

Abstract
Background
The main purpose of this research was to design a self-nanoemulsifying drug delivery system (SNEDDS) for improving the bioavailability of cyclovirobuxine D as a poorly water-soluble drug.
Materials and methods
Solubility trials, emulsifying studies, and pseudo-ternary phase diagrams were used to screen the SNEDDS formulations. The optimized drug-loaded SNEDDS was prepared at a mass ratio of 3:24:38:38 for cyclovirobuxine D, oleic acid, Solutol SH15, and propylene glycol, respectively. The optimized formulation was characterized in terms of physicochemical and pharmacokinetic parameters compared with marketed cyclovirobuxine D tablets.
Results
The optimized cyclovirobuxine-D-loaded SNEDDS was spontaneously dispersed to form a nanoemulsion with a globule size of 64.80±3.58 nm, which exhibited significant improvement of drug solubility, rapid absorption rate, and enhanced area under the curve, together with increased permeation and decreased efflux. Fortunately, there was a nonsignificant cytotoxic effect toward Caco-2 cells. The relative bioavailability of SNEDDS was 200.22% in comparison with market tablets, in rabbits.
Conclusion
SNEDDS could be a potential candidate for an oral dosage form of cyclovirobuxine D with improved bioavailability.
Keywords: self-nanoemulsifying drug delivery, bioavailability, cyclovirobuxine D
Introduction
Cyclovirobuxine D, (3β, 5α, 16α, 20S)-4, 4, 14-trimethyl-3, 20-bis (methylamino)-9, 19-cyclopregnan-16-ol, is a steroidal alkaloid obtained from traditional Chinese medicinal herbs, such as Buxus microphylla. It has been widely used as a blood-regulating agent for treatment of arrhythmias, angina pectoris, coronary heart disease, heart failure, and other cardiovascular disorders in the People’s Republic of China.1–3 It is available in the form of oral tablets (0.5 mg and 1.0 mg) for current clinical applications.
Cyclovirobuxine D is a small molecule with a poor water solubility of ~0.06 mg/mL, which limits its effective absorption after oral administration. In view of the fact that enhancing dissolution rate is the most common way to improve bioavailability, we utilized solid dispersion technology to enhance the in vitro dissolution rate of cyclovirobuxine D in preliminary studies. However, we observed that the disintegrating tablets did not have a considerably higher value of bioavailability of cyclovirobuxine D in in vivo research, which was in accordance with other studies.4,5 Consequently, improving dissolution may not be a completely feasible way for improvement of bioavailability. No increase in bioavailability may be linked to low permeability, high P-glycoprotein (P-gp) efflux, and high first-pass hepatic metabolism of the lipophilic drug.
Various techniques have been developed to improve the bioavailability of poorly water-soluble drugs, such as liposomes, nanoparticles, and self-nanoemulsifying drug delivery systems (SNEDDS). Because of its simple preparation and intestinal absorption-enhancing effects,6 SNEDDS has recently been paid much attention to improve the oral bioavailability of poorly soluble drugs. SNEDDS is an isotropic mixture of drug, oil, and surfactant/cosurfactant, which forms a fine oil-in-water nanoemulsion with a droplet size of <100 nm when introduced into an aqueous medium, such as gastrointestinal (GI) fluids, under mild agitation. It is reported that the nanoemulsion helps to increase the bioavailability of various drugs via one/combination of the following mechanisms: increase in effective drug solubility, protection against enzymatic hydrolysis, modifications in the biochemical barrier function of the GI tract through the use of surfactants, and inhibition of P-gp efflux of drug.7,8 Selection of a suitable formulation of SNEDDS depends upon the solubility of the drug in various media, emulsifying ability of surfactant/cosurfactant, area of the self-emulsifying region, and droplet size distribution of the resultant nanoemulsion.9,10
Extensive survey of literature and patent databases did not reveal any cyclovirobuxine-D-loaded SNEDDS designed for improvement of oral absorption. The current study discusses the optimization of SNEDDS for improved bioavailability of cyclovirobuxine D. Transmission electron microscopy (TEM) and laser particle size analyzer were used to determine the shape and size of the resultant nanoemulsion. In vivo and in vitro studies were carried out to ascertain the release and absorption characteristics of the drug from SNEDDS compared with that of commercially available tablets.
Materials and methods
Materials
Cyclovirobuxine D with 99% purity was purchased from Anhui New LongHai Pharmaceutical Co., Ltd (Suzhou, People’s Republic of China). Maisine 35-1, oleic acid, castor oil, ethyl oleate, and isopropyl myristate were purchased from East Sea Pharmaceutical Co., Ltd (Qingdao, People’s Republic of China). Labrafac CM10 and Solutol SH15 were obtained from Gattefosse (Saint-Priest Cedex, France). Tween-80, glycerin, ethanol, and propylene glycol were procured from Sinopharm Chemical Reagent Co., Ltd (Beijing, People’s Republic of China). Market tablets (Huangyangnin pian) were purchased from Xiaoying Pharmaceutical Co., Ltd (Nanjing, People’s Republic of China). All the other chemicals used were of analytical reagent grade. Double-distilled water was used throughout the whole study.
Optimization and preparation of cyclovirobuxine-D-loaded SNEDDS
Solubility studies
Various oils, surfactant, and cosurfactant were screened for their ability to dissolve cyclovirobuxine D. An excess quantity of cyclovirobuxine D powder (~1.0 g) was added to each excipient (Maisine 35-1, oleic acid, castor oil, ethyl oleate, isopropyl myristate, Labrafac CM10, Tween-80, Solutol SH15, glycerin, ethanol, propylene glycol), vortexed, and shaken in a water bath at 37°C for 48 hours to attain the equilibrium. The centrifugation was performed at 5,000 rpm for 20 minutes to separate the undissolved drug. The supernatant was diluted with methanol and passed through 0.22 μm membrane filter. The concentration of dissolved cyclovirobuxine D was determined in each of the excipients by validated high-performance liquid chromatography (HPLC)-evaporative light scattering detector (ELSD) method described in the following section.
HPLC–ELSD analysis
The HPLC analysis was established and carried out on an Agilent Alliance 1200 equipped with a vacuum degasser, a high pressure quaternary pump, an auto-sampler, and an Alltech 2000 ELSD. The separation of sample solution was performed on an Alltima C18 column (250×4.6 mm, 5 μm); methanol–water–acetonitrile (80:15:5) was used as the mobile phase with a flow rate of 1.0 mL/min; the column was maintained at 40°C; and 10 μL sample solution was injected in each run. The effluent was introduced into the Alltech 2000 ELSD in which the drift tube temperature was 85°C, and the gas flow rate was 1.8 L/min.11 The method was validated for linearity, repeatability, and recovery.
Screening of surfactants and cosurfactants for emulsifying ability
Surfactants and cosurfactants were further studied for their self-emulsifying properties. Optimal surfactant and cosurfactant were screened using a reported method with minor modification.12 For surfactant screening, in brief, 0.5 g of each surfactant (Labrafac CM10, Solutol SH15, and Tween-80) was mixed with 0.5 g oleic acid. The mixture was gently heated at 37°C to homogenize the components. Each mixture (0.2 g) was reconstituted with 20 mL deionized water in a volumetric flask. A clear or slight blue appearance was visually observed for nanoemulsion formation by noting the number of flask inversion. Briefly, for cosurfactant selecting, 0.25 g of each cosurfactant (glycerin, ethanol, propylene glycol) was mixed with 0.25 g Solutol SH15, and 0.5 g oleic acid was added, and the mixture was kept at 37°C to homogenize the components. The formed mixtures were used to select cosurfactant by the method introduced in surfactant screening. A minimum value of flask inversion indicates a best emulsifying ability.
Ternary phase diagram
To identify the region of nanoemulsion visually, ternary phase diagrams were constructed for the selected oil, surfactant/cosurfactant of different mass ratios (Km=2:1, 1:1, and 1:2), each representing an apex of triangles. For every phase diagram, oil and surfactant/cosurfactant at specific ratio were mixed uniformly in nine proportions (1:9, 2:8, 3:7, 4:6, 5:5, 6:4, 7:3, 8:2, 9:1). A small amount of water in 0.5% w/w increment was added into the mixture under agitation at 600 rpm and allowed to equilibrate. Titration was stopped and the water volume was recorded when a nanoemulsion appeared clear or slightly bluish. As the sum was 100%, the mass percents of oil, surfactant/cosurfactant, and water were noted at these end points.13 The size of nanoemulsion region in the phase diagrams was compared; the larger the size the better the self-nanoemulsifying property.
Effect of oil content on globule size
On the premise of the same amount of surfactant and cosurfactant, the globule of emulsion is directly influenced by the oil content in the system.14 A series of blank SNEDDS were prepared in accordance with formulations shown in Table 1 to determine the relationship between oil content and mean droplet diameter. Briefly, oil, surfactant, and cosurfactant were mixed to form homogeneous mixtures. Then, 0.1 g of each mixture was dispersed in 10 mL deionized water, and the globule size was determined immediately after dilution. On the basis of nanoemulsion droplet size of <100 nm, it was found that the higher the oil content, the larger the drug-loading (DL) capability.
Table 1.
Compositions of blank SNEDDS formulations varying in oil content
| Formulation | F1 | F2 | F3 | F4 | F5 | F6 |
|---|---|---|---|---|---|---|
| Solutol SH15 + propylene glycol (g) | 22 | 21 | 20 | 19 | 18 | 17 |
| Oleic acid (g) | 3 | 4 | 5 | 6 | 7 | 8 |
| Oil content (%) | 12 | 16 | 20 | 24 | 28 | 32 |
Abbreviation: SNEDDS, self-nanoemulsifying drug delivery system.
Effect of cyclovirobuxine D loading on globule size
Based on the result of blank SNEDDS studies, the relationship of DL and globule size of nanoemulsion was investigated. Briefly, a series of cyclovirobuxine-D-loaded SNEDDS were prepared with different mass concentrations of cyclovirobuxine D (1.0%, 2.0%, 3.0%, 4.0%, 5.0%, 6.0%). Then, 0.5 g of each drug-loaded SNEDDS was evenly dispersed in 50 mL deionized water to form nanoemulsion, and the globule size was measured immediately after dilution. Based on the suitable size of droplets, the DL was selected for the preparation of SNEDDS.
Preparation of optimized cyclovirobuxine-D-loaded SNEDDS
On the basis of the solubility, phase diagram, oil content, and DL studies, oleic acid, Solutol SH15, and propylene glycol were selected as oil, surfactant, and cosurfactant, respectively. The optimized SNEDDS was prepared as reported earlier.15 Briefly, weighed quantity of cyclovirobuxine D (0.15 g) was dissolved in a mixture (5 g) of 24% oleic acid, 38% Solutol SH15, and 38% propylene glycol. The resulting mixture was vortexed for 5 minutes to obtain a homogeneous colloidal dispersion. The prepared optimized SNEDDS was stored in sealed transparent glass bottles at 25°C until used.
Physical characterization of the prepared SNEDDS
Droplet size measurement and zeta potential study All the prepared SNEDDS were diluted (0.5 g to 50 mL) 100 times with deionized water under mild agitation.16 The droplet size and zeta potential of the nanoemulsion were determined by dynamic light scattering techniques using a particle sizer (Malvern Zetasizer 3000 HS; Malvern Instruments, Malvern, UK).
Measurement of percent transmittance
The percent transmittance of the nanoemulsion was measured after diluting each of them 100 times (0.5 g to 50 mL) with deionized water using an ultraviolet spectrophotometer at 450 nm, using deionized water as blank.17
Drug entrapment efficiency and DL
Entrapment efficiency (EE) and DL of cyclovirobuxine D nanoemulsion were estimated as per the procedure established by Bu et al. In brief, the content of free cyclovirobuxine D was separated from nanoemulsion by the ultrafiltration method (3,500 Da) with centrifugation at 3,000 g for 5 minutes. The drug amount in the filtrate and nanoemulsion was quantified by HPLC–ELSD. The EE and DL were calculated as follows:
| (1) |
| (2) |
where Wt is the total amount of cyclovirobuxine D in the nanoemulsion, Wf is the amount of free cyclovirobuxine D, and Wc is the total ingredients in the nanoemulsion.18
Cloud point measurement
Optimized SNEDDS was diluted 100 times with deionized water and placed into a water bath under gradual warming. The temperature at which a sudden appearance of cloudiness occurs was recorded as cloud point.19 After the temperature exceeded the cloud point, the sample was cooled and heated once again to check the reproducibility of the experiment.
Transmission electron microscopy
The morphology of the formed nanoemulsion (1:100 dilution) was determined using a TEM (JEM-1200EX; JEOL, Kanagawa, Japan). The nanoemulsion was negatively stained with 2% phosphotungstic acid, kept for 3 minutes at room temperature, and allowed to dry before observation under the electron microscope.
Stability studies
The optimized cyclovirobuxine-D-loaded SNEDDS was kept in airtight glass vials at the accelerated condition (60°C±2°C/75%±5% relative humidity) and was sampled periodically at the time intervals of 0 days, 5 days, and 10 days.20 The nanoemulsion originated from the 100-times dilution of sample SNEDDS was studied for any change in droplet size and drug content, and observed for appearance when subjected to six cycles of heating (40°C) and cooling (4°C) with 48-hour storage at each temperature.
In vitro dissolution and cellular permeability studies
In vitro drug release
In order to clarify the drug-release behavior in vitro, market tablets (Huangyangning pian; Nanjing Xiaoying Inc., Nanjing, People’s Republic of China) and SNEDDS (each containing 2 mg of drug) were studied by an USP 35/711 dissolution type I apparatus with 900 mL of simulated intestinal fluid (pH 6.8 phosphate buffer solution) as the dissolution medium, kept at 37°C±0.5°C and the paddle speed was set at 50 rpm.21 Equilibrated dialysis bags (3,500 Da) were used to obtain the amount of real free drug without interference of the unreleased drug from the SNEDDS. At predetermined time intervals (0 hour, 0.5 hour, 1 hour, 2 hours, 3 hours, 4 hours, 6 hours, 8 hours), an aliquot (1.0 mL) of the samples was collected and replaced with fresh dissolution medium. The collected samples were suitably diluted with mobile phase and subjected to drug analysis using HPLC–ELSD method. All measurements were performed in triplicate.
Cellular permeability and post-cytotoxicity tests in Caco-2 cell monolayer
Considering fast release in vitro does not mean fast absorption, the permeability of formed nanoemulsion was further evaluated in Caco-2 cell monolayer, which has been widely approved to simulate the human intestinal epithelial absorption. Caco-2 cells were seeded at a density of 1.0×105 cells/well on 12 mm Transwell polycarbonate membrane inserts with 0.4 mm pores and cultured for 21 days. Inserts with a higher transepithelial electrical resistance value than 300 Ω/cm2 were washed three times with Hank’s Balanced Salt Solution (pH 6.5) before the transcellular transport assessment. Transport buffer was put into both the apical (A, 0.5 mL) and basolateral sides (B, 1.5 mL). The cyclovirobuxine D solution was obtained by dissolving the drug of the market tablets with 0.1% dimethyl sulfoxide. The solution and SNEDDS were added into the apical and basolateral sides of the cell inserts, respectively, to reach a content of 20 μM. Then, 0.1 mL of the basolateral or apical side solutions were sampled at predetermined times (1 hour, 2 hours, 3 hours, 4 hours) and replaced with the equivalent volume of fresh transport media.22 Samples were filtrated and analyzed by the established liquid chromatography (LC)–mass spectrometry (MS) method. Apparent permeability coefficient (Papp) was calculated as follows:
| (3) |
where ΔQ is the transfer amount (nmol), A is the filter surface area (cm2), t is the time of incubation(s), and C0 is the initial concentration (μM).
After incubation, the Caco-2 cells were treated with 80 μL of 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium (MTS)-based CellTiter 96® AQueous One Solution Cell Proliferation Assay Reagent in 5% CO2 atmosphere condition at 37°C for 4 hours. The absorbance was detected at 490 nm wavelength by EMax precision microplate reader.23 The experimental protocols for the cell research were approved by the ethics committee affiliated to Nanjing University of Chinese Medicine.
In vivo absorption study
Administration and blood collection
All animal experiments complied with the requirements of the National Act on the Use of Experimental Animals (People’s Republic of China) and were approved by the ethics committee affiliated to Nanjing University of Chinese Medicine. Twelve New Zealand rabbits (1.0–1.5 kg) were randomly divided into two groups and were kept in animal cages for a week. The animals were fasted 24 hours prior to the experiment but had free access to water. The two groups were orally given SNEDDS or suspension at a dose of 10 mg/kg; cyclovirobuxine D suspension was prepared by dispersing market tablets powder in 5% sodium carboxymethyl cellulose solution. Serial 2.0 mL blood samples were collected from the marginal ear vein at specified intervals. The heparinized blood samples were immediately centrifuged at 40,000 rpm for 15 minutes, and the separated plasma was stored at −20°C until analysis.24
LC-MS analysis of plasma samples
On the basis of previous studies,25 an Agilent 1200 series HPLC system and a Finnigan LCQ Advantage ion-trap mass spectrometer (Thermo Fisher Scientific, Waltham, MA, USA) equipped with an electrospray ionization source operating in positive ion mode were used for the analysis of plasma samples. Separation was achieved by isocratic elution using an Alltima C18 column (250×4.6 mm, 5 μm), and the injection volume was 20 μL. The solvent used for separation was methanol/water (88/12, v/v) containing 0.4% formic acid. A flow rate of 1.0 mL/min and a temperature of 30°C were used. The mass spectrometer was operated in the positive ionization mode using the following settings: spray voltage, 5,000 V; sheath gas (N2) pressure, 35 psi; auxiliary gas (N2) pressure, 5 psi; and capillary temperature, 350°C. Under these conditions, ions detected were m/z 403.0 (parent)→380.0 (product) for cyclovirobuxine D and m/z 325.0 (parent)→234.0 (product) for citalopram (internal standard). The linear regressions of the peak area ratios vs concentrations for cyclovirobuxine D were fitted in the concentration range of 2–200 ng/mL.
Plasma sample extraction and analysis
Prior to extraction, frozen plasma samples were thawed at ambient temperature. Rabbit plasma (0.5 mL) was mixed with 0.2 mL of sodium hydroxide (3.0 mol/L). After adding 5.0 mL tert butyl ether–dichloromethane (1:1), the mixture was vortexed for 3 minutes. The samples were centrifuged at 3,000 rpm for 10 minutes, and the supernatant was collected and evaporated to dryness under a nitrogen stream at 40°C.24 The residue was reconstituted in 100 μL methanol, and 20 μL of the supernatant was injected into the LC-MS system using the same conditions as mentioned earlier.
Pharmacokinetic data analysis
After oral administration of SNEDDS and market tablets, the maximum cyclovirobuxine D plasma concentration (Cmax) and time to maximum plasma concentration (tmax) were determined from the individual plasma concentration vs time curves using Kinetica 4.4 software. Area under the curve (AUC) was calculated using the trapezoidal equation. The elimination rate constant (Ke) was calculated by linear regression of the terminal portion in the curve of natural logarithm of concentration with time and the elimination half-life (t1/2) was calculated. Finally, the relative bioavailability was calculated to illustrate that SNEDDS could be a potential candidate oral dosage form of cyclovirobuxine D.
Statistical analysis
All data were expressed as mean ± SD. Data were statistically analyzed by analysis of variance and Student’s t-test. A P-value of <0.05 was considered statistically significant.
Results and discussion
Formulation optimization
Solubility study
The formulation enhances solubility and fraction of lipophilic drugs transported through the intestinal lymphatic system, thus increasing absorption through the GI tract. Hence, it is necessary to ascertain the component showing maximum solubility. The solubility of cyclovirobuxine D was determined using the following equation:
| (4) |
where y represents the peak area and x represents the concentration (μg/mL). The method was found to be linear in the range of 26.4–132.4 μg/mL, and the interday and intraday relative SD values were calculated to be 0.84% and 1.76% respectively, indicating the precise method for the estimation of cyclovirobuxine D. The recovery value was found to be 99.78%, suggesting that the developed HPLC–ELSD method is accurate for the determination of cyclovirobuxine D.
The solubility of cyclovirobuxine D in various excipients is shown in Table 2. As oleic acid showed the highest solubility in each category, it was selected as the oil phase; Tween-80 and propylene glycol were preliminarily selected as surfactant and cosurfactant, respectively.
Table 2.
Solubility results of cyclovirobuxine D in various excipients (mean ± SD; n=3)
| Oil phase | Solubility (mg/g) | Surfactants and cosurfactants | Solubility (mg/g) |
|---|---|---|---|
| Maisine 35-1 | 65.64±4.09 | Labrafac CM10 | 3.15±0.48 |
| Oleic acid | 124.32±8.53 | Tween-80 | 7.86±0.57 |
| Castor oil | 73.38±4.04 | Solutol SH15 | 7.18±0.64 |
| Ethyl oleate | 40.16±3.38 | Glycerin | 5.87±0.45 |
| Isopropyl myristate | 64.65±2.52 | Ethanol | 3.84±0.29 |
| Propylene glycol | 9.57±1.38 |
Screening of surfactant and cosurfactant for emulsifying ability
Tween-80 is shown to possess the best solubilization capacity of the three surfactants; however, surfactant and cosurfactant were selected based on not only dissolution properties but also their emulsifying abilities.26 Good emulsifying property is favorable for the spontaneous formation of nanoemulsion; therefore, the selection of surfactant and cosurfactant was governed by their emulsification efficiency, which was consistent with the approach of others.6,27 Various surfactants were investigated, and the results are shown in Table 3. Obviously, Solutol SH15 showed the highest emulsifying ability associated with a maximum value of transmittance (96.8%) and a minimum value of flask inversion (9) while Tween-80 and Labrafac CM10 were difficult to disperse. The results for cosurfactant screening are presented in Table 4. In all the cosurfactants screened, propylene glycol was found to possess the best emulsifying ability. Consequently, Solutol SH15 and propylene glycol were selected as surfactant and cosurfactant, respectively, for further study.
Table 3.
Emulsification efficacy of various surfactants
| Oil | Surfactant | Number of flask inversions | Transmittance (%) | Visual observation |
|---|---|---|---|---|
| Oleic acid | Labrafac CM10 | 13 | 82.4 | Difficult to disperse |
| Oleic acid | Solutol SH15 | 9 | 96.8 | Spontaneous dispersion |
| Oleic acid | Tween-80 | 15 | 77.5 | Difficult to disperse |
Table 4.
Emulsification abilities of various cosurfactants
| Oil | Surfactant | Cosurfactant | Number of flask inversions | Transmittance (%) | Visual observation |
|---|---|---|---|---|---|
| Oleic acid | Solutol SH15 | Glycerin | 7 | 93.5 | Spontaneous dispersion |
| Oleic acid | Solutol SH15 | Ethanol | 7 | 92.1 | Spontaneous dispersion |
| Oleic acid | Solutol SH15 | Propylene glycol | 3 | 97.6 | Spontaneous dispersion |
Pseudo-ternary phase diagram study
Ternary phase diagrams were constructed to determine the nanoemulsion area and to quantify the probable ratio of surfactant/cosurfactant to develop a stable SNEDDS. The phase diagram consists of oil, surfactant/cosurfactant, and water in each corner with 100% of each component; the wider dark area indicates the greater nanoemulsifying ability (Figure 1).
Figure 1.

Pseudo-ternary phase diagram depicting the nanoemulsion region (NE).
Notes: (A) Km=1:2; (B) Km=1:1; (C) Km=2:1.
Abbreviations: S/Cos, surfactant/cosurfactant; Km, surfactant/cosurfactant of different mass ratios.
In the current study, oleic acid was tested for phase behavior studies with Solutol SH15 and propylene glycol as the surfactant/cosurfactant mixture of various ratios. According to different ratios, all the components were converted to mass percent (w%) before using Oringe 6.0 software to construct the phase diagrams, as shown in Figure 1. It was observed that nanoemulsion area was highest at Km=1, and either a higher or lower Km resulted in a smaller nanoemulsion area, suggesting that an appropriate ratio of surfactant/cosurfactant is important for the rapid formation of stable nanoemulsion due to their mutual coordination in improving the hydrophilicity of the oil and reducing the globule size.28
Effect of oil content on globule size
It is widely accepted that nanoemulsion with a high content of oil may not be necessarily stable and may result in increasing droplet size or phase separation.29 Hence, SNEDDS with diverse oil content were formulated to screen the optimum oil concentration that can yield nanoemulsion of a desired droplet size. The effect of oil concentration on the globule size is shown in Figure 2.
Figure 2.

Effect of oil content and cyclovirobuxine D loading on globule size.
Notes: (A) *P<0.05, **P<0.01 vs 10% oil; (B) *P<0.05, **P<0.01 vs 1% cyclovirobuxine D.
It is observed that average globule sizes of nanoemulsion were kept to less than 60 nm when the concentration of oleic acid was <24% (formulations F1, F2, F3, F4). However, when the oil content was increased up to 28%, formulations F5 and F6 exhibited an average globule size of >100 nm. Based on this study, formulation F3 that contained 24% oleic acid was selected for further study.
Effect of cyclovirobuxine D loading on globule size
The amount of DL can affect the state of nanoemulsion, and high DL could cause the increase in droplet size and drug precipitation.30 The effect of cyclovirobuxine D loading on globule size is shown in Figure 2. It was found that incorporation of cyclovirobuxine D did not have a great influence on the globule size of nanoemulsion when the concentration was relatively low (1%, 2%, 3%), and each nanoemulsion obtained from SNEDDS had a globule size of <70 nm. However, particle size was observed at more than 100 nm when cyclovirobuxine D loading was increased to 4% and above, so 3% cyclovirobuxine D loading (equal to 30 mg/g) was selected for SNEDDS formulation. In addition, saturation solubility of cyclovirobuxine D in optimized SNEDDS was found to be 36.57±2.29 mg/g, which indicates a significant enhancement in cyclovirobuxine D solubility (580-fold) in optimized blank SNEDDS compared with the solubility in water (0.063±0.004 mg/g). As cyclovirobuxine D loading of 30 mg/g in SNEDDS is equal to 83% saturation solubility, the result was consistent with the DL between 75% and 90% saturation solubility in some other research.31–33
Physical characterization
The optimized SNEDDS autonomously formed a fine oil-in-water nanoemulsion when introduced into an aqueous medium (1:100 dilution) under mild agitation.
Droplet size analysis and zeta potential analysis
The average globule size and polydispersity index value of nanoemulsion were found to be 64.80±3.58 nm and 0.36±0.07 nm, respectively. The low polydispersity index value indicates the narrow size distribution of globules. The zeta potential of the nanoemulsion was found to be −25.5 mV, and the absolute value of >20.0 indicates a good stability of nanoemulsion droplets with low possibility of coagulation due to sufficient repulsion,34 and the negative value of zeta potential may be originated from free fatty acids.35
Measurement of transmittance
It was observed that the formed nanoemulsion was clear and transparent, and the transmittance was determined to be 98.5%±0.03% (n=3), suggesting that particle size of nanoemulsion was fine and the solution was homogeneous.
Drug EE and DL
EE and DL are two important indicators for evaluating the DL capacity of nanopharmaceutics; the larger the value the better the DL capability. The EE of cyclovirobuxine D in nanoemulsion was 98.52%±0.46%, and the DL was 2.96%±0.08%.
Cloud point measurement
Cloud point is an important parameter for the evaluation of nanoemulsion stability. For a desired nanoemulsion, the cloud point should be >37°C. The optimized formulation showed a cloud point of 62°C±1°C, indicating the formed nanoemulsion remains stable in vivo.
Transmission electron microscopy
The microscopy of nanoemulsion was examined using TEM micrographs. Figure 3 reveals that nanodroplets were formed with sizes varying from 60 nm to 70 nm. These experimental data agreed with dynamic light scattering results confirming the feasibility of the optimized SNEDDS formulation. The nanodroplets appear as dark, and the surroundings were found to be bright.36
Figure 3.

TEM images of the nanoemulsion in the different scale of 0.2 μm (A) and 0.05 μm (B).
Abbreviations: SNEDDS, self-nanoemulsifying drug delivery system; TEM, transmission electron microscopy.
Stability studies
Because phase separation or drug precipitation can be observed in the case of unstable formulation, thermodynamic stability studies have a vital role in developing self-emulsifying formulation. Hence, the nanoemulsion obtained from optimized SNEDDS was examined by centrifugation and heating–cooling cycle; phase separation and drug precipitation were not observed in the sample solution during these studies. Subsequently, on the fifth day and the tenth day of the acceleration experiment, the average droplet size of formed nanoemulsion was 65.33±3.79 nm and 65.76±3.17 nm (n=3), respectively; the remaining percentages of cyclovirobuxine D were 99.78%±0.28% and 99.69%±0.36% (n=3), respectively, indicating good stability of the optimized SNEDDS loaded with cyclovirobuxine D.
In vitro dissolution and cellular permeability studies
In vitro dissolution studies
The dissolution profiles of cyclovirobuxine D from different formulations with the corresponding amount of drug are shown in Figure 4. The percentage drug release in 2 hours for the optimized SNEDDS and market tablets (Huangyangning pian; Nanjing Xiaoying Inc.) was found to be 50.24%±2.38% and 86.11%±3.34%, respectively. It has been proved that the oil/surfactant/cosurfactant was spontaneously dispersed to entrap cyclovirobuxine D to form nanoscale droplets, and finally resulted in a condition of sustained release.18,24 The sustained release of optimized SNEDDS was likely due to the stronger interaction between cyclovirobuxine D and the oleic acid/Solutol SH15/propylene glycol colloidal particles;37 the relative faster release of market tablet was perhaps induced by the solubilization of sodium lauryl sulfate contained in the tablets.
Figure 4.
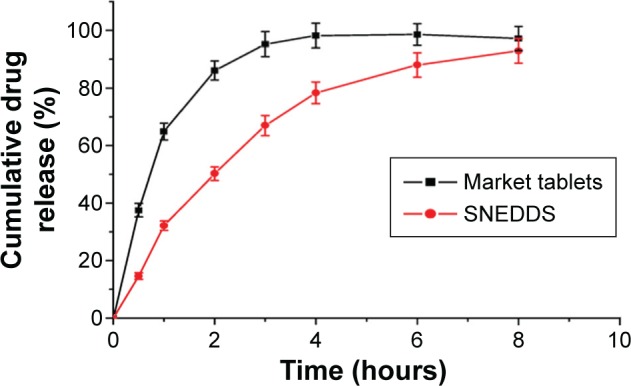
In vitro release profiles of SNEDDS and market tablets.
Abbreviation: SNEDDS, self-nanoemulsifying drug delivery system.
In vitro permeation study and post-cytotoxicity tests
To study the effect of optimized SNEDDS on the transport of cyclovirobuxine D across the intestinal membrane, in vitro permeation research was performed in Caco-2 cell monolayers. As shown in Figure 5A, the optimized SNEDDS exhibited 1.54 times higher drug permeation amount and 0.57 times lower drug excretion amount than that of market tablets at 4 hours (P<0.01). Papp (A–B) of the optimized SNEDDS and market tablets is 6.07×10−6 cm/s and 3.54×10−6 cm/s, respectively, indicating that SNEDDS has better intestinal permeability, which may be due to endocytosis of nanodroplets; Papp (B–A) of the optimized SNEDDS and market tablets is 3.30×10−6 cm/s and 6.87×10−6 cm/s, respectively, indicating that SNEDDS has reductive efflux, which may be due to P-gp inhibition of lipid nanoemulsion. Briefly, SNEDDS significantly improves bioavailability by promoting intestinal permeability and inhibiting the efflux of cyclovirobuxine D.
Figure 5.

Transport amount of cyclovirobuxine D in 4 hours (A) and Caco-2 cell viability after transport study (B) #P>0.05 vs market tablets.
Abbreviations: SNEDDS, self-nanoemulsifying drug delivery system; A–B, from apical side to basolateral side; B–A, from basolateral side to apical side.
After the permeation study, MTS-based assay was performed to evaluate the cytotoxic effects on the Caco-2. To better understand the cytotoxicity of the prepared SNEDDS, the relationship between the Caco-2 cell viability and different formulations is plotted in Figure 5B. It was found that the cell viability was lower in SNEDDS (91.32%±1.57%) compared with the market tablets (92.57%±1.26%; P>0.05). It can be inferred that SNEDDS formulation has no significant negative effect.
In this study, Solutol SH15 was used as a surfactant and has been reported for its permeation-enhancing effect.38 Propylene glycol is also known for its permeation-enhancing effect.39 Moreover, it is assumed that the characteristics of nanoemulsion, such as small droplet size and the interaction between the surfactants and cellular membrane, may result in the permeation enhancement of insoluble drug. Decreasing drug efflux should be related to inhibition of the P-gp pump function.40 These findings indicate an increase in transport of cyclovirobuxine D via nontoxic nanoemulsion with a simultaneous decrease in the efflux, which is of immense importance for the clinical application.
In vivo study
Pharmacokinetic parameters of cyclovirobuxine D calculated after single oral dose of optimized SNEDDS and market tablets in rabbits are listed in Table 5. The mean plasma concentration time curves for the study are shown in Figure 6. The Cmax and AUC0−t of cyclovirobuxine D obtained after oral administration of SNEDDS (224.72±9.88 ng/mL and 2,820.52±127.06 ng⋅h/mL, respectively) were significantly higher than those obtained from the market tablets (Huangyangnin pian) in rabbits (108.42±3.79 ng/mL and 1,408.72±49.84 ng⋅h/mL, respectively), which showed that SNEDDS afford us a higher bioavailability of cyclovirobuxine D. The elimination half-life (t1/2) showed no significant differences between the SNEDDS and market tablets in rabbits (P>0.05). The time to reach maximum plasma concentration (Tmax) and the absorption rate (ka) of cyclovirobuxine D obtained from optimized SNEDDS (2.87±0.13 h, 0.459±0.02 h−1) were obviously different from those of the market tablets (3.56±0.12 h, 0.373±0.02 h−1), indicating that SNEDDSs provide a significantly faster rate of absorption (P<0.05), in accordance with the result of in vitro permeation study and other active components studied.41 The relative bioavailability of optimized SNEDDS was 200.22% in comparison with market tablets in rabbits.
Table 5.
Pharmacokinetic parameters of cyclovirobuxine D following oral administration to rabbits (mean ± SD, n=6)
| Parameters | Market tablets | SNEDDS |
|---|---|---|
| AUC (ng⋅h/mL) | 1,408.72±49.84 | 2,820.52±127.06a |
| Tmax (h) | 3.56±0.12 | 2.87±0.13a |
| Cmax (ng/mL) | 108.42±3.79 | 224.72±9.88a |
| ka (h−1) | 0.373±0.02 | 0.459±0.02a |
| ke (h−1) | 0.055±0.002 | 0.058±0.002 |
| t1/2 (h) | 12.65±0.56 | 11.94±0.33 |
Note:
Significant differences with parameters obtained from market tablets.
Abbreviations: AUC, area under the curve; Tmax, time to reach maximum plasma concentration; Cmax, maximum plasma concentration; ke, elimination rate; ka, absorption rate; t1/2, elimination half-life; SNEDDS, self-nanoemulsifying drug delivery system; SD, standard deviation.
Figure 6.
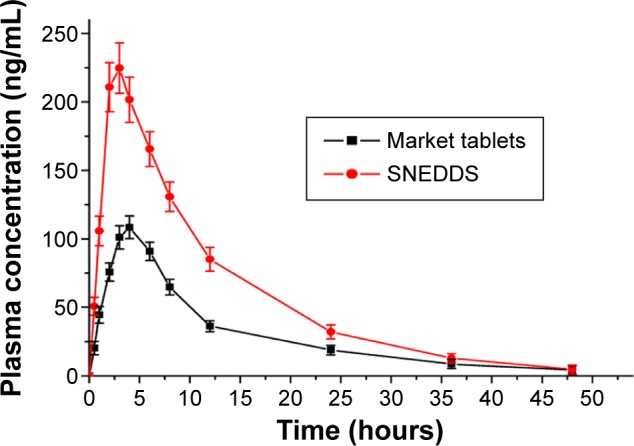
Plasma concentrations vs time profile after oral administration of SNEDDS and market tablets (mean ± SD, n=6).
Abbreviations: SNEDDS, self-nanoemulsifying drug delivery system; SD, standard deviation.
However, the in vivo absorption property was not well associated with in vitro release behavior, and SNEDDS slowed down cyclovirobuxine D release in vitro, but it promoted the absorption into blood. The main mechanism might be as follows: the long-chain unsaturated fatty acid of oleic acid supports the whole endocytosis and lymphatic absorption of drug molecules entrapped in the nanodroplets.35
Conclusion
In this research, an SNEDDS was optimized for cyclovirobuxine D containing oleic acid (oil), Solutol SH15 (surfactant), and propylene glycol (cosurfactant). Compared with market tablets, the developed SNEDDS exhibited significant improvement of drug solubility, rapid dissolution rate together with the enhanced permeation, and decreased efflux, and it is fortunately observed to have no significant cytotoxicity to the Caco-2 cell. More than 200% of the relative bioavailability was obtained, and the positive effect on the promotion of intestinal absorption was confirmed. However, there is no evidence to support the endocytosis of drug molecules entrapped in the nanodroplets, and the influence on the first-pass hepatic metabolism was not explicit. Therefore, further research needs to be carried out for the absorption mechanism, as well as the relationship between SNEDDS formulation and first-pass hepatic metabolism.
Acknowledgments
The authors gratefully acknowledge the financial support for this study by the National Natural Science Fund (No 81573620, 81274088, People’s Republic of China) and Anhui Universities Provincial Natural Science Key Research Project (No KJ2014A242, People’s Republic of China).
Footnotes
Disclosure
The authors report no conflicts of interest in this work.
References
- 1.Hu D, Liu XY, Wang YY, et al. Cyclovirobuxine D ameliorates acute myocardial ischemia by KATP channel opening, nitric oxide release and anti-thrombosis. Eur J Pharmacol. 2007;569(1–2):103–109. doi: 10.1016/j.ejphar.2007.04.038. [DOI] [PubMed] [Google Scholar]
- 2.Yu B, Fang TH, Lü GH, Xu HQ, Lu JF. Beneficial effect of Cyclovirobuxine D on heart failure rats following myocardial infarction. Fitoterapia. 2011;82(6):868–877. doi: 10.1016/j.fitote.2011.04.016. [DOI] [PubMed] [Google Scholar]
- 3.Chinese Pharmacopoeia Commission . Pharmacopoeia of People’s Republic of China, Part I. 2015. Beijing: Chemical Industry Press; 2015. [Google Scholar]
- 4.Hao GT, Zheng ZJ, Jiang B, et al. Bioavailability and bioequivalence of cyclovirobuxine-D dispersible tablets in humans. Chun Shih I Hsueh Ko Hsueh Yuan Yuan Kan. 2008;32(2):141–143. [Google Scholar]
- 5.Yu P, Di B, Liu WY, et al. Pharmacokinetics and relative bioavailability of cyclovirobuxine D orally disintegrating tablets in healthy volunteers. Chin J Clin Pharmacol Ther. 2007;12(7):824–827. [Google Scholar]
- 6.Basalious EB, Shawky N, Badr-Eldin SM. SNEDDS containing bioenhancers for improvement of dissolution and oral absorption of lacidipine I: development and optimization. Int J Pharm. 2010;391(1–2):203–211. doi: 10.1016/j.ijpharm.2010.03.008. [DOI] [PubMed] [Google Scholar]
- 7.Date AA, Desai N, Dixit R, et al. Self nanoemulsifying drug delivery systems: formulation insights, applications and advances. Nanomedicine. 2010;5(10):1595–1616. doi: 10.2217/nnm.10.126. [DOI] [PubMed] [Google Scholar]
- 8.Lind ML, Jacobsen J, Holm R, Müllertz A. Intestinal lymphatic transport of halofantrine in rats assessed using a chylomicron flow blocking approach: the influence of polysorbate 60 and 80. Eur J Pharm Sci. 2008;35(3):211–218. doi: 10.1016/j.ejps.2008.07.003. [DOI] [PubMed] [Google Scholar]
- 9.Seo YG, Kim DH, Ramasamy T, et al. Development of docetaxel-loaded solid self-nanoemulsifying drug delivery system (SNEDDS) for enhanced chemotherapeutic effect. Int J Pharm. 2013;452(1–2):412–420. doi: 10.1016/j.ijpharm.2013.05.034. [DOI] [PubMed] [Google Scholar]
- 10.Singh B, Singh R, Bandyopadhyay S, Kapil R, Garg B. Optimized nanoemulsifying systems with enhanced bioavailability of carvedilol. Colloid Surface B. 2013;101:465–474. doi: 10.1016/j.colsurfb.2012.07.017. [DOI] [PubMed] [Google Scholar]
- 11.Ke ZC, Zhou YQ, Fang C. Improvement on quality standard of Cyclovirobuxine D tablets. Chin Tradit Pat Med. 2007;29(5):Sul3–Sul4. [Google Scholar]
- 12.Date AA, Nagarsenker MS. Design and evaluation of self-nanoemulsifying drug delivery systems (SNEDDS) for cefpodoxime proxetil. Int J Pharm. 2007;329(1–2):166–172. doi: 10.1016/j.ijpharm.2006.08.038. [DOI] [PubMed] [Google Scholar]
- 13.Zhu L, Li M, Dong J, Jin Y. Dimethyl silicone dry nanoemulsion inhalations: formulation study and anti-acute lung injury effect. Int J Pharm. 2015;491(1–2):292–298. doi: 10.1016/j.ijpharm.2015.06.041. [DOI] [PubMed] [Google Scholar]
- 14.Hoscheida J, Outukia PM, Kleinubinga SA, et al. Development and characterization of Pterodon pubescens oil nanoemulsions as a possible delivery system for the treatment of rheumatoid arthritis. Colloid Surf A. 2015;484:19–27. [Google Scholar]
- 15.Quan Q, Kim DW, Marasini N, et al. Physicochemical characterization and in vivo evaluation of solid self-nanoemulsifying drug delivery system for oral administration of docetaxel. J Microencapsul. 2013;30(4):307–314. doi: 10.3109/02652048.2012.726280. [DOI] [PubMed] [Google Scholar]
- 16.Zhang JJ, Peng Q, Shi SJ, et al. Preparation, characterization, and in vivo evaluation of a self-nanoemulsifying drug delivery system (SNEDDS) loaded with morin-phospholipid complex. Int J Nanomed. 2011;6:3405–3414. doi: 10.2147/IJN.S25824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Harwansh RK, Mukherjee PK, Bahadur S, Biswas R. Enhanced permeability of ferulic acid loaded nanoemulsion based gel through skin against UVA mediated oxidative stress. Life Sci. 2015;141:202–211. doi: 10.1016/j.lfs.2015.10.001. [DOI] [PubMed] [Google Scholar]
- 18.Bu H, He X, Zhang Z, Yin Q, Yu H, Li Y. A TPGS-incorporating nanoemulsion of paclitaxel circumvents drug resistance in breast cancer. Int J Pharm. 2014;471(1–2):206–213. doi: 10.1016/j.ijpharm.2014.05.039. [DOI] [PubMed] [Google Scholar]
- 19.Bandyopadhyay S, Katare OP, Singh B. Optimized self nano-emulsifying systems of ezetimibe with enhanced bioavailability potential using long chain and medium chain triglycerides. Colloid Surf B. 2012;100:50–61. doi: 10.1016/j.colsurfb.2012.05.019. [DOI] [PubMed] [Google Scholar]
- 20.Zhang Y, Gao J, Zheng H, Zhang R, Han Y. The preparation of 3, 5-dihydroxy-4-isopropylstilbene nanoemulsion and in vitro release. Int J Nanomed. 2011;6:649–657. doi: 10.2147/IJN.S17242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fofaria NM, Qhattal HSS, Liu XL, et al. Nanoemulsion formulations for anti-cancer agent piplartine – characterization, toxicological, pharmacokinetics and efficacy studies. Int J Pharm. 2016;498(1–2):12–22. doi: 10.1016/j.ijpharm.2015.11.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Welling SH, Clemmensen LK, Buckley ST, Hovgaard L, Brockhoff PB, Refsgaard HH. In silico modelling of permeation enhancement potency in Caco-2 monolayers based on molecular descriptors and random forest. Eur J Pharm Biopharm. 2015;94:152–159. doi: 10.1016/j.ejpb.2015.05.012. [DOI] [PubMed] [Google Scholar]
- 23.Doh HJ, Jung Y, Balakrishnan P, Cho HJ, Kim DD. A novel lipid nanoemulsion system for improved permeation of granisetron. Colloid Surface B. 2013;101:475–480. doi: 10.1016/j.colsurfb.2012.07.019. [DOI] [PubMed] [Google Scholar]
- 24.Thomas N, Holm R, Mullertz A, Rades T. In vitro and in vivo performance of novel supersaturated self-nanoemulsifying drug delivery systems (super-SNEDDS) J Control Release. 2012;160(1):25–32. doi: 10.1016/j.jconrel.2012.02.027. [DOI] [PubMed] [Google Scholar]
- 25.Morais JM, Burgess DJ. In vitro release testing methods for vitamin E nanoemulsions. Int J Pharm. 2014;475(1–2):393–400. doi: 10.1016/j.ijpharm.2014.08.063. [DOI] [PubMed] [Google Scholar]
- 26.Balakrishnan P, Lee BJ, Oh DH, et al. Enhanced oral bioavailability of dexibuprofen by a novel solid self-emulsifying drug delivery system (SEDDS) Eur J Pharm Biopharm. 2009;72(3):539–545. doi: 10.1016/j.ejpb.2009.03.001. [DOI] [PubMed] [Google Scholar]
- 27.Tran TH, Guo Y, Song D, Bruno RS, Lu X. Quercetin-containing self-nanoemulsifying drug delivery system for improving oral bioavailability. J Pharm Sci. 2014;103(3):840–852. doi: 10.1002/jps.23858. [DOI] [PubMed] [Google Scholar]
- 28.Bazylinska U, Kulbacka J, Wilk KA. Dicephalic ionic surfactants in fabrication of biocompatible nanoemulsions: factors influencing droplet size and stability. Colloid Surf A. 2014;460:312–320. [Google Scholar]
- 29.Mostafa DM, El-Alim SHA, Asfour MH, et al. Transdermal nanoemulsions of Foeniculum vulgare mill. essential oil: preparation, characterization and evaluation of antidiabetic potential. J Drug Deliv Sci Technol. 2015;29:99–106. [Google Scholar]
- 30.Tayel SA, El-Nabarawi MA, Tadros MI, Abd-Elsalam WH. Promising ion-sensitive in situ ocular nanoemulsion gels of terbinafine hydrochloride: design, in vitro characterization and in vivo estimation of the ocular irritation and drug pharmacokinetics in the aqueous humor of rabbits. Int J Pharm. 2013;433(1–2):293–305. doi: 10.1016/j.ijpharm.2012.12.049. [DOI] [PubMed] [Google Scholar]
- 31.Pund S, Shete Y, Jagadale S. Multivariate analysis of physicochemical characteristics of lipid based nanoemulsifying cilostazol-quality by design. Colloid Surf B. 2014;115:29–36. doi: 10.1016/j.colsurfb.2013.11.019. [DOI] [PubMed] [Google Scholar]
- 32.Shakeel F, Haq N, Badry M, et al. Ultra fine super self-nanoemulsifying drug delivery system (SNEDDS) enhanced solubility and dissolution of indomethacin. J Mol Liq. 2013;180:89–94. [Google Scholar]
- 33.Dash RN, Mohammed H, Humaira T, Ramesh D. Design, optimization and evaluation of glipizide solid self-nanoemulsifying drug delivery for enhanced solubility and dissolution. Saudi Pharm J. 2015;23(5):528–540. doi: 10.1016/j.jsps.2015.01.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.El-Laithy HM, Basalious EB, El-Hoseiny BM, Adel MM. Novel self-nanoemulsifying self-nanosuspension (SNESNS) for enhancing oral bioavailability of diacerein: simultaneous portal blood absorption and lymphatic delivery. Int J Pharm. 2015;490(1–2):146–154. doi: 10.1016/j.ijpharm.2015.05.039. [DOI] [PubMed] [Google Scholar]
- 35.McClements DJ. Food Emulsions: Principles, Practice, and Techniques. 2nd ed. Boca Raton, FL: CRC Press; 2005. [Google Scholar]
- 36.Bello V, Mattei G, Mazzoldi P, et al. Transmission electron microscopy of lipid vesicles for drug delivery: comparison between positive and negative staining. Microsc Microanal. 2010;16(4):456–461. doi: 10.1017/S1431927610093645. [DOI] [PubMed] [Google Scholar]
- 37.Liu H, Xu H, Jiang Y, et al. Preparation, characterization, in vivo pharmacokinetics, and biodistribution of polymeric micellar dimethoxycurcumin for tumor targeting. Int J Nanomedicine. 2015;10(1):6395–6410. doi: 10.2147/IJN.S91961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Edmund AR, Kambalapally S, Wilson TA, Nicolosi RJ. Encapsulation of cadmium selenide quantum dots using a self-assembling nanoemulsion (SANE) reduces their in vitro toxicity. Toxicol In Vitro. 2011;25(1):185–190. doi: 10.1016/j.tiv.2010.10.017. [DOI] [PubMed] [Google Scholar]
- 39.Saberi AH, Fang Y, McClements DJ. Fabrication of vitamin E-enriched nanoemulsions by spontaneous emulsification: effect of propylene glycol and ethanol on formation, stability, and properties. Food Res Int. 2013;54(1):812–820. [Google Scholar]
- 40.Choudhury H, Gorain B, Karmakar S, et al. Improvement of cellular uptake, in vitro antitumor activity and sustained release profile with increased bioavailability from a nanoemulsion platform. Int J Pharm. 2014;460(1–2):131–143. doi: 10.1016/j.ijpharm.2013.10.055. [DOI] [PubMed] [Google Scholar]
- 41.Villar AM, Naveros BC, Campmany AC, Trenchs MA, Rocabert CB, Bellowa LH. Design and optimization of self-nanoemulsifying drug delivery systems (SNEDDS) for enhanced dissolution of gemfibrozil. Int J Pharm. 2012;431(1–2):167–175. doi: 10.1016/j.ijpharm.2012.04.001. [DOI] [PubMed] [Google Scholar]