mol. Microbiol. 50, 1199–1213 [DOI] [PubMed] [Google Scholar]
- 60.Maisonneuve E., Shakespeare L. J., Jørgensen M. G., and Gerdes K. (2011) Bacterial persistence by RNA endonucleases. Proc. Natl. Acad. Sci. U.S.A. 108, 13206–13211 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 61.Verstraeten N., Knapen W. J., Kint C. I., Liebens V., Van den Bergh B., Dewachter L., Michiels J. E., Fu Q., David C. C., Fierro A. C., Marchal K., Beirlant J., Versées W., Hofkens J., Jansen M., et al. (2015) Obg and membrane depolarization are part of a microbial bet-hedging strategy that leads to antibiotic tolerance. Mol. Cell 59, 9–21 [DOI] [PubMed] [Google Scholar]
- 62.Goeders N., and Van Melderen L. (2014) Toxin-antitoxin systems as multilevel interaction systems. Toxins 6, 304–324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Schuster C. F., and Bertram R. (2013) Toxin-antitoxin systems are ubiquitous and versatile modulators of prokaryotic cell fate. FEMS Microbiol. Lett. 340, 73–85 [DOI] [PubMed] [Google Scholar]
- 64.Yamaguchi Y., Park J. H., and Inouye M. (2011) Toxin-antitoxin systems in bacteria and archaea. Annu. Rev. Genet. 45, 61–79 [DOI] [PubMed] [Google Scholar]
- 65.Keren I., Shah D., Spoering A., Kaldalu N., and Lewis K. (2004) Specialized persister cells and the mechanism of multidrug tolerance in Escherichia coli. J. Bacteriol. 186, 8172–8180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Rotem E., Loinger A., Ronin I., Levin-Reisman I., Gabay C., Shoresh N., Biham O., and Balaban N. Q. (2010) Regulation of phenotypic variability by a threshold-based mechanism underlies bacterial persistence. Proc. Natl. Acad. Sci. U.S.A. 107, 12541–12546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Shah D., Zhang Z., Khodursky A., Kaldalu N., Kurg K., and Lewis K. (2006). Persisters: a distinct physiological state of E. coli. BMC Microbiol. 6, 53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Van Acker H., Sass A., Dhondt I., Nelis H. J., and Coenye T. (2014) Involvement of toxin-antitoxin modules in Burkholderia cenocepacia biofilm persistence. Pathog. Dis. 71, 326–335 [DOI] [PubMed] [Google Scholar]
- 69.Wang X., and Wood T. K. (2011) Toxin-antitoxin systems influence biofilm and persister cell formation and the general stress response. Appl. Environ. Microbiol. 77, 5577–5583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Dörr T., Vulić M., and Lewis K. (2010). Ciprofloxacin causes persister formation by inducing the TisB toxin in Escherichia coli. PLos Biol. 8, e1000317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ren D., Bedzyk L. A., Thomas S. M., Ye R. W., and Wood T. K. (2004) Gene expression in Escherichia coli biofilms. Appl. Microbiol. Biotechnol. 64, 515–524 [DOI] [PubMed] [Google Scholar]
- 72.González Barrios A. F., Zuo R., Hashimoto Y., Yang L., Bentley W. E., and Wood T. K. (2006) Autoinducer 2 controls biofilm formation in Escherichia coli through a novel motility quorum-sensing regulator (MqsR, B3022). J. Bacteriol. 188, 305–316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kim Y., Wang X., Ma Q., Zhang X. S., and Wood T. K. (2009) Toxin-antitoxin systems in Escherichia coli influence biofilm formation through YjgK (TabA) and fimbriae. J. Bacteriol. 191, 1258–1267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Michel B. (2005) After 30 years of study, the bacterial SOS response still surprises us. PLoS Biol. 3, e255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Bernier S. P., Lebeaux D., DeFrancesco A. S., Valomon A., Soubigou G., Coppée J. Y., Ghigo J. M., and Beloin C. (2013). Starvation, together with the SOS response, mediates high biofilm-specific tolerance to the fluoroquinolone ofloxacin. PLoS Genet. 9, e1003144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Li Y., and Zhang Y. (2007) PhoU is a persistence switch involved in persister formation and tolerance to multiple antibiotics and stresses in Escherichia coli. Antimicrob. Agents Chemother. 51, 2092–2099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Ma C., Sim S., Shi W., Du L., Xing D., and Zhang Y. (2010) Energy production genes sucB and ubiF are involved in persister survival and tolerance to multiple antibiotics and stresses in Escherichia coli. FEMS Microbiol. Lett. 303, 33–40 [DOI] [PubMed] [Google Scholar]
- 78.Orman M. A., and Brynildsen M. P. (2013) Dormancy is not necessary or sufficient for bacterial persistence. Antimicrob. Agents Chemother. 57, 3230–3239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Orman M. A., and Brynildsen M. P. (2015) Inhibition of stationary phase respiration impairs persister formation in E. coli. Nat. Commun. 6, 7983; Correction (2016) Nat. Commun.7, 10756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Mok W. W., Park J. O., Rabinowitz J. D., and Brynildsen M. P. (2015) RNA futile cycling in model persisters derived from MazF accumulation. mBio 6, e01588–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Cohen N. R., Lobritz M. A., and Collins J. J. (2013) Microbial persistence and the road to drug resistance. Cell Host Microbe 13, 632–642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Wood T. K., Knabel S. J., and Kwan B. W. (2013) Bacterial persister cell formation and dormancy. Appl. Environ. Microbiol. 79, 7116–7121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Amato S. M., Fazen C. H., Henry T. C., Mok W. W., Orman M. A., Sandvik E. L., Volzing K. G., and Brynildsen M. P. (2014) The role of metabolism in bacterial persistence. Front. Microbiol. 5, 70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Prax M., and Bertram R. (2014) Metabolic aspects of bacterial persisters. Front. Cell Infect. Microbiol. 4, 148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Völzing K. G., and Brynildsen M. P. (2015) Stationary-phase persisters to ofloxacin sustain DNA damage and require repair systems only during recovery. mBio 6, e00731–00715 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.