

Summary
Endoplasmic reticulum (ER) stress has been reported to be involved in many cardiovascular diseases such as atherosclerosis, diabetes, myocardial ischemia, and hypertension that ultimately result in heart failure. XBP1 is a key ER stress signal transducer and an important pro‐survival factor of the unfolded protein response (UPR) in mammalian cells. The aim of this study was to establish a role for XBP1 in the deregulation of pro‐angiogenic factor VEGF expression and potential regulatory mechanisms in hypertrophic and failing heart. Western blots showed that myocardial XBP1s protein was significantly increased in both isoproterenol (ISO)‐induced and pressure‐overload‐induced hypertrophic and failing heart compared to normal control. Furthermore, XBP1 silencing exacerbates ISO‐induced cardiac dysfunction along with a reduction of myocardial capillary density and cardiac expression of pro‐angiogenic factor VEGF‐A in vivo. Consistently, experiments in cultured cardiomyocytes H9c2 (2‐1) cells showed that UPR‐induced VEGF‐A upregulation was determined by XBP1 expression level. Importantly, VEGF‐A expression was increased in failing human heart tissue and blood samples and was correlated with the levels of XBP1. These results suggest that XBP1 regulates VEGF‐mediated cardiac angiogenesis, which contributes to the progression of adaptive hypertrophy, and might provide novel targets for prevention and treatment of heart failure.
Keywords: angiogenesis, cardiac hypertrophy, unfolded protein response, vascular endothelial growth factor‐A, XBP1
Abbreviations
- ATF
activating transcription factor
- ER
endoplasmic reticulum
- Grp
glucose‐regulated protein
- IRE
inositol‐requiring kinase
- ISO
isoproterenol
- TAC
thoracic aorta constriction
- TG
thapsigargin
- TM
tunicamycin
- UPR
unfolded protein response
- VEGF‐A
vascular endothelial growth factor‐A
- XBP
X‐box binding protein
Introduction
Heart failure is the most common reason for hospitalization in the aging population and is a leading cause of mortality worldwide (Shah & Mann, 2011). Heart failure is the final common pathway of various cardiovascular diseases, including hypertension, myocardial ischemia or infarction, valvular heart disease, and inherited or acquired cardiomyopathies (Heineke & Molkentin, 2006). Cardiac hypertrophy occurs to enhance cardiac function as an adaptive response to increased workload, but prolonged cardiac hypertrophy causes heart failure (Heineke & Molkentin, 2006). During the development of cardiac hypertrophy, it has been postulated that cardiac angiogenesis, which is induced in the early adaptive phase, may be sufficient to reduce the disparity between the number of capillaries and the size of cardiomyocytes that develop to maintain cardiac function (Tomanek, 1990). In the maladaptive phase, the expression of angiogenic factors such as vascular endothelial growth factor‐A (VEGF‐A) is reduced (Giordano et al., 2001; Sano et al., 2007). In parallel, the number of CD31‐positive microvessels is reduced from compensatory hypertrophy with intact systolic function to symptomatic heart failure with depressed systolic function in patients with isolated valvular aortic stenosis and differing degrees of left ventricular systolic dysfunction (Hein et al., 2003). Thus, insufficient angiogenesis is considered as the key step for the transition of hypertrophic heart to heart failure (Shiojima et al., 2005). However, it remains poorly characterized that in hypertrophic hearts, the factors instigate angiogenesis and how angiogenesis becomes insufficient to maintain cardiac function in failing hearts.
The endoplasmic reticulum (ER) is a crucial organelle with essential roles in multiple cellular processes including intracellular calcium homeostasis, protein secretion, and lipid biosynthesis. When subjected to multiple stimulus and inadequate extracellular conditions including hypoxia, oxidative injury, cytotoxic drugs, etc., the resultant accumulation of unfolded proteins triggers an evolutionarily conserved series of signal transduction events termed the unfolded protein response (UPR) (Groenendyk et al., 2010). The UPR initiates three molecular compensatory mechanisms to promote cell survival and restore homeostasis to the ER (Okada et al., 2004). One of the major signaling pathways in the UPR is the IRE1α‐XBP1 pathway. Phosphorylation of IRE1α activates the endoribonuclease activity to splice XBP1 mRNA, induced by ATF6α, generating spliced XBP1 (XBP1s) mRNA encoding a potent transcription factor (Yoshida et al., 2001). XBP1s, is a key signal transducer in the ER stress response, also is a member of the CREB/ATF basic region‐leucine zipper family of transcription factors, which regulate genes involved in protein folding, glycosylation, ER‐associated degradation (ERAD), autophagy, lipid biogenesis, insulin secretion (Koong et al., 2006; Glimcher, 2010). XBP 1 is essential for survival under hypoxic conditions and is required for tumor growth (Romero‐Ramirez et al., 2004; Thuerauf et al., 2006). Specifically, accumulating evidence suggests that XBP1s exerts potential regulator effects in several cardiovascular diseases, including such atherosclerosis (Zeng et al., 2009), diabetes (Park et al., 2010; Lee et al., 2011; Zhou et al., 2011), myocardial ischemia (Wang et al., 2014) that ultimately results in heart failure. But so far, the expression trend and pathophysiological functions of XBP1 in cardiac hypertrophy and heart failure are still unknown. Recently, XBP1, as a key UPR‐inducible transcription factor (Glimcher, 2010), binds to the VEGF‐A promoter in response to ER stress in cancer cells (Roybal et al., 2005; Ghosh et al., 2010; Pereira et al., 2010). Furthermore, transient activation of XBP1 splicing may increase endothelial cell (EC) proliferation (Zeng et al., 2009), and endothelial cell‐specific knockout of XBP1 impaired the angiogenesis triggered by ischemia in mice (Zeng et al., 2013). Nevertheless, whether or not UPR such as XBP1 contributes to cardiac angiogenesis in hypertrophic hearts and heart failure remains to be determined.
Here, we hypothesized that ER stress transducer XBP1 may contribute to cardiac VEGF expression and then promote angiogenesis during the progression of cardiac hypertrophy. To test this hypothesis, we examined cardiac XBP1 expression and found that XBP1 is activated in animal hypertrophic heart model. Furthermore, our results have indicated that XBP1 activation is required for VEGF expression in vitro and consequent angiogenesis in the early stages of hypertrophic hearts in vivo, resulting in the progression of cardiac hypertrophy.
Results
Increased expression of XBP1s and ER stress in isoproterenol‐induced hypertrophic and failing heart
The mice hypertrophy model was established using isoproterenol (ISO) infusion. We first characterized this model using morphological, hemodynamic, and echocardiographic analysis. As depicted in Fig. 1A–B, heart weight/body weight (HW/BW) and cross‐sectional area (CSA) of cardiomyocytes significantly increased after ISO infusion. Consistently, hemodynamic analysis confirmed a reduction of dP/dtmax (mmHg/s) and dP/dtmin (mmHg/s) as early as 1 week after ISO infusion (Fig. 1C). Further, echocardiographic analysis showed that LV posterior wall thickness (LVPW) was increased after ISO infusion (Fig. 1D). Consistently, myocardial atrial natriuretic peptide (ANP) mRNA, a marker associated with cardiac hypertrophy and heart failure, was also significantly increased in mouse hearts after ISO infusion (Fig. 1E). Taken together, these results indicate that cardiac hypertrophy gradually developed from 1 to 2 weeks after ISO infusion and cardiac function declined thereafter. Then, we further determined whether ISO infusion caused ER stress in hypertrophic hearts in vivo. As depicted in Fig. 1F, protein levels of the ER chaperone GRP78 and UPR transcription factor XBP1 were increased as early as 1 week and continue to rise at 2 weeks after ISO infusion (Fig. 1F), suggesting that ISO infusion caused aberrant ER stress in the early phase of cardiac hypertrophy.
Figure 1.
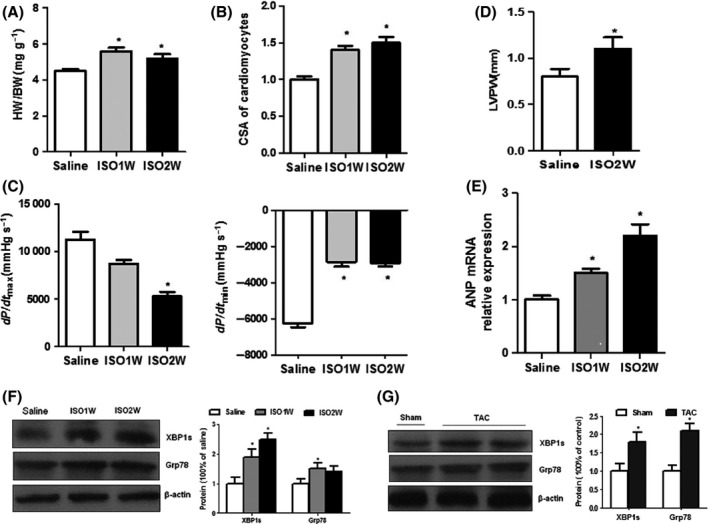
Cardiac XBP1 expression is increased in ISO infusion‐induced hypertrophic and failing heart. (A) Heart weight/body weight (HW/BW, grams) after ISO infusion. (B) Cross‐sectional area (CSA) of cardiomyocytes after ISO infusion. (C) Hemodynamic analysis of mice at 1 and 2 weeks of ISO infusion: (left) dP/dtmax (mmHg/s) and (right) dP/dtmin (mmHg/s). (D) Echocardiographic analysis of mice at 2 weeks of ISO infusion (LVPW, LV posterior wall thickness). (E) ANP mRNA expression in ISO‐treated mice. (F) Western blots of Grp78 and XBP1s in mouse hearts at 1 and 2 weeks of ISO infusion. (G) Western blot of Grp78 and XBP1s in mouse hearts after TAC treatment. Error bars indicate SEM. n = 6 for A–E. The blot is representative of at least four blots from four independent experiments; *P < 0.05 compared with control.
Thoracic aorta constriction (TAC) is another well established model to study hypertrophic and failing hearts. We next assayed the levels of ER stress, XBP1s in TAC‐induced hypertrophy in mice. As shown in Fig. 1G, cardiac expression of Grp78 and XBP1s were increased in the mice model at 2 week after TAC treatment (Fig. 1G). The levels of XBP1s in hearts were increased, as seen in ISO‐infused hypertrophic hearts. Taken together, our results suggest that XBP1 may play an important role in cardiac hypertrophy and heart failure.
AAV9‐mediated XBP1 gene silencing exacerbates ISO‐induced cardiac dysfunction
Because XBP1 was markedly induced in hypertrophic and failing heart, we investigated the significance of XBP1 induction and its pathophysiological role in the development of heart failure. To address this issue, we silenced XBP1 by tail vein injection of the myocardium‐tropic adeno‐associated virus serotype 9 (AAV9) vector‐carrying XBP1 shRNA. Two weeks after tail intravenous injection of AAV9, the mice were subjected to ISO infusion to evaluate cardiac hypertrophy and cardiac functions. As expected, injection of XBP1 siRNA virus inhibited XBP1 expression in vivo (Fig. 2A). However, silencing of XBP1 did not alter basal cardiac size and function (Fig. 2B–G).
Figure 2.
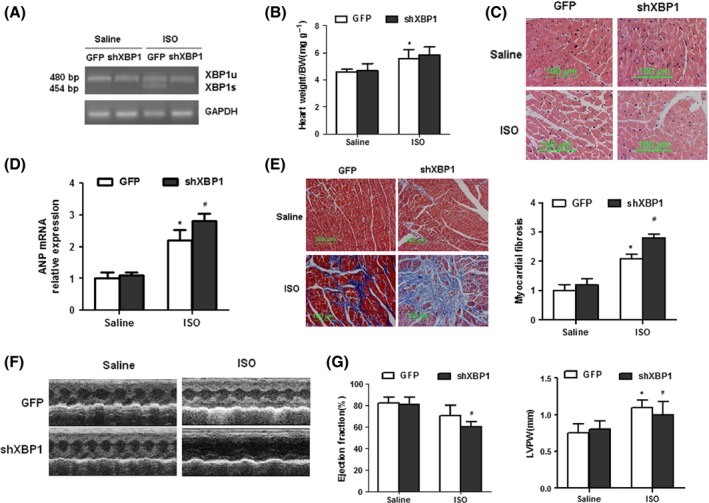
AAV9‐mediated‐inhibition expression of XBP1 exacerbates cardiac hypertrophy induced by ISO infusion. (A) Semiquantitative RT–PCR analysis of cardiac XBP1 mRNA expression in ISO‐infused mouse hearts after AAV‐shRNA XBP1 treatment. (B) Heart weight/body weight (HW/BW) in ISO‐treated mice after AAV‐XBP1‐shRNA treatment. (C) HE staining in ISO‐treated hearts after AAV‐shRNA XBP1 injection. (D) ANP mRNA expression in ISO‐treated mice after AAV‐XBP1‐shRNA treatment. (E) Representative sections for interstitial fibrosis (Meson‐stained). (F and G) Echocardiographic analysis of ISO‐treated mice after AAV‐shRNA XBP1 injection (LVPW, LV posterior wall thickness). Error bars indicate SEM. *P < 0.05 compared with GFP or #P < 0.05 compared with ISO+GFP. n = 8 for A–E.
It was important to determine if XBP1 silencing altered the morphological and echocardiographic analysis in the ISO‐infused mice in vivo. Treatment with XBP1 silencing had no significant difference on heart size and myocyte size (Fig. 2B–C); however, XBP1‐silenced mice exacerbated cardiac hypertrophy after ISO infusion as evidenced by ANP mRNA expression and interstitial fibrosis (Fig. 2D–E). Meanwhile, ISO infusion also impaired both contractile functions and ventricular dilatation in control group mice. XBP1‐silenced animals developed significantly greater contractile impairment than the control group, despite reduced cardiac wall thickness (Fig. 2F–G). Taken together, these results indicate that an increase in myocardial XBP1 expression is protective against the detrimental consequences of ISO infusion.
Myocardial capillary density and cardiac expression of pro‐angiogenic factor VEGF‐A is decreased in ISO‐treated mice after AAV9‐XBP1 shRNA treatment
Angiogenesis is essential in maintenance of cardiac function in hypertrophic hearts (Sano et al., 2007). Immunohistochemical analysis showed that the numbers of CD31‐positive cells were significantly lowered in XBP1‐silenced hearts (Fig. 3A), indicating that myocardial capillary density was impaired after XBP1 inhibition. Previous studies show that VEGF is an essential angiogenic factor to promote cardiac angiogenesis and maintain cardiac functions in hypertrophic hearts (Tomanek, 1990; Giordano et al., 2001; Sano et al., 2007). To address the potential role of XBP1 in VEGF‐A expression, the levels of VEGF‐A in ISO‐infused mice with or without AAV9‐XBP1 shRNA treatment were determined. Concomitantly with dramatic expression of XBP1, protein and mRNA expression of VEGF‐A was markedly increased after ISO infusion, but was ablated in XBP1‐silenced mice (Fig. 3B–C). Furthermore, secreted VEGF‐A in serum have same trend after XBP1 silencing treatment (Fig. 3D). These results suggest that XBP1 induction during stress is required to protect against ISO‐induced cardiac dysfunction by controlling the compensatory increase in cardiac VEGF‐A expression and angiogenesis.
Figure 3.
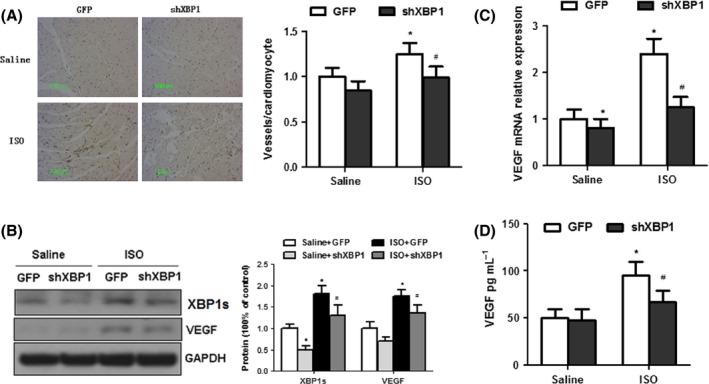
Myocardial capillary density and cardiac expression of pro‐angiogenic factor VEGF‐A is decreased in ISO‐treated mice after AAV9‐XBP1 shRNA treatment. (A) The number of microvessels per cardiomyocyte in ISO‐treated mice after AAV9‐XBP1 shRNA treatment. (B) Western blot analysis of VEGF and XBP1 expression in ISO‐treated hearts, with or without infection of AAV‐shRNA XBP1. n = 4. (C) Real‐time PCR analysis of VEGF mRNA expression in ISO‐treated hearts, with or without infection of AAV‐shRNA XBP1. n = 8. (D) ELISA of VEGF in blood sample of ISO‐infused mouse hearts after AAV‐shRNA XBP1 treatment. Error bars indicate SEM. n = 8 per group. The blot is representative of at least four blots from four independent experiments; *P < 0.05 compared with GFP or #P < 0.05 compared with ISO+GFP. n = 4.
ER stress increases both XBP1 and VEGF‐A expression in cardiomyocytes
To investigate the possible role of UPR‐induced XBP1 in inducting pro‐angiogenic factor VEGF in vitro, we treated rat embryonic heart‐derived myogenic H9c2 (2‐1) cells with two pharmacological UPR inducers, thapsigargin (TG, a Ca2+ ATPase inhibitor that promotes ER stress by depletion of lumenal calcium stores) and tunicamycin (TM, which blocks protein glycosylation) for 24 h. Incubation of H9c2 cells with TM or TG markedly increased expression levels, as well as mRNA levels, of Grp78, spliced XBP1, and of VEGF‐A in cultured cardiac myocytes (Fig. 4A–C). We next determined the effects of ISO on XBP1 and VEGFA in cultured cardiomyocytes. As shown in Fig. 4D, ISO‐induced UPR attained similar results in H9c2 (2‐1) cells. Collectively, these results suggest that UPR synchronously induced upregulation of VEGF‐A and XBP1.
Figure 4.
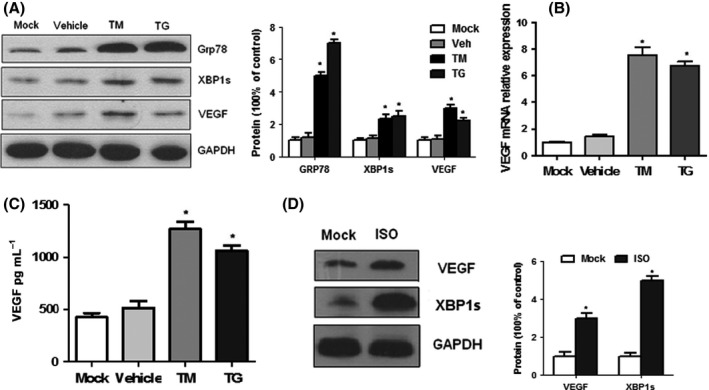
UPR increased VEGF‐A expression in H9C2 (2‐1) cells. (A) Western blots of XBP1s and VEGF in untreated and TM/TG‐treated H9C2 (2‐1) cells. (B) Real‐time PCR analysis of VEGF in H9C2 (2‐1) cells. (C) VEGF levels in culture supernatant from untreated and treated cells by ELISA. (D) Western blots of XBP1s and VEGF in untreated and ISO‐treated H9C2 (2–1) cells. Values are means ± SEM. The blot is representative of at least four blots from four independent experiments; *P < 0.05 compared with control. n = 4.
UPR‐induced VEGF‐A upregulation depends on XBP1 in vitro
To further test whether XBP1 is involved in ER stress‐mediated VEGF‐A induction, siRNA targeting rat XBP1 (siRNA‐XBP1) was transfected into H9c2 (2‐1) cells with or without TG treatment. The results showed that the siRNA targeting XBP1 also synchronously reduced the mRNA and protein levels of spliced XBP1 and VEGF‐A (Fig. 5A–B). This induction of XBP1 and VEGF of ISO treatment is similar to that of TG treatment. Conversely, we next determined if ectopic expression of XBP1 altered VEGF expression in H9c2 (2‐1) cells. Compared to those transfected with control vectors, overexpression of XBP1s by transient transfection of pCMV6‐XL5‐XBP1s plasmid markedly increased VEGF expression in cultured H9c2 (2‐1) cells (Fig. 5D). Recently, XBP1 was reported to bind to the promoter regions of VEGF‐A in cancer cells (Roybal et al., 2005; Pereira et al., 2010). To confirm whether XBP1 is a transcription factor of VEGF in cardiomyocytes, we performed the Chromatin immunoprecipitation (ChIP) analyses and found that one putative XBP1s‐binding site (~1.9 kp) in VEGFA gene promoter was positively amplified after immunoprecipitation with primary antibody to XBP1, and TG increased this binding in H9C2 cells (Fig. 5E). Taken together, these data indicate that VEGF is a direct transcription target of XBP1 in cardiomyocytes. Furthermore, to characterize the relevance of the above observations with mouse cell line, we performed the ChIP assay and detected similar effects of XBP1 on VEGF regulation in mouse embryonic fibroblast (MEF) (Fig. 5F).
Figure 5.
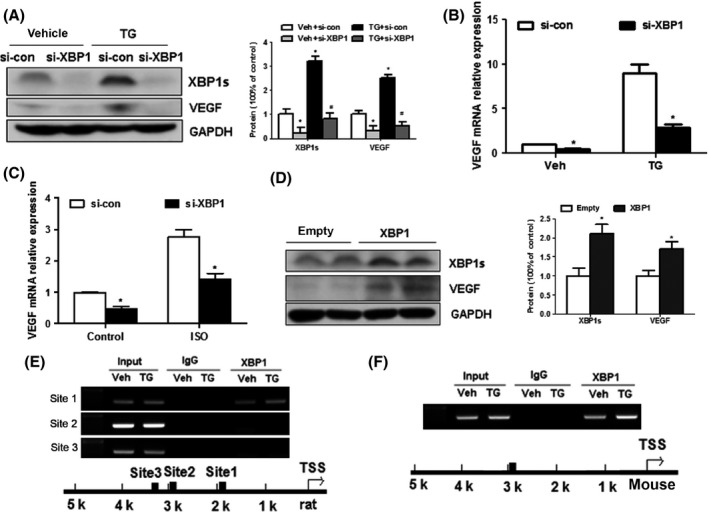
UPR‐induced VEGF‐A upregulation depends on XBP1 in H9C2 (2‐1) cells. (A) Western blots of XBP1s and VEGF in untreated and TG‐treated H9C2 (2‐1) cells after XBP1 siRNA transfection. (B), XBP1 siRNA (100 nM) knockdown of VEGF mRNA, with or without TG treatment in H9C2 (2‐1) cells, confirmed by real‐time PCR analysis. (C) XBP1 siRNA (100 nM) knockdown of VEGF mRNA, with or without ISO treatment in H9C2 (2‐1) cells, confirmed by real‐time PCR analysis. (D) XBP1s reintroduction by pCMV6‐XL5‐XBP1s vector in H9C2 (2‐1) cells increased VEGF and XBP1s expression. (E) ChIP assays of extracts from H9C2 cells treated with TG using anti‐XBP1s antibody (site 1: 1930bp). (F) ChIP assays of XBP1s on mouse VEGF promoter in untreated and TG‐treated MEFs (binding site: 2876 bp). Values are means ± SEM. The blot is representative of at least four blots from four independent experiments; *P < 0.05 compared with control. n = 4.
Upregulated VEGF‐A is correlated with XBP1s in human hypertrophic and failing heart
Finally, to characterize the relevance of the above observations of animal model and cardiomyocytes, we further measured the expression of UPR transcription factor XBP1, the ER chaperone GRP78, and the pro‐angiogenic factor VEGF in human myocardial samples by Western blot analysis. And the results showed that ER stress marker GRP78 and XBP1 were significantly increased in all failing human hearts compared to normal hearts, as well as ANP, a marker associated with cardiac hypertrophy and heart failure (Fig. 6A). As expected, the level of VEGF also was significantly elevated in failing hearts when compared to donor normal hearts (Table 1). The increase of myocardial VEGF was significantly correlated with the increase of XBP1 but not Grp78 (Fig. 6B–C). Then, we collected plasma samples from 120 patients with heart failure and 72 healthy adults to investigate circulating VEGF levels and found that the circulating expression levels of VEGF measured by ELISA were significantly increased in HF patients when compared to normal control (Fig. 6D).
Figure 6.
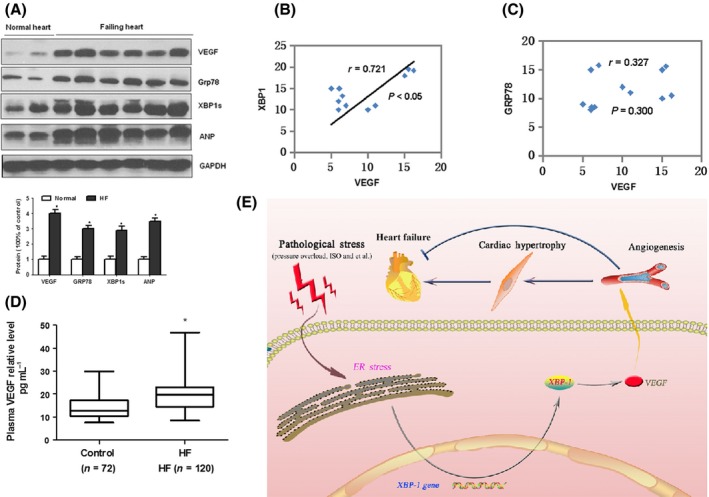
Upregulated VEGF in human heart failure heart samples is correlated with the increases of XBP1. (A) Western blot analysis of cardiac XBP1s, Grp78, VEGF, and ANP expression in human failing heart samples. (B–C) Increased VEGF expression in human heart failure heart samples is correlated with the increases of ER stress markers XBP1s but not Grp78. (D) ELISA of VEGF in blood sample of heart failure patients. The blot is representative of at least four blots from four independent experiments; *P < 0.05 compared with control. (E) Proposed mechanism underlying the transition from cardiac hypertrophy to heart failure. Deregulation of XBP1 suppresses VEGF‐mediated angiogenesis, which may contribute to the progression from adaptive hypertrophy to heart failure.
Table 1.
Characteristics for the study population
| Characteristics | HF Cases(n = 120) | Normal(n = 72) |
|---|---|---|
| Age (years) | 65 ± 15 | 60 ± 10 |
| Male/female (n/n) | 83/37 | 40/32 |
| Current smoking, n (%) | 36 (30%) | 18 (25%) |
| Hypertension, n (%) | 68 (56.67%) | 15 (20.8%) |
| Fasting glucose (mm) | 6.808 ± 1.36 | 5.2 ± 0.68 |
| SBP (mmHg) | 137.7 ± 24 | 125.2 ± 18 |
| DBP (mmHg) | 76.6 ± 12 | 65.5 ± 7 |
| Ejection fraction (%) | 46.2 ± 9.2 | 62 ± 7.8 |
SBP, systolic blood pressure; DBP, diastolic blood pressure.
Mean ± SD.
Taken together, these results raise the intriguing possibility that increased expression of XBP1 actually causes accumulation of VEGF protein and myocardial angiogenesis and contribute to the progression of cardiac hypertrophy (Fig. 6E).
Discussion
In the present study, we have demonstrated that cardiac expression of ER stress transcription factor XBP1 was upregulated in pressure‐overload‐ and ISO‐induced cardiac hypertrophic mice. In addition, we found that increased XBP1 promotes VEGF‐A expression while silencing XBP1 inhibits VEGF‐A expression in cardiomyocytes. Furthermore, genetic inhibition of XBP1 inhibits cardiac VEGF expression and angiogenesis and exacerbates ISO‐induced cardiac dysfunction in vivo. Overall, our results indicate that an abnormal increase of XBP1 in the early stage of hypertrophic and failing heart is important in regulation of both the pro‐angiogenic factor VEGF and cardiac angiogenesis in vivo.
One of the most important findings of the present study is that XBP1 is essential for VEGF‐A upregulation and cardiac angiogenesis. The evidence can be summarized as follows: First, the levels of VEGF‐A and angiogenesis were correlated with the levels of XBP1s and ER stress in ISO‐infused hypertrophic hearts and failing human hearts. Second, inhibited expression of XBP1s decreased VEGF‐A expression and the number of CD31‐positive cell in hypertrophic and failing heart, demonstrating the pro‐angiogenesis property of XBP1s in vivo. Third, ISO increases both XBP1 and VEGF‐A expression in cardiomyocytes and silencing of XBP1 blocks ER stress‐induced VEGF expression in cardiomyocytes and reduces endothelial cell proliferation, migration, tube formation, and angiogenesis (Duan et al., 2015b), supporting that XBP1 silencing decreased cardiac capillary‐like network formation. Certainly, future research using AAV‐mediated restoration of XBP1 in maladaptive cardiac hypertrophy is required to provide further evidence of the biological role of XBP1 in the development of heart failure. Recent research further shows that XBP1 is a coregulator of HIF1α and controls the HIF1α transcriptional program in breast cancer (Chen et al., 2014). Interestingly, our recent study further found that cardiac expression of VEGF‐A, is correlated with XBP1s, was increased in the early adaptive phase, but decreased in the maladaptive phase in rat pressure‐overload‐ and isoproterenol‐induced hypertrophic and failing heart (Duan et al., 2015a). These researches support our finding that XBP1 is essential for VEGF induction in heart tissue.
On the other hand, a recent study reported that VEGF activates XBP1 mRNA splicing in endothelial cells via the AKT/GSK/β‐catenin/E2F2 signal pathway to promote endothelial cell proliferation and angiogenesis (Zeng et al., 2013), which supports and explains our findings on how secreted VEGF from cardiomyocyte regulates endothelial cell angiogenesis. There may be a XBP1‐VEGF‐XBP1 regulatory circuit from cardiomyocyte to endothelial cell in the process of cardiac angiogenesis. Importantly, Xbp1s was shown to be sufficient and necessary to protect heart from ischemia/reperfusion (I/R) injury in vivo (Wang et al., 2014). Our data are consistent with other investigations and also demonstrate that XBP1 is essential for tissue angiogenesis under physiological or pathological conditions (Romero‐Ramirez et al., 2009; Zeng et al., 2009, 2013; Ghosh et al., 2010; Ruan et al., 2013; Miyagi et al., 2013; Wang et al., 2014) and that XBP1 is an important regulator of vascular function and cardiac angiogenesis. In recent years, previous studies have shown that as a key stress‐inducible transcription factor in mammalian cells, XBP1 splicing plays an important role in the regulation of cell survival (Thuerauf et al., 2006), inflammation (Martinon et al., 2010), insulin sensitivity (Ozcan et al., 2004), glucose homeostasis (Ozcan et al., 2004; Zhou et al., 2011), lipogenesis (Lee et al., 2008; So et al., 2012), and autophagy (Margariti et al., 2013). Here, uncovering the exact molecular mechanisms of XBP1s‐induced cardioprotection will require further investigation.
Vascular endothelial growth factor (VEGF) is an essential angiogenic factor to promote angiogenesis and neovascularization and regulate all types of vascular growth and has thus received much attention regarding their potential use for therapeutic vascular growth in cardiovascular diseases (Ng et al., 2006). Previous studies have shown that VEGF‐B gene transfer resulted in prevention of the angiotensin II‐induced diastolic dysfunction associated with induction of the Akt pathway (Serpi et al., 2011), while VEGF blockade promotes the transition from compensatory cardiac hypertrophy to failure in response to pressure overload (Izumiya et al., 2006). Our data also show that the circulating expression levels of VEGF were significantly increased in heart failure patients, suggesting the plasma concentration of VEGF can be a potential indicator for heart failure. Certainly, further research is warranted to establish whether plasma levels of VEGF were related to different cardiac function. Interestingly, it is confused about that there is an unmatched phenotype between cardiac hypertrophy and cardiac function in mice with VEGF‐assisted treatment. Possible explanation was that the upregulation of VEGF‐A expression increases the capillary/myocyte ratio, but still leads to a net reduction in capillary density (capillaries mm−2), because the increase in capillarization (capillaries/myocyte) cannot keep match with myocyte growth (myocyte cross‐sectional area). When inhibition of cardiac angiogenesis further reduces capillary/myocyte ratio and leads to a greater reduction in coronary capillary density, contractile function, increased LVED dimension, ANP expression, and interstitial fibrosis contributed to a rapid transition to heart failure (Izumiya et al., 2006).
Previous studies have shown that XBP1s and VEGF were involved, respectively, in diverse cellular functions and processes (Ng et al., 2006; Glimcher, 2010). Now, our study linked these two different pathways and offered a new insight to investigate the physiologic and pathophysiologic significance of the XBP1/VEGF axis in multiple human diseases. Base our present study, XBP1s/VEGF‐A was correlated with cardiac angiogenesis in the progression from adaptive hypertrophy to heart failure. In the future, we will continue to check the contribution of this pathway in the aging heart disease and myocardial infarction. In additional, both XBP1s and VEGF‐A can regulate the development of retinal vasculature (Zeng et al., 2013), may offer a potential therapeutic target in the treatment of pathological angiogenesis for retinal/choroidal diseases. Certainly, the XBP1s/VEGF‐A pathway also may have important implications in developing antiangiogenic tumor treatment strategies in cancer diseases (Koong et al., 2006). Importantly, as a novel regulator of metabolic homeostasis (Lee et al., 2008; Zhou et al., 2011; Wu et al., 2014), the proposed XBP1 regulation of VEGF also broadens our knowledge on the pathologies of metabolic syndrome and sheds light on the intervention of diabetes mellitus and obesity‐related metabolic diseases.
In summary, our study has for the first time established that XBP1 is an important pro‐angiogenic factor, required to maintain normal cardiac function in the early stage of hypertrophy which results in the transition of hypertrophic hearts to heart failure. Thus, modulation of XBP1 might be a valid therapeutic target to prevent or delay heart failure.
Experimental procedures
Materials and reagents
Antibodies against XBP1, Grp78, VEGF, GAPDH, and β‐actin were purchased from Santa Cruz Biotechnology Inc. (Santa Cruz, CA, USA); antibodies against CD31 were purchased from Abcam Inc. (Cambridge, MA, USA). Nonspecific negative control oligonucleotides and specific siRNA against human or rat/mice XBP1 were obtained from RiboBio (Guangzhou, China). All other chemicals and reagents were purchased from Sigma‐Aldrich China Inc. (Shanghai, China), unless otherwise specified.
Cell lines
The embryonic rat heart–derived myogenic cell line H9c2 (2‐1) and mouse embryonic fibroblast (MEF) were obtained from the American Type Culture Collection (ATCC, Manassas, VA, USA). The H9c2 (2‐1) cells and MEFs were grown in Gibco DMEM medium supplemented with 10% fetal bovine serum (Gibco, Invitrogen, Carlsbad, CA, USA), streptomycin 100 μg mL−1, and penicillin 100 U mL−1 (all obtained from Sigma, St Louis, MO, USA). All cells were grown at 37 °C in an atmosphere of 5% CO2.
Animal models
Eight‐week‐old male C57BL/6 mice were subjected to thoracic aorta constriction (TAC) or sham operation as described previously (Okada et al., 2004). Pressure‐overload‐induced cardiac hypertrophy and heart failure was created by TAC for 2 weeks. C57BL/6 mice were continuously infused with isoproterenol hydrochloride (15 mg kg−1 day−1; Sigma‐Aldrich China Inc.) over a period of 14 days with implanted minipumps. Mini‐osmotic pumps (Alzet model 1007D and 1002; DURECT Corp., Cupertino, CA, USA) were implanted as described previously (Nienaber et al., 2003). Mice were anesthetized with pentobarbital sodium at a dose of 80 mg kg−1 body weight intraperitoneally. Animals were euthanized via an anesthetic overdose (200 mg kg−1 of pentobarbital sodium delivered by intraperitoneal injection). All animal studies were approved by the Animal Research Committee of Tongji College and were carried out according to the guidelines of the National Institutes of Health (NIH).
Echocardiography
Echocardiography was performed on mice with a VisualSonics Vevo 2100 System echocardiograph (VisualSonics, Toronto, CA, USA) using a 35‐MHz probe as described previously (Lin et al., 2009; Wang et al., 2011).
Heart tissue samples
Human heart tissue samples were studied according to the protocol approved by the Clinical Research Committees of Tongji Medical College and the guidelines of the National Institutes of Health (NIH). The investigation also conforms to the principles outlined in the Declaration of Helsinki. The subjects recruited to the study provided informed consent. We collected heart samples from six recipients of heart transplantation who suffered end‐stage heart failure. The normal hearts were from victims of traffic accidents. Tissue samples were frozen at −80 °C until used for extraction of protein.
Vector construction
Recombinant adeno‐associated virus vectors rAAV9‐GFP and rAAV9‐shXBP1 (rAAV9 generating shRNA to silence XBP1) were produced by a 2‐plasmid protocol described previously (Suckau et al., 2009) and then prepared and purified according to a published protocol (Inagaki et al., 2006). The siRNA sequences for mice XBP1 were: 5′‐CAC CCU GAA UUC AUU GUC U‐3′. The plasmid constructs were verified by sequencing.
Gene delivery study in vivo
In C57BL/6 mice study, rAAV9 was used as carrier. C57BL/6 mice were injected with rAAV‐GFP or rAAV‐XBP1 shRNA (n = 8 per group) through a sublingual vein injection 2 weeks before ISO infusion as described previously (Masson et al., 1998; Feng & Li, 2010). Two weeks after ISO infusion, mice were subjected to echocardiography examination, followed by morphological and immunohistological tests.
Immunohistochemical analysis
For immunohistochemical examination of CD31, paraffin‐embedded rat heart tissues were cut into 5‐μm sections, and assays were performed as previously described (Jiang et al., 2005). Antibodies for CD31 were used as the primary antibody.
ChIP assay for XBP1 and VEGFA gene promoter binding
ChIP assays were performed according to the manufacturer's protocol using a ChIP kit (17‐295; Millipore, Temecula, CA 92590). The primers used for ChIP in H9C2 cells were described previously (Pereira et al., 2010). The primers used for ChIP in MEFs were as follows: 5′‐GCG TCA CTG CTC TGG CTC CCT GT‐3′ and the reverse primer 5′‐CGG GCC TAC AGA AGA GGG ACT CAG TA‐3′.
Cell transfection
H9c2 cells were seeded in 6‐well plates at 1.5 × 105 cells/well, 24 h before transfection. Cells were transfected with siRNA after seeding using Lipofectamine 2000 (Invitrogen) and transfected with plasmid using X‐tremeGENE HP DNA Transfection Reagent (Roche, Indianapolis, IN, USA), according to the manufacturer's recommendations. Transfections contained 50 or 100 nm small interfering RNAs (siRNAs) against XBP1 and siRNA control (RiboBio) in a final volume of 2 mL. The siRNA sequences for rat XBP1 were as follows: 5′‐GAG AAA GCG CUG CGG AGG A‐3′ (Sawada et al., 2010). Cells were collected 48 h after transfection for protein and RNA extraction.
VEGF secretion assay
After cells were exposed to 24 h of TM or TG, the VEGF‐A protein concentrations in the culture medium were measured by ELISA using a human or rat VEGF‐A ELISA kit (R&D Systems, Abingdon, UK) according to the manufacturer's protocol.
RNA extraction and real‐time quantitative PCR
Total RNA was extracted using TRIzol, according to the manufacture's protocol. Reverse transcription reactions were performed starting from equal amounts of total RNA/sample (1 μg) using EasyScript First‐Strand cDNA Synthesis SuperMix (TransGen Biotech, Beijing, China). Rat VEGF mRNA levels were determined using the forward primer 5′‐CAG CTA TTG CCG TCC AAT TGA‐3′ and the reverse primer 5′‐CCA GGG CTT CAT CAT TGC A‐3′ (Romero‐Ramirez et al., 2009); mice VEGF mRNA levels were determined using the forward primer 5′‐TGT ACC TCC ACC ATG CCA AGT‐3′ and the reverse primer 5′‐TGG AAG ATG TCC ACC AGG GT‐3′; β‐actin or GAPDH was used as an internal reference. Results of the real‐time quantitative PCR were analyzed and expressed as relative mRNA levels of the cycle threshold (CT) value, which was then converted to fold change.
Western blot analysis
Proteins from cell lysates (20 μg) were separated by 10% SDS–polyacrylamide gel electrophoresis and transferred to a polyvinylidene difluoride membrane. After blocking in 5% nonfat milk, protein blots were incubated with a specific antibody followed by incubation with a peroxidase‐conjugated secondary antibody in blocking buffer. The bands were visualized using the enhanced chemiluminescence method, according to manufacturer's instructions (Beyotime Institute of Biotechnology, Haimen, China).
Semiquantitative reverse transcription–PCR
To detect rat unspliced and spliced XBP1, the primers were 5′‐AAA CAG AGT AGC AGC ACA GAC TGC‐3′ and 5′‐TCC TTC TGG GTA GAC CTC TGG GAG‐3′ (Lai et al., 2008). After 35 cycles, PCR products for rat unspliced (XBP1u, 480 bp) and spliced (XBP1s, 454 bp) XBP1 fragments were separated by 2% agarose gels stained with GoldView. The relative intensity of XBP1 compared with β‐actin was calculated for each sample by densitometry.
Statistical analysis
The data are expressed as mean values ± SD. Differences between groups were evaluated for significance using Student's t‐test of unpaired data or one‐way analysis of variance (anova) and Bonferroni post‐test. P < 0.05 was considered significant.
Funding info
This study was supported by grants from the National Natural Science Foundation of China (30770882, 81470519, 31400998, 91439203, and 31130031) and National Basic Research Program of China (973 Program, No. 2007CB512004). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Author contributions
The author(s) have made the following declarations about their contributions: Quanlu Duan and Dao Wen Wang conceived and designed the experiments. Quanlu Duan, Lei Yang, Ben Ma, and Wei Gong performed the experiments. Quanlu Duan, Chen Chen, and Li Ni analyzed the data. Quanlu Duan, Peihua Wang, and Zhejun Cai contributed reagents/materials/analysis tools. Quanlu Duan, Dao Wen Wang, and Ming‐Hui Zou wrote the manuscript.
Conflict of interest
The authors have declared that no competing interests exist.
Acknowledgments
None.
References
- Chen X, Iliopoulos D, Zhang Q, Tang Q, Greenblatt MB, Hatziapostolou M, Lim E, Tam WL, Ni M, Chen Y, Mai J, Shen H, Hu DZ, Adoro S, Hu B, Song M, Tan C, Landis MD, Ferrari M, Shin SJ, Brown M, Chang JC, Liu XS, Glimcher LH (2014) XBP1 promotes triple‐negative breast cancer by controlling the HIF1alpha pathway. Nature 508, 103–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan Q, Chen C, Yang L, Li N, Gong W, Li S, Wang DW (2015a) MicroRNA regulation of unfolded protein response transcription factor XBP1 in the progression of cardiac hypertrophy and heart failure in vivo. J. Transl. Med. 13, 363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan Q, Yang L, Gong W, Chaugai S, Wang F, Chen C, Wang P, Zou MH, Wang DW (2015b) MicroRNA‐214 is upregulated in heart failure patients and suppresses XBP1‐mediated endothelial cells angiogenesis. J. Cell. Physiol. 230, 1964–1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng W, Li W (2010) The study of ISO induced heart failure rat model. Exp. Mol. Pathol. 88, 299–304. [DOI] [PubMed] [Google Scholar]
- Ghosh R, Lipson KL, Sargent KE, Mercurio AM, Hunt JS, Ron D, Urano F (2010) Transcriptional regulation of VEGF‐A by the unfolded protein response pathway. PLoS ONE 5, e9575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giordano FJ, Gerber HP, Williams SP, VanBruggen N, Bunting S, Ruiz‐Lozano P, Gu Y, Nath AK, Huang Y, Hickey R, Dalton N, Peterson KL, Ross J Jr, Chien KR, Ferrara N (2001) A cardiac myocyte vascular endothelial growth factor paracrine pathway is required to maintain cardiac function. Proc. Natl Acad. Sci. USA 98, 5780–5785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glimcher LH (2010) XBP1: the last two decades. Ann. Rheum. Dis. 69(Suppl 1), i67–i71. [DOI] [PubMed] [Google Scholar]
- Groenendyk J, Sreenivasaiah PK, Kim dH, Agellon LB, Michalak M (2010) Biology of endoplasmic reticulum stress in the heart. Circ. Res. 107, 1185–1197. [DOI] [PubMed] [Google Scholar]
- Hein S, Arnon E, Kostin S, Schonburg M, Elsasser A, Polyakova V, Bauer EP, Klovekorn WP, Schaper J (2003) Progression from compensated hypertrophy to failure in the pressure‐overloaded human heart: structural deterioration and compensatory mechanisms. Circulation 107, 984–991. [DOI] [PubMed] [Google Scholar]
- Heineke J, Molkentin JD (2006) Regulation of cardiac hypertrophy by intracellular signalling pathways. Nat. Rev. Mol. Cell Biol. 7, 589–600. [DOI] [PubMed] [Google Scholar]
- Inagaki K, Fuess S, Storm TA, Gibson GA, McTiernan CF, Kay MA, Nakai H (2006) Robust systemic transduction with AAV9 vectors in mice: efficient global cardiac gene transfer superior to that of AAV8. Mol. Ther. 14, 45–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izumiya Y, Shiojima I, Sato K, Sawyer DB, Colucci WS, Walsh K (2006) Vascular endothelial growth factor blockade promotes the transition from compensatory cardiac hypertrophy to failure in response to pressure overload. Hypertension 47, 887–893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang JG, Chen CL, Card JW, Yang S, Chen JX, Fu XN, Ning YG, Xiao X, Zeldin DC, Wang DW (2005) Cytochrome P450 2J2 promotes the neoplastic phenotype of carcinoma cells and is up‐regulated in human tumors. Cancer Res. 65, 4707–4715. [DOI] [PubMed] [Google Scholar]
- Koong AC, Chauhan V, Romero‐Ramirez L (2006) Targeting XBP‐1 as a novel anti‐cancer strategy. Cancer Biol. Ther. 5, 756–759. [DOI] [PubMed] [Google Scholar]
- Lai E, Bikopoulos G, Wheeler MB, Rozakis‐Adcock M, Volchuk A (2008) Differential activation of ER stress and apoptosis in response to chronically elevated free fatty acids in pancreatic beta‐cells. Am. J. Physiol. Endocrinol. Metab. 294, E540–E550. [DOI] [PubMed] [Google Scholar]
- Lee AH, Scapa EF, Cohen DE, Glimcher LH (2008) Regulation of hepatic lipogenesis by the transcription factor XBP1. Science 320, 1492–1496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J, Sun C, Zhou Y, Gokalp D, Herrema H, Park SW, Davis RJ, Ozcan U (2011) p38 MAPK‐mediated regulation of Xbp1s is crucial for glucose homeostasis. Nat. Med. 17, 1251–1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Z, Murtaza I, Wang K, Jiao J, Gao J, Li PF (2009) miR‐23a functions downstream of NFATc3 to regulate cardiac hypertrophy. Proc. Natl Acad. Sci. USA 106, 12103–12108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margariti A, Li H, Chen T, Martin D, Vizcay‐Barrena G, Alam S, Karamariti E, Xiao Q, Zampetaki A, Zhang Z, Wang W, Jiang Z, Gao C, Ma B, Chen YG, Cockerill G, Hu Y, Xu Q, Zeng L (2013) XBP1 mRNA splicing triggers an autophagic response in endothelial cells through BECLIN‐1 transcriptional activation. J. Biol. Chem. 288, 859–872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinon F, Chen X, Lee AH, Glimcher LH (2010) TLR activation of the transcription factor XBP1 regulates innate immune responses in macrophages. Nat. Immunol. 11, 411–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masson S, Arosio B, Luvara G, Gagliano N, Fiordaliso F, Santambrogio D, Vergani C, Latini R, Annoni G (1998) Remodelling of cardiac extracellular matrix during beta‐adrenergic stimulation: upregulation of SPARC in the myocardium of adult rats. J. Mol. Cell. Cardiol. 30, 1505–1514. [DOI] [PubMed] [Google Scholar]
- Miyagi H, Kanemoto S, Saito A, Asada R, Iwamoto H, Izumi S, Kido M, Gomi F, Nishida K, Kiuchi Y, Imaizumi K (2013) Transcriptional regulation of VEGFA by the endoplasmic reticulum stress transducer OASIS in ARPE‐19 cells. PLoS ONE 8, e55155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng YS, Krilleke D, Shima DT (2006) VEGF function in vascular pathogenesis. Exp. Cell Res. 312, 527–537. [DOI] [PubMed] [Google Scholar]
- Nienaber JJ, Tachibana H, Naga Prasad SV, Esposito G, Wu D, Mao L, Rockman HA (2003) Inhibition of receptor‐localized PI3K preserves cardiac beta‐adrenergic receptor function and ameliorates pressure overload heart failure. J Clin Invest. 112, 1067–1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okada K, Minamino T, Tsukamoto Y, Liao Y, Tsukamoto O, Takashima S, Hirata A, Fujita M, Nagamachi Y, Nakatani T, Yutani C, Ozawa K, Ogawa S, Tomoike H, Hori M, Kitakaze M (2004) Prolonged endoplasmic reticulum stress in hypertrophic and failing heart after aortic constriction: possible contribution of endoplasmic reticulum stress to cardiac myocyte apoptosis. Circulation 110, 705–712. [DOI] [PubMed] [Google Scholar]
- Ozcan U, Cao Q, Yilmaz E, Lee AH, Iwakoshi NN, Ozdelen E, Tuncman G, Gorgun C, Glimcher LH, Hotamisligil GS (2004) Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes. Science 306, 457–461. [DOI] [PubMed] [Google Scholar]
- Park SW, Zhou Y, Lee J, Lu A, Sun C, Chung J, Ueki K, Ozcan U (2010) The regulatory subunits of PI3K, p85alpha and p85beta, interact with XBP‐1 and increase its nuclear translocation. Nat. Med. 16, 429–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pereira ER, Liao N, Neale GA, Hendershot LM (2010) Transcriptional and post‐transcriptional regulation of proangiogenic factors by the unfolded protein response. PLoS ONE 5, e12521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romero‐Ramirez L, Cao H, Nelson D, Hammond E, Lee AH, Yoshida H, Mori K, Glimcher LH, Denko NC, Giaccia AJ, Le QT, Koong AC (2004) XBP1 is essential for survival under hypoxic conditions and is required for tumor growth. Cancer Res. 64, 5943–5947. [DOI] [PubMed] [Google Scholar]
- Romero‐Ramirez L, Cao H, Regalado MP, Kambham N, Siemann D, Kim JJ, Le QT, Koong AC (2009) X box‐binding protein 1 regulates angiogenesis in human pancreatic adenocarcinomas. Transl Oncol. 2, 31–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roybal CN, Hunsaker LA, Barbash O, Vander Jagt DL, Abcouwer SF (2005) The oxidative stressor arsenite activates vascular endothelial growth factor mRNA transcription by an ATF4‐dependent mechanism. J. Biol. Chem. 280, 20331–20339. [DOI] [PubMed] [Google Scholar]
- Ruan Q, Xi L, Boye SL, Han S, Chen ZJ, Hauswirth WW, Lewin AS, Boulton ME, Law BK, Jiang WG, Jiang HB, Cai J (2013) Development of an anti‐angiogenic therapeutic model combining scAAV2‐delivered siRNAs and noninvasive photoacoustic imaging of tumor vasculature development. Cancer Lett. 332, 120–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sano M, Minamino T, Toko H, Miyauchi H, Orimo M, Qin Y, Akazawa H, Tateno K, Kayama Y, Harada M, Shimizu I, Asahara T, Hamada H, Tomita S, Molkentin JD, Zou Y, Komuro I (2007) p53‐induced inhibition of Hif‐1 causes cardiac dysfunction during pressure overload. Nature 446, 444–448. [DOI] [PubMed] [Google Scholar]
- Sawada T, Minamino T, Fu HY, Asai M, Okuda K, Isomura T, Yamazaki S, Asano Y, Okada K, Tsukamoto O, Sanada S, Asanuma H, Asakura M, Takashima S, Kitakaze M, Komuro I (2010) X‐box binding protein 1 regulates brain natriuretic peptide through a novel AP1/CRE‐like element in cardiomyocytes. J. Mol. Cell. Cardiol. 48, 1280–1289. [DOI] [PubMed] [Google Scholar]
- Serpi R, Tolonen AM, Huusko J, Rysa J, Tenhunen O, Yla‐Herttuala S, Ruskoaho H (2011) Vascular endothelial growth factor‐B gene transfer prevents angiotensin II‐induced diastolic dysfunction via proliferation and capillary dilatation in rats. Cardiovasc. Res. 89, 204–213. [DOI] [PubMed] [Google Scholar]
- Shah AM, Mann DL (2011) In search of new therapeutic targets and strategies for heart failure: recent advances in basic science. Lancet 378, 704–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiojima I, Sato K, Izumiya Y, Schiekofer S, Ito M, Liao R, Colucci WS, Walsh K (2005) Disruption of coordinated cardiac hypertrophy and angiogenesis contributes to the transition to heart failure. J Clin Invest. 115, 2108–2118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- So JS, Hur KY, Tarrio M, Ruda V, Frank‐Kamenetsky M, Fitzgerald K, Koteliansky V, Lichtman AH, Iwawaki T, Glimcher LH, Lee AH (2012) Silencing of lipid metabolism genes through IRE1alpha‐mediated mRNA decay lowers plasma lipids in mice. Cell Metab. 16, 487–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suckau L, Fechner H, Chemaly E, Krohn S, Hadri L, Kockskamper J, Westermann D, Bisping E, Ly H, Wang X, Kawase Y, Chen J, Liang L, Sipo I, Vetter R, Weger S, Kurreck J, Erdmann V, Tschope C, Pieske B, Lebeche D, Schultheiss HP, Hajjar RJ, Poller WC (2009) Long‐term cardiac‐targeted RNA interference for the treatment of heart failure restores cardiac function and reduces pathological hypertrophy. Circulation 119, 1241–1252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thuerauf DJ, Marcinko M, Gude N, Rubio M, Sussman MA, Glembotski CC (2006) Activation of the unfolded protein response in infarcted mouse heart and hypoxic cultured cardiac myocytes. Circ. Res. 99, 275–282. [DOI] [PubMed] [Google Scholar]
- Tomanek RJ (1990) Response of the coronary vasculature to myocardial hypertrophy. J. Am. Coll. Cardiol. 15, 528–533. [DOI] [PubMed] [Google Scholar]
- Wang JX, Jiao JQ, Li Q, Long B, Wang K, Liu JP, Li YR, Li PF (2011) miR‐499 regulates mitochondrial dynamics by targeting calcineurin and dynamin‐related protein‐1. Nat. Med. 17, 71–78. [DOI] [PubMed] [Google Scholar]
- Wang ZV, Deng Y, Gao N, Pedrozo Z, Li DL, Morales CR, Criollo A, Luo X, Tan W, Jiang N, Lehrman MA, Rothermel BA, Lee AH, Lavandero S, Mammen PP, Ferdous A, Gillette TG, Scherer PE, Hill JA (2014) Spliced X‐box binding protein 1 couples the unfolded protein response to hexosamine biosynthetic pathway. Cell 156, 1179–1192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu LE, Meoli CC, Mangiafico SP, Fazakerley DJ, Cogger VC, Mohamad M, Pant H, Kang MJ, Powter E, Burchfield JG, Xirouchaki CE, Mikolaizak AS, Stockli J, Kolumam G, van Bruggen N, Gamble JR, Le Couteur DG, Cooney GJ, Andrikopoulos S, James DE (2014) Systemic VEGF‐A neutralization ameliorates diet‐induced metabolic dysfunction. Diabetes 63, 2656–2667. [DOI] [PubMed] [Google Scholar]
- Yoshida H, Matsui T, Yamamoto A, Okada T, Mori K (2001) XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell 107, 881–891. [DOI] [PubMed] [Google Scholar]
- Zeng L, Zampetaki A, Margariti A, Pepe AE, Alam S, Martin D, Xiao Q, Wang W, Jin ZG, Cockerill G, Mori K, Li YS, Hu Y, Chien S, Xu Q (2009) Sustained activation of XBP1 splicing leads to endothelial apoptosis and atherosclerosis development in response to disturbed flow. Proc. Natl Acad. Sci. USA 106, 8326–8331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng L, Xiao Q, Chen M, Margariti A, Martin D, Ivetic A, Xu H, Mason J, Wang W, Cockerill G, Mori K, Li JY, Chien S, Hu Y, Xu Q (2013) Vascular endothelial cell growth‐activated XBP1 splicing in endothelial cells is crucial for angiogenesis. Circulation 127, 1712–1722. [DOI] [PubMed] [Google Scholar]
- Zhou Y, Lee J, Reno CM, Sun C, Park SW, Chung J, Fisher SJ, White MF, Biddinger SB, Ozcan U (2011) Regulation of glucose homeostasis through a XBP‐1‐FoxO1 interaction. Nat. Med. 17, 356–365. [DOI] [PMC free article] [PubMed] [Google Scholar]