

Summary
Aging is associated with progressive telomere shortening, resulting in the formation of dysfunctional telomeres that compromise tissue proliferation. However, dysfunctional telomeres can limit tumorigenesis by activating p53‐dependent cellular senescence and apoptosis. While activation of both senescence and apoptosis is required for repress tumor formation, it is not clear which pathway is the major tumor suppressive pathway in vivo. In this study, we generated Eμ‐myc; Pot1b ∆/∆ mouse to directly compare tumor formation under conditions in which either p53‐dependent apoptosis or senescence is activated by telomeres devoid of the shelterin component Pot1b. We found that activation of p53‐dependent apoptosis plays a more critical role in suppressing lymphoma formation than p53‐dependent senescence. In addition, we found that telomeres in Pot1b ∆/∆ ; p53 −/− mice activate an ATR‐Chk1‐dependent DNA damage response to initiate a robust p53‐independent, p73‐dependent apoptotic pathway that limited stem cell proliferation but suppressed B‐cell lymphomagenesis. Our results demonstrate that in mouse models, both p53‐dependent and p53‐independent apoptosis are important to suppressing tumor formation.
Keywords: apoptosis, cellular senescence, DNA damage, molecular biology of aging, stem cells, telomeres
Introduction
Aging is due in part to increased accumulation of damaged DNA in highly proliferative tissues, leading to compromised tissue homeostasis, disease, and frailty (Kirkwood, 2008). One source of endogenous DNA damage comes from telomeres, protein‐TTAGGG repetitive DNA complexes that cap the ends of eukaryotic chromosomes (Greider, 1996). Telomeres play important roles in maintaining genome stability by preventing the activation of DNA damage checkpoints that induce p53‐dependent cell cycle arrest and apoptosis (Chin et al., 1999; Artandi et al., 2000; Gonzalez‐Suarez et al., 2000). They are synthesized by the enzyme telomerase, and interact with a number of telomere‐specific binding proteins that form a complex, termed shelterin, which protects telomeres from inappropriately activating DNA damage responses (Deng et al., 2008; Palm & de Lange, 2008; Martinez & Blasco, 2010). Shelterin components that interact with double‐stranded telomeric DNA include TRF1 and TRF2‐RAP1, while the single‐stranded (ss) telomere DNA‐binding protein POT1 forms a heterodimer with TPP1. TIN2 serves to bridge the POT1‐TPP1 complex with TRF1 and TRF2 and stabilizes POT1‐TPP1 to promote genome stability (Baumann & Cech, 2001; O'Sullivan & Karlseder, 2010).
POT1 homologs are highly conserved evolutionarily and have been identified in almost all eukaryotes. The mouse possesses two POT1 proteins, Pot1a and Pot1b while humans possess a single POT1 protein (He et al., 2006; Hockemeyer et al., 2006; Wu et al., 2006). All POT1 proteins bind to the 3′ ss TTAGGG repeats that mark the ends of mammalian telomeres with high affinity (Loayza et al., 2004; He et al., 2006; Wu et al., 2006; Palm et al., 2009; Nandakumar et al., 2010). In addition, both human POT1 and mouse Pot1b modulates the 5′ nucleolytic processing of the telomeric C‐strand and consequently regulates the formation of the 3′ ss overhang (He et al., 2006; Hockemeyer et al., 2008; Palm et al., 2009). The ability of POT1 to interact and limit the generation of the 3′ overhang is essential to prevent the activation of the ataxia telangiectasia mutated and Rad3‐related protein (ATR)‐kinase‐dependent DNA damage signaling at telomeres. Removal of POT1 results in the recruitment of replication protein A (RPA), the sensor of the ATR pathway, to telomeres. It also promotes the activation of downstream signaling kinases, including Chk1, that then signal additional effector proteins that these dysfunctional telomeres now resemble double‐stranded DNA breaks (Wu et al., 2006; Denchi & de Lange, 2007; Guo et al., 2007; Flynn et al., 2011; Thanasoula et al., 2012). In mouse models, dysfunctional telomeres due to Pot1b loss activate p53‐dependent DNA damage checkpoint responses that compromise stem cell proliferation, leading to premature aging phenotypes and compromised organismal lifespan (Wang et al., 2011, 2013). These results reveal an essential role for POT1 in preventing the inappropriate activation of the ATR‐mediated, p53‐dependent DNA damage response at telomeres of highly proliferative cells.
The POT1‐TPP1 complex also prevents inappropriate repair of dysfunctional telomeres, manifested as chromosome fusions in metaphase spreads. Both activation of the ATR kinase, and the generation of 3′ overhangs, are essential to promote alternative, nonhomologous end joining (A‐NHEJ)‐mediated repair of mouse telomeres devoid of Pot1a/b (Rai et al., 2010, 2011; Sfeir & de Lange, 2012). A‐NHEJ is a microhomology based error‐prone repair pathway that promotes gross chromosomal abnormalities, including the generation of large deletions, insertions, and translocations, resulting in elevated genome instability (Bennardo et al., 2008; Simsek & Jasin, 2010; Symington & Gautier, 2011). Importantly, point mutations in human POT1 predicted to inactivate its telomere protective functions have been found in both Chronic Lymphocytic Lymphoma and familial melanoma (Quesada et al., 2012; Ramsay et al., 2013; Robles‐Espinoza et al., 2014; Shi et al., 2014). Coupled with observations that deletion of Pot1a or Pot1b promotes end‐to‐end chromosome fusions that is tumor promoting in p53 mutant tumor prone mouse models (Wu et al., 2006; Hockemeyer et al., 2008; He et al., 2009; Wang et al., 2011), these results highlight the importance of POT1 in promoting genome stability to prevent the onset of cancer.
The p53 tumor suppressor is functionally inactivated in the majority of human cancers. Double strand breaks including those generated by the expression of oncogenes initiate p53‐dependent cellular checkpoint responses to induce a tumorigenic barrier (Halazonetis et al., 2008). Consequently, mice with critically shortened telomeres are resistant to tumor development due to the activation of p53‐dependent cellular senescence and apoptosis programs. Activation of p53‐dependent downstream targets, including the cyclin‐dependent kinase inhibitor CDKN1A/p21WAF1/CIP1 (p21) by dysfunctional telomeres, results in G1 cell cycle arrest and the onset of cellular senescence to potently suppresses tumor initiation and progression in vivo (Cosme‐Blanco et al., 2007; Feldser & Greider, 2007). In addition, dysfunctional telomeres also activate p53‐dependent apoptotic proteins, including BAX and PUMA, to limit stem cell proliferation and suppress tumorigenesis (Chin et al., 1999; Gonzalez‐Suarez et al., 2000; Rudolph et al., 2001; Begus‐Nahrmann et al., 2009; Sperka et al., 2012a). While dysfunctional telomere‐induced p53‐dependent apoptosis and senescence are both required for repress tumor formation, it is not known which pathway is the dominant tumor suppressive pathway in vivo.
In this study, we generated Eμ‐myc; Pot1b ∆/∆ mouse to directly compare tumor formation under conditions in which either p53‐dependent apoptosis or cellular senescence is activated by dysfunctional telomeres. Overexpression of the Myc oncogene in B cells results in high penetrance formation of B‐cell lymphomas within 5 months, thus providing an excellent model to test mechanisms that limit tumor growth (Harris et al., 1988). We found that p53‐dependent apoptosis plays a more important role in lymphoma suppression than p53‐dependent senescence. In addition, we also discovered that dysfunctional telomeres in Pot1b ∆/∆ ; p53 −/− mice can activate a robust p53‐independent, the p53 ortholog p73‐dependent apoptotic program to negatively impact upon tissue proliferative capacity but is able to repress B‐cell lymphomagenesis. In addition to uncovering the importance of dysfunctional telomere‐induced apoptosis in tumor suppression, our results also reveal a previously unappreciated role for p73 in mediating p53‐independent suppression of B‐cell lymphomas.
Results
Near complete suppression of lymphomagenesis in Eμ‐myc; Pot1b ∆/∆ mice
We crossed Pot1b ∆/∆ mice with Eμ‐myc transgenic mice and monitored cohorts of Pot1b ∆/∆ , Eμ‐myc and Eμ‐myc; Pot1b ∆/∆ mice for lymphoma development. While B‐cell lymphomas were never observed in Pot1b ∆/∆ mice, Eμ‐myc mice rapidly succumbed to fulminant B‐cell lymphoma with a median onset 19.5 weeks. In sharp contrast, 14 of 17 Eμ‐myc; Pot1b ∆/∆ mice remained tumor free by 55 weeks of age (P < 0.0001, Fig. 1A). H&E staining revealed that while malignant B cells from Eμ‐myc mice completely infiltrated the spleens and livers, lymphoma infiltrate was undetectable in Eμ‐myc; Pot1b ∆/∆ mice (Fig. 1B). To understand why deletion of Pot1b potently suppressed Myc‐induced lymphomagenesis, we used the B‐cell marker B220 to monitor the number of B cells in the bone marrows (BM) of similarly aged mouse cohorts. Compared to wild‐type (WT) mice, tumor‐free 50‐week‐old Eμ‐myc; Pot1b ∆/∆ mice displayed significant decrease in the percentage of B cells (22.4% ± 3.4% vs. 13.7% ± 1.1%, P = 0.02) (Fig. 1C). We postulated that this diminished B‐cell count could be due to either a block in Eμ‐myc; Pot1b ∆/∆ B‐cell differentiation or due to reduced cellular proliferation. To distinguish between these two possibilities, we first investigated the status of the B‐cell population in the BM of our mouse cohorts. Of the three stages of B cells (pre‐B, immature, and mature) that were evaluated, a significant expansion of the pre‐B‐cell compartment was detected in the BM from 6‐week‐old Eμ‐myc mice (Fig. 1D and Table 1). Expansion of this compartment is characteristic of Myc‐induced B‐cell activation (Harris et al., 1988) and was not observed in either WT or Pot1b ∆/∆ mice. A similar expansion of the pre‐B‐cell compartment was also observed in Eμ‐myc; Pot1b ∆/∆ mice, suggesting that malignant B‐cell clones, while present in Eμ‐myc; Pot1b ∆/∆ mice, were incapable of forming lymphomas (Fig. 1D and Table 1). This hypothesis was further reinforced when we deleted a single allele of p53 in this mouse cohort. In this genetic setting, fulminant B‐cell lymphoma infiltrating distant organs was detected in 100% of Eμ‐myc; Pot1b ∆/∆ ; p53 +/− mice, revealing that activation of p53‐dependent checkpoint by dysfunctional telomeres is essential to repress lymphomagenesis (Fig. S1A, Supporting information). We next asked whether pre‐B cells in the bone marrow of our mice were able to form colonies in vitro, a robust measure of cellular proliferative capacity. Compared to the number of colonies formed by Eμ‐myc B cells, Eμ‐myc; Pot1b ∆/∆ B cells formed significantly fewer number of colonies (265 ± 11 vs. 74 ± 4, P = 1.1 × 10−5) (Fig. 1E). In fact, the number of colonies formed by Eμ‐myc; Pot1b ∆/∆ B cells was similar to those formed by WT and Pot1b ∆/∆ B cells (WT: 67.6 ± 3.2; Pot1b ∆/∆: 51.3 ± 4.0). These data suggest that the potent suppression of lymphomagenesis in Eμ‐myc; Pot1b ∆/∆ mice was not due to diminished production of malignant pre‐B‐cell clones, but rather due to decreased proliferative capacity.
Figure 1.
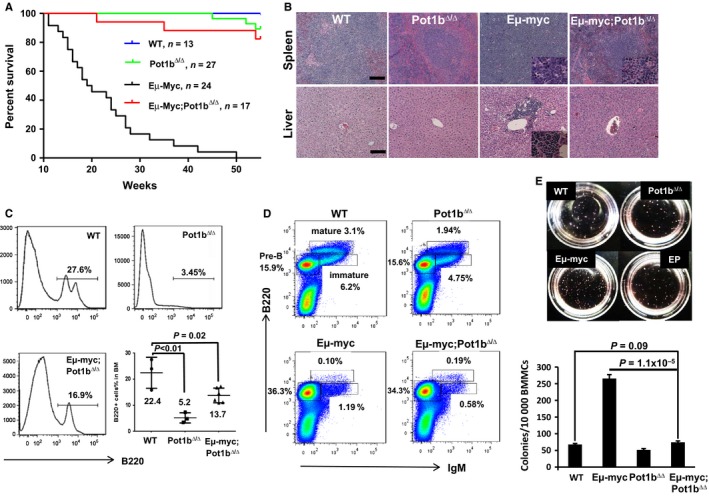
Inhibition of tumorigenesis in Eμ‐myc; Pot1b Δ/Δ mice promotes lifespan extension. (A) Kaplan–Meier analysis of tumor free survival of WT, Pot1b Δ/Δ, Eμ‐myc and Eμ‐myc; Pot1b Δ/Δ mice. All mouse cohorts were monitored for at least 55 weeks and sacrificed when moribund. A significant increase in lifespan of Eμ‐myc; Pot1b Δ/Δ over Eμ‐myc mice was observed (P < 0.0001). The log‐rank test was used to calculate statistical significance. (B) Hematoxylin and eosin (H&E) staining of tissues from Eμ‐myc mice demonstrates malignant B‐cell infiltration of the spleen and metastasis to the liver. Mice of other genotypes show normal spleen and liver architectures (magnification × 10). Scale bar: 25 μm. Insets show magnified (40 ×) malignant cells. (C) First three panels: representative FACS analysis of B220 positive cells in whole bone marrow (WBM) of 55 weeks old mice of the indicated genotypes. Significantly reduced number of B220+ cells were found in the Eμ‐myc Pot1b Δ/Δ mice compared with those of WT (P = 0.02). Last panel: summary of results. A two‐tailed Student's t test was used to calculate statistical significance. WT: n = 3. Pot1bΔ/Δ: n = 4. Eμ‐myc Pot1bΔ/Δ: n = 6. (D) Representative FACS analysis of pre‐B (B220+, IgM−), immature B (B220 lower, IgM+) and mature B cells (B220 higher, IgM+) in WBM from mice of the indicated genotypes. A summary of the data is shown in Table 1. Each experiment was repeated at least three times. (E) (Top) Representative images of colony forming assays after 12 days in culture in M3630 media specific for pre‐B cells growth. Cell genotypes are indicated. EP: Eμ‐myc; Pot1b Δ/Δ. (Bottom) Quantification of the number of colonies. No significant differences exist between WT and Eμ‐myc; Pot1b Δ/Δ cells (P = 0.09). There is a significant difference between Eμ‐myc and Eμ‐myc; Pot1b Δ/Δ cells (P = 1.1 × 10−5). Results are mean from three experiments, and error bars represent SEM. A two‐tailed Student's t test was used to calculate statistical significance.
Table 1.
B‐cell differentiation status in the bone marrow
| Genotype | Pre‐B | Immature | Mature | P value |
|---|---|---|---|---|
| WT | 17.2% ± 1.1% | 6.6% ± 0.81% | 3.7% ± 0.67% | |
| Pot1b∆/∆ | 15.4% ± 0.82% | 5.6% ± 1.51% | 2.7% ± 0.59% | 0.95a |
| Eμ‐myc | 37.6% ± 3.1% | 1.4% ± 0.45% | 0.10% ± 0.07% | 0.0023a |
| Eμ‐myc; Pot1b∆/∆ | 33.5% ± 4.0% | 0.86% ± 0.26% | 0.20% ± 0.06% | 0.0012a; 0.99b |
Compared with WT.
Compared with Eμ‐myc (by two‐way ANOVA).
Elevated telomere damage and activation of p53‐dependent cellular checkpoints in Eμ‐myc; Pot1b ∆/∆ B cells
To test the hypothesis that dysfunctional telomeres present in Eμ‐myc; Pot1b ∆/∆ B cells activate a p53‐dependent DNA damage response (DDR) to suppress proliferation of precursor oncogenic B‐cell clones, we sorted B220 + cells from the BM and used the dysfunctional telomere‐induced DNA damage (TIF) assay (d'Adda di Fagagna et al., 2003; Takai et al., 2003) to determine the functional status of telomeres in 6‐week‐old mice. As expected, the DNA damage marker γ‐H2AX did not associate appreciably with telomeres in B cells isolated from WT or Eμ‐myc mice (Fig. 2A). In contrast, a dramatic increase in the number of TIFs was observed in B cells from both Pot1b ∆/∆ and Eμ‐myc; Pot1b ∆/∆ mice. In total, 36% of Pot1b ∆/∆ B cells displayed 2–5 TIFs while 15% displayed more than 6 TIFs. In Eμ‐myc; Pot1b ∆/∆ B cells, 34% displayed 2–5 TIFs while 25% displayed more than 6 TIFs (P < 0.0001, Fig. 2B). As dysfunctional telomeres activate a p53‐dependent DDR, we next determined the expression of downstream targets of p53, including the cyclin‐dependent kinase inhibitor CDKN1A/p21WAF1/CIP1 (p21), BAX, and PUMA, in isolated B cells by real‐time RT–PCR. Compared to WT and Eμ‐myc B cells, where only a slight increase in the expression of p21, PUMA, and BAX was observed, a dramatic increase in the expression of p21, PUMA, and BAX was evident in Eμ‐myc; Pot1b ∆/∆ B cells (40.4‐fold increase for p21, 6.97‐fold increase for PUMA, and 5.5‐fold increase for BAX) (Figs 2C,D). Taken together, these results suggest that the chronic activation of both p53‐dependent cell cycle arrest/cellular senescence and apoptotic programs by dysfunctional telomeres lacking Pot1b may be responsible for the potent suppression of B‐cell lymphomagenesis observed in Eμ‐myc; Pot1b ∆/∆ mice.
Figure 2.
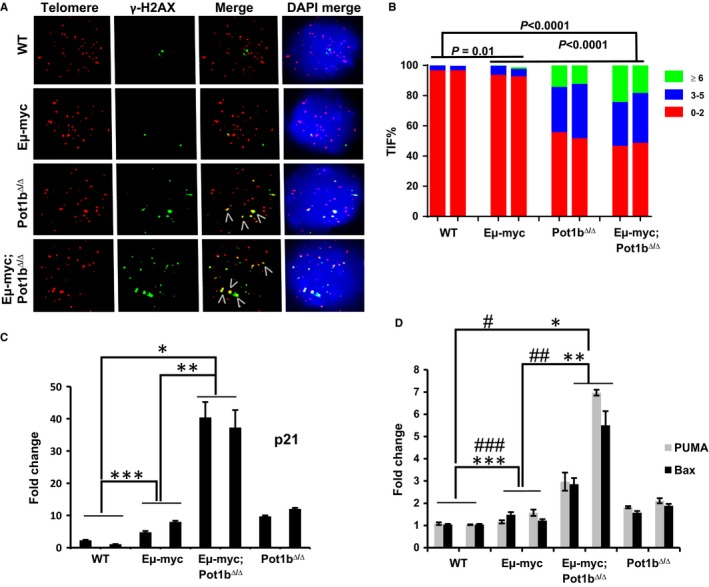
Enhanced DNA damage response in Eμ‐myc; Pot1b Δ/Δ B220+ cells. (A) Representative TIF images of B‐ cells from mice of the indicated genotypes. Cells were stained with anti‐ γ‐H2AX antibody (green), telomere PNA‐FISH probe [Tam‐OO‐(CCCTAA)4 (red)] and DAPI (blue). B‐cells were harvested from at least three 6–8 weeks old mice per genotype. (B) Quantification of percentage sorted B220+ cells displaying γ‐H2AX‐positive TIFs. A minimum of 150 nuclei were scored per genotype. A higher frequency of TIFs was detected in Eμ‐myc; Pot1b Δ/Δ B220+ cells compared with Eμ‐myc cells (P < 0.001). 2‐way ANOVA was performed to calculate statistical significance. (C) and (D) Real‐time PCR quantification of mRNA expression levels of p21 (C), PUMA and Bax (D) in sorted B220+ cells from 6 to 8 weeks old mice of the indicated genotypes. Each experiment was repeated in triplicate. Error bars represent SEM. 2‐way ANOVA was performed to calculate statistical significance. For (C), *P = 4.7 × 10−8, **P = 2.4 × 10−7 and ***P = 1.5 × 10−4. For (D), PUMA: # P = 1.5 × 10−4; ## P = 2.7 × 10−3; ### P = 1.4 × 10−2. BAX: *P = 6.7 × 10−4; **P = 1.4 × 10−3; ***P = 2.8 × 10−3.
Cellular proliferation is compromised in Pot1b ∆/∆ ; p53 R172P mice
To determine which antitumor program (apoptosis or cellular senescence) activated by dysfunctional telomeres is more important to suppress Eμ‐myc‐induced lymphomagenesis, we first generated and characterized the Pot1b∆/∆; p53R172P/R172P mice. The p53R172P point mutation substitutes the wild‐type p53 alleles with mutant p53R172P, in which arginine at position 172 is replaced with a proline residue. p53R172P (abbreviated as p53P) is the ortholog of a human p53 mutant that is completely defective for p53‐dependent apoptosis but still able to mediate cell cycle arrest/cellular senescence (Liu et al., 2004b; Post et al., 2010). Pot1b∆/∆; p53P/P mice do not develop spontaneous tumors but instead die prematurely (median lifespan of ~40 weeks, P < 0.0001) (Fig. 3A). Preliminary characterizations of aging phenotypes revealed that these mice were born smaller than WT, Pot1b ∆/∆ or p53 P/P littermate controls and display premature age‐related phenotypes, including diminished body size, kyphosis, and alopecia (Fig. S2A, Supporting information). Examination of total nucleated bone marrow (TBM) cells revealed that Pot1b ∆/∆ ; p53 P/P mice displayed significantly fewer proliferative TBM cells than those from WT littermates (6.92 ± 0.3 × 107 cells for WT; 4.03 ± 0.59 × 107 cells for Pot1b ∆/∆ ; p53 P/P, P = 2.1 × 10−5) (Fig. S3A, Supporting information). To investigate whether this proliferative defect was due to loss of hematopoietic stem cell populations, we examined Lin−, Sca‐1+, c‐kit+ (LSK) cells, a population enriched in hematopoietic stem cells and multipotent progenitors. FACS analysis revealed that 25.6% of Pot1b ∆/∆ and 23.4% of Pot1b ∆/∆ ; p53 P/P LSK cells were in S‐phase due to increased cellular proliferation as a consequence of enhanced depletion of this cellular compartment from telomere dysfunction‐induced apoptosis (16). In total, 11.4% of Pot1b ∆/∆ ; p53 P/P LSK cells, compared to 4.6% of Pot1b ∆/∆ LSK cells, were found in the G2/M phase of the cell cycle (Figs 3B,C and S3B). To ascertain whether uncapped telomeres contributed to this phenotype, we examined metaphase chromosome spreads isolated from WT, Pot1b ∆/∆; p53 P/P and Pot1b ∆/∆ ; p53 P/P TBM for signs of telomere dysfunction. Telomere PNA‐FISH analysis of age‐matched Pot1b ∆/∆ ; p53 P/P TBM metaphase spreads revealed a significant increase in the number of elevated end‐to‐end chromosome fusions and telomere signal‐free chromosome ends (Figs 3D,E and S3C). SA‐β‐galactosidase staining revealed significantly increased number of senescent Pot1b ∆/∆ ; p53 P/P BM cells (Fig. S2B,C). Western and RT–PCR analysis revealed significantly increased expression of p21 in Pot1b ∆/∆ ; p53 P/P LSK cells, splenocytes, and kidneys (Figs 3F,G and S2D). In addition, we observed increased p16 expression in Pot1b ∆/∆ ; p53 P/P LSK BM cells (Fig. 3H). Taken together, these results suggest that highly proliferative cells unable to activate p53‐dependent apoptosis respond to Pot1b deletion by promoting cellular senescence to compromise cellular proliferation.
Figure 3.
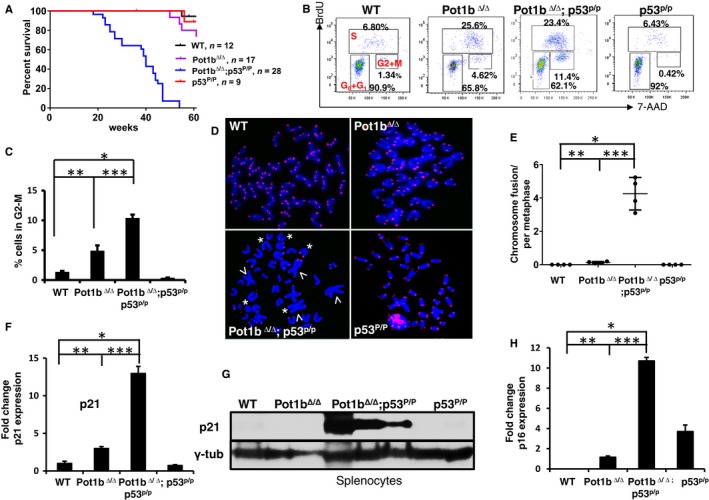
Deletion of Pot1b promotes cell cycle arrest and limits cellular proliferation. (A) Kaplan–Meier survival analysis of mice of the indicated genotypes. All mice were monitored for 60 weeks and sacrificed when moribund. A log‐rank test was used to calculate statistical significance. The lifespan of Pot1b Δ/Δ ; p53 P/P mice was significantly shorter than that of p53 P/P mice (P < 0.0001). (B) BrdU cell‐cycle analysis of LSK cells isolated from 30 to 35 weeks old mice of the indicated genotypes. Representative FACS plots from experiments with at least three mice in each genotype are shown. Percentage of cells in various phases of the cell cycle are indicated. For example, Pot1b Δ/Δ ; p53 P/P mice have 11.4% BM LSK cells in the G2/M phase of the cell cycle. (C) Quantification of cells in (B) undergoing G2/M cell cycle arrest. A two‐tailed student's t test was used to calculate statistical significance. P values: *P = 1.08 × 10−5; **P = 2.4 × 10−3; ***P = 8.1 × 10−4. (D) Telomere PNA‐FISH of metaphase spreads showing end‐to‐end chromosome fusions (arrowheads) and telomere signal‐free ends (SFEs) (*). Only representative SFEs are indicated. At least four mice were used for each genotype, and a minimum of 40 metaphases scored per genotype. (E) Quantification of chromosome fusions in (D). A two‐tailed student's t test was used to calculate statistical significance. P values: *P = 2.0 × 10−4; **P = 1.6 × 10−2; ***P = 1.0 × 10−4. (F) Real‐time RT‐PCR analysis of p21 expression levels in sorted LSK cells from 30 to 35 weeks old indicated mice. Each experiment was repeated in triplicate. Error bars represent SEM. A two‐tailed student's t test was used to calculate statistical significance. *P = 4.5 × 10−5; **P = 4.8 × 10−4; ***P = 2.4 × 10−5. (G) Western blot for p21expression in splenocytes from mice of indicated genotypes. γ‐tubulin was used as the loading control. (H) Real‐time RT‐PCR analysis of p16 expression levels in sorted LSK cells from 30 to 35 weeks old indicated mice. Each experiment was repeated in triplicate. Error bars represent SEM. A two‐tailed student's t test was used to calculate statistical significance. *P = 1.6 × 10−8; **P = 1.1 × 10−4; ***P = 7.9 × 10−7.
Complete suppression of lymphomagenesis in Eμ‐myc; Pot1b ∆/∆ mice requires the activation of both p53‐dependent cell cycle arrest/cellular senescence and apoptosis
To ascertain the importance of the p53‐dependent cell cycle arrest/cellular senescence pathway in the suppression of Eμ‐myc lymphomas, we crossed Eμ‐myc; Pot1b ∆/∆ mice with p21 −/− mice to generate Eμ‐myc; Pot1b ∆/∆ ; p21 +/− and Eμ‐myc; Pot1b ∆/∆ ; p21 −/− mice and monitored the formation of B‐cell lymphomas. Compared with Eμ‐myc; Pot1b∆/∆ mice, both Eμ‐myc; Pot1b ∆/∆ ; p21 +/− and Eμ‐myc; Pot1b ∆/∆ ; p21 −/− mice displayed significantly reduced tumor‐free survival, although the tumor‐free survival between the two p21 cohorts was not significant (Fig. 4A). This result shows that both alleles of p21 are required to completely repress lymphomagenesis observed in Eμ‐myc; Pot1b ∆/∆ mice. Telomere PNA‐FISH revealed that compared to Eμ‐myc lymphomas, ~35% of Eμ‐myc; Pot1b ∆/∆ ; p21 +/− chromosomes displayed telomere‐free ends and ~1.5 end‐to‐end chromosome fusions per metaphase, both indicative of increased telomere dysfunction (Fig. 4B,C). p53 expression was undetectable or aberrant in all Eμ‐myc; Pot1b ∆/∆ ; p21 +/− lymphomas examined, suggesting that p53 is mutated in these tumors (Fig. 4D). These results reinforce the notion that activation of cell cycle arrest/cellular senescence in Eμ‐myc; Pot1b ∆/∆ mice by uncapped telomeres is important to suppress B‐cell lymphomagenesis induced by Eμ‐myc oncogene expression.
Figure 4.
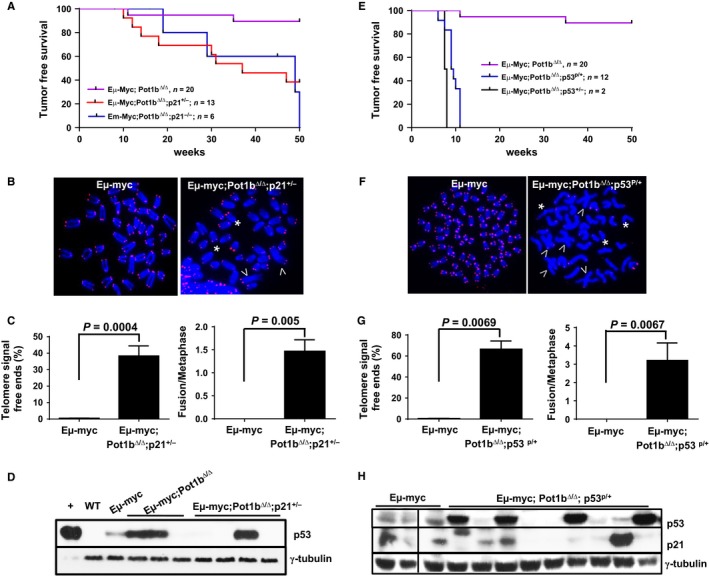
Both p53 dependent apoptosis and cellular senescence are required for complete tumor suppression in Eμ‐myc; Pot1b Δ/Δ mice. (A) Kaplan–Meier survival analysis of mice of the indicated genotypes, monitored over 50 weeks and sacrificed when moribund. Compared to the lifespan of Eμ‐myc; Pot1b Δ/Δ mice, the lifespan of Eμ‐myc; Pot1b Δ/Δ ; p21 +/− and Eμ‐myc; Pot1b Δ/Δ ; p21 −/− mice were significantly reduced (both P values < 0.01). The survival between Eμ‐myc; Pot1b Δ/Δ ; p21 +/− and Eμ‐myc; Pot1b Δ/Δ ; p21 −/− mice was not significantly different (P = 0.08). (B) Telomere FISH analysis of metaphase chromosome spreads from Eμ‐myc and Eμ‐myc; Pot1b Δ/Δ ; p21 +/− lymphomas using Tam‐OO‐(CCCTAA)4 (red) and DAPI (blue). A minimum of 50 metaphases were analyzed per genotype. Arrows indicate fused chromosomes; *indicates signal free chromosome ends. Only a few SFEs are shown. (C) Quantification of percentage of telomere SFEs (left) and chromosome fusions (right) in tumor metaphases shown in (B). Metaphases from lymphomas isolated from at least three different mice were examined. A two‐tailed Student's t test was used to calculate statistical significance. (D) Western blot for p53 expression in tumors isolated from mice of the indicated genotype. +: aphidocolin treated WT MEF cells, loaded at 20% the level of other samples. WT: normal splenocytes. γ‐tubulin was used as loading control. (E) Kaplan–Meier survival analysis of mice of the indicated genotypes, monitored over 50 weeks and sacrificed when moribund. The difference in survival between the Eμ‐myc; Pot1b Δ/Δ ; Trp53 P/+ mice and Eμ‐myc; Pot1b Δ/Δ mice was highly significant (P < 0.001). (F) Telomere FISH analysis of metaphase chromosome spreads of Eμ‐myc and Eμ‐myc; Pot1b Δ/Δ ; Trp53 P/+ lymphomas using Tam‐OO‐(CCCTAA)4 PNA (red) and DAPI (blue). A minimum of 50 metaphases were analyzed per genotype. Arrowheads indicate fused chromosomes; *indicate telomere SFEs. (G) (left) Quantification of percentage of telomere SFEs (left) and chromosome fusions (right) in tumor metaphases shown in (F). Metaphases from lymphomas isolated from at least three different mice were examined. A two‐tailed Student's t test was used to calculate statistical significance. (H) Western blot for p53 and p21 expression in tumors isolated from mice of the indicated genotypes. γ‐tubulin was used as loading control.
We next examined how activation of p53‐dependent apoptosis in Eμ‐myc; Pot1b ∆/∆ mice contributes to the suppression of lymphomagenesis. As indicated previously, Eμ‐myc; Pot1b ∆/∆ ; p53 +/− mice died by 8 weeks of age from fulminant, infiltrating B‐cell lymphoma, indicating that one allele of WT p53 was insufficient for complete tumor suppression in this genetic setting (Figs 4E and S1A). We next generated Eμ‐myc; Pot1b ∆/∆ ; p53 R172P/+ cohorts (we were unable to generate Eμ‐myc; Pot1b ∆/∆ ; p53 P/P cohorts despite repeated attempts). Given the tetrameric nature of the p53 complex, a combination of p53P and p53WT proteins will result in only 1 of 16 of the tetramers with fully WT p53 function, and 15 of 16 of the tetramers compromised in p53‐mediated apoptosis (but still competent to mediate cell cycle arrest/cellular senescence). Compared to Eμ‐myc; Pot1b ∆/∆ mice, Eμ‐myc; Pot1b ∆/∆ ; p53 R172P/+ mice exhibited dramatically reduced tumor‐free survival, with 12 of 12 mice succumbing to B‐cell lymphomas by 11 weeks of age (P < 0.001) (Figs 4E and S1B). All Eμ‐myc; Pot1b ∆/∆ ; p53 R172P/+ lymphomas examined displayed dysfunctional telomeres, manifested as telomere‐free chromosome ends involving ~70% of all chromosome ends and ~3 end‐to‐end chromosome fusions (Fig. 4F,G). Western analyses of 9 Eμ‐myc; Pot1b ∆/∆ ; p53 R172P/+ lymphomas revealed that the p53 is aberrantly expressed in 4 of 9 tumors and minimally expressed in the remaining 5. Sequence analysis revealed the presence of mutant p53 allele in 9 of 9 tumors, explaining the loss of the ability to regulate p21 expression (Fig. 4H and Table 2). Taken together, these results suggest that while complete suppression of Eμ‐myc‐induced lymphomagenesis in the setting of uncapped telomeres requires the activation of both p53‐dependent apoptosis and cellular senescence programs, p53‐dependent apoptosis plays a more prominent role for suppression of B‐cell lymphoma development in Eμ‐myc; Pot1b ∆/∆ mice.
Table 2.
DNA sequencing and protein expression of p53 in lymphomas
| Mouse number | Mouse genotype | p53 status in lymphoma | p53 protein expression |
|---|---|---|---|
| 169 | Eμ‐myc | p53+/+ | Low |
| 594 | Eμ‐myc | p53+/+ | Low |
| 1027 | Eμ‐myc | p53+/+ | Low |
| 1028 | Eμ‐myc | p53+/+ | Low |
| 6033 | Eμ‐myc; Pot1b∆/∆; p53p/+ | p53p/− | Very high |
| 6007 | Eμ‐myc; Pot1b∆/∆; p53p/+ | p53+/− | Very low |
| 5000 | Eμ‐myc; Pot1b∆/∆; p53p/+ | p53p/+ | Very high |
| 6011 | Eμ‐myc; Pot1b∆/∆; p53p/+ | p53+/− | No expression |
| 5320 | Eμ‐myc; Pot1b∆/∆; p53p/+ | p53+/− | No expression |
| 3171 | Eμ‐myc; Pot1b∆/∆; p53p/+ | p53p/+ | Very high |
| 2719 | Eμ‐myc; Pot1b∆/∆; p53p/+ | p53+/− | Low |
| 4036 | Eμ‐myc; Pot1b∆/∆; p53p/+ | p53+/− | Low |
| 2098 | Eμ‐myc; Pot1b∆/∆; p53p/+ | p53p/+ | Very high |
Telomeres without Pot1b activate p53‐independent apoptosis in highly proliferative organs
While chronic activation of p53‐dependent apoptosis and cell cycle arrest/cellular senescence programs by uncapped telomeres is potently tumor suppressive, we have previously shown that an undesirable consequence of Pot1b deletion is compromised stem cell proliferation, resulting in systemic multi‐organ failure and the onset of premature aging phenotypes, presumably due to activation of p53‐dependent proliferative checkpoints (Wang et al., 2011, 2013). We therefore postulated that deleting p53 in the setting of Pot1b deficiency should rescue these adverse phenotypes. Deletion of Pot1b did not impact upon the incidence of tumor formation in p53 −/− mice (Fig. 5A). However, we were surprised to find that compared to age‐matched Pot1b ∆/∆ mice, Pot1b ∆/∆ ; p53 −/− mice exhibited profound cellular proliferative defects in highly proliferative organs, including the testes and intestines. We found that compared to age‐matched Pot1b ∆/∆ mice, both the size and weight of testes from Pot1b ∆/∆ ; p53 −/− mice were significantly reduced (Figs 5B and S4A,B, Supporting information). Testicular weight reduction was even greater when the weights of age‐matched WT and Pot1b ∆/∆ ; p53 −/− testes were compared. Histological analyses revealed that Pot1b ∆/∆ ; p53 −/− testes displayed a complete absence of spermatogenesis in all seminiferous tubules examined. In contrast, most tubules in Pot1b ∆/∆ testes contained germ cells at various developmental stages (Figs 5C, S4C). Analysis of Pot1b ∆/∆ ; p53 P/P testes revealed a nearly identical phenotype of testicular dysfunction (Figs 5C and S4B,C). TUNEL staining revealed significantly increased number of apoptotic cells in both Pot1b ∆/∆ ; p53 −/− and Pot1b ∆/∆ ; p53 P/P testes compared with testes from both WT and Pot1b ∆/∆ mice (Fig. 5C). Examination of the highly proliferative intestinal epithelia by TUNEL and Caspase 3 staining revealed that compared to Pot1b ∆/∆ intestines, Pot1b ∆/∆ ; p53 −/− and Pot1b ∆/∆ ; p53 P/P intestinal crypt epithelia possess significantly increased number of apoptotic cells (Figs 5D,E and S4D). Finally, examination of TBM cells revealed increased apoptosis in cells isolated from Pot1b ∆/∆ ; p53 P/P mice (Figs 5F and S5A, Supporting information). In addition, RT–PCR analysis revealed that BAX expression was increased sixfold in Pot1b ∆/∆ ; p53 P/P cells (Fig. 5G). Together, these results reveal that in addition to the activation of p53‐dependent cell cycle checkpoints, telomeres devoid of Pot1b also activate p53‐independent apoptosis to limit the proliferative capacity of rapidly dividing tissues.
Figure 5.
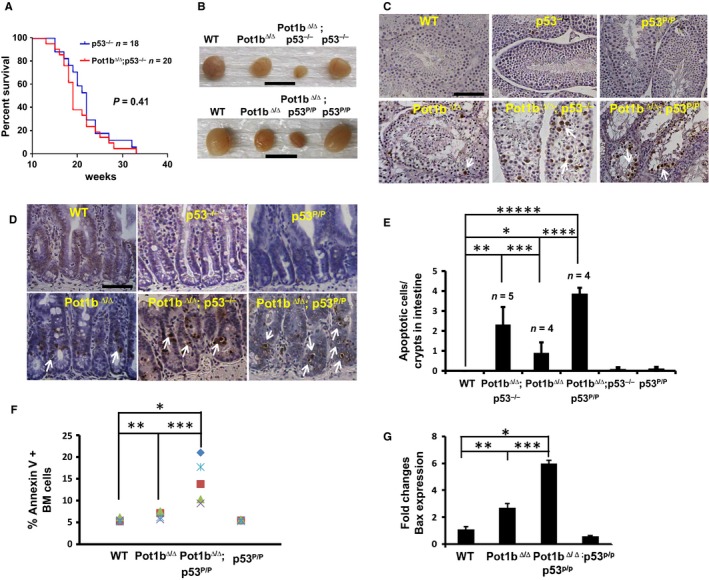
Loss of functional p53 accelerates proliferative defects in Pot1b Δ/Δ ; p53 −/− mice. (A) Kaplan–Meier survival analysis showing the percent survival of p53 −/− and Pot1b Δ/Δ ; p53 −/− mice. All mice were monitored for 40 weeks and sacrificed when moribund. The log‐rank test was used to calculate statistical significance. No significant survival difference was observed between the two cohorts (P = 0.41). (B) Testes from mice of the indicated genotypes. Scale bar: 10 mm. (C) TUNEL staining of testicular sections from mice of indicated genotypes. Magnification: 20×, scale bar: 50 μm. Representative TUNEL positive cells are indicated by arrowheads. (D) Representative TUNEL stained intestinal sections from mice of the indicated genotypes. Arrowheads indicate apoptotic cells. Scale bar: 25 μm. (E) Quantification of apoptotic cells in basal crypts of intestines from (D). A two‐tailed student's t test was used to calculate statistical significance. *P = 4.2 × 10−2; **P = 4.4 × 10−3; ***P = 4.6 × 10−2; ****P = 4.9 × 10−4; *****P = 4.6 × 10−6. (F) Annexin V staining of bone marrow cells isolated from indicated mouse cohorts at 30–35 weeks of age. BMs from a minimum of five mice per genotype were analyzed. A two‐tailed student's t test was used to calculate statistical significance. *P = 2.1 × 10−4; **P = 4.0 × 10−3; ***P = 9.0 × 10−3. (G) Real‐time RT‐PCR quantification of mRNA expression levels of Bax in sorted LSK cells from 30 to 35 weeks old mice of indicated genotypes. Each experiment was repeated in triplicate. Error bars represent SEM. A two‐tailed student's t test was used to calculate statistical significance. *P = 1.21 × 10−5; **P = 1.8 × 10−3; ***P = 1.0 × 10−4.
DNA damage and apoptosis in Pot1b ∆/∆ ; p53 mutant cells requires p73
Rb functions as a tumor suppressor and prevents cell cycle progression by binding to the E2F family of transcription factors. Inactivation of Rb can initiate E2F1‐mediated transactivation of pro‐apoptotic genes, resulting in p53‐independent apoptosis (Nahle et al., 2002; Polager & Ginsberg, 2008). Western analysis revealed that both E2F1 and the hypophosphorylated, active form of Rb are highly expressed in Pot1b ∆/∆ ; p53 −/− and Pot1b ∆/∆ ; p53 P/P splenocytes, suggesting that the Rb‐E2F1 pathway is functional in these cells (Fig. 6A,B). This notion is supported by increased p16 expression, which could promote the formation of active Rb‐E2F1 complexes to repress pro‐apoptotic genes. We therefore turned our attention to other pathways that activate p53‐independent apoptosis. In response to DNA damage, the checkpoint kinases Chk1 and Chk2, together with E2F1 and the p38 MAP kinase, all promote the activation and stabilization of the p53‐related tumor suppressor p73 (Sanchez‐Prieto et al., 2002; Urist et al., 2004). Like p53, p73 plays important roles in DNA damage‐initiated apoptosis (Flores et al., 2002, 2005). Western analysis of Pot1b ∆/∆ ; p53 −/− and Pot1b ∆/∆ ; p53 P/P splenocytes revealed robust up‐regulation of Chk1, E2F1, p73, p38 MAP kinase, suggesting that p73 is active in these cells (Fig. 6A,B). The level of transcriptional targets of p73, including BAX, PUMA, and Caspase 3, is also significantly increased at the RNA level in the absence of functional p53 (Fig. 6C). In contrast, anti‐apoptotic genes, including Bcl‐2 and Bcl‐XL, are not induced in Pot1b∆/∆; p53−/− splenocytes of following Pot1b deletion (Fig. S5B Supporting information). Importantly, control experiments reveal that expression of p21 was undetectable in Pot1b ∆/∆ ; p53 −/− splenocytes (Fig. S5C Supporting information), suggesting that cellular senescence is not prominently activated in this setting.
Figure 6.
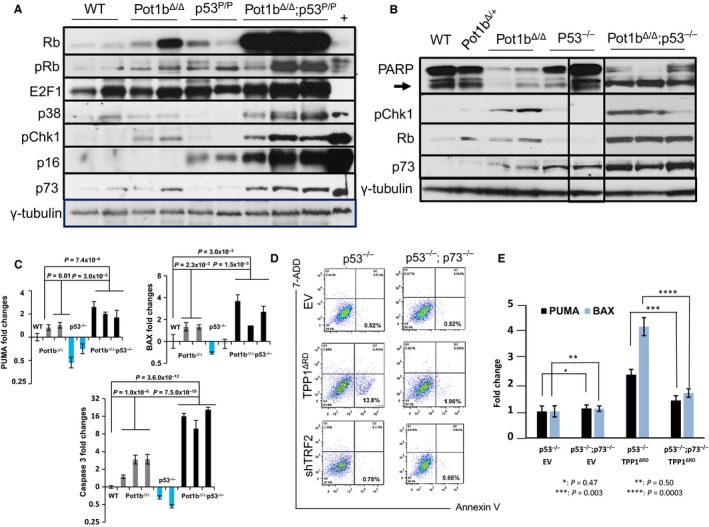
Activation of p53 independent apoptosis in Pot1b Δ/ ; p53 −/− mice. (A) Immunoblotting for total Rb, phosphorylated‐Rb, E2F1, p38 MAP kinase, phosphorylated‐Chk1, p16 and p73 expression in mouse spleen cells of the indicated genotypes. γ‐tubulin was used as the loading control. (B) Western analysis for PARP, cleaved PARP (arrow), phospho‐Chk1, total Rb and p73 expression in mouse spleen cells of the indicated genotypes. γ‐tubulin was used as the loading control. Samples were run on the same gel but the lanes were not all contiguous. (C) Real‐time RT‐PCR results showing the expression of PUMA (upper left), BAX (upper right) and Caspase 3 (lower) in spleen cells from WT, Pot1b Δ/Δ , p53 −/− and Pot1bΔ/Δ ; p53 −/− mice. Each experiment was repeated in triplicate. Error bars represent SEM. (D) Representative FACS analysis of Annexin V and 7‐AAD expression in p53 −/− and p53 −/− ; p73 −/− MEFs 72 h after treatment with empty vector (EV), TPP1ΔRD or shTRF2. p53 −/− MEFs treated with TPP1ΔRD displayed increased number of Annexin‐V+, 7‐AAD‐ cells, which was significantly reduced in p53 −/− ;p73 −/− MEFs. (E) Real‐time (RT)‐PCR analysis of PUMA and BAX expression profiles in p53 −/− and p53 −/− ; p73 −/− MEFs after 72 h of treatment with EV or TPP1ΔRD. Compared with p53−/− MEFs expressing TPP1ΔRD, PUMA and BAX expression was largely abrogated in p53 −/− ; p73 −/− MEFs expressing TPP1ΔRD. Each experiment was repeated in triplicate. Error bars represent SEM. A 2‐way ANOVA analysis was performed to calculate statistical significance.
Our results suggest that p53‐independent apoptosis is induced in diverse murine cell types by the removal of Pot1b from telomeres. In addition, a recent report revealed that deletion of p53 potently activates p73‐ via E2F‐1‐mediated transcription, suggesting that up‐regulation of p73 in p53−/− cells could play an important role in mediating p53‐independent apoptosis (Tophkhane et al., 2012). To further test this hypothesis, we utilized p53 −/− and p53 −/− ; p73 −/− MEFs (Flores et al., 2002) to ascertain the role of p73 in p53‐independent apoptosis. We rendered telomeres dysfunctional either by removing Pot1a/b from telomeres using the dominant negative molecule TPP1ΔRD (Deng et al., 2009) or by removing the Shelterin component TRF2. In contrast to removal of Pot1a/b, which generates single‐stranded telomeric G‐overhangs and preferentially activates an ATR‐Chk1‐dependent DDR, removal of TRF2 generates chromosome ends resembling double‐stranded DNA breaks and only activates an ATM‐Chk2‐dependent DDR (Denchi & de Lange, 2007; Guo et al., 2007; Rai et al., 2011). Cellular proliferation, as measured by both low density seeding and NIH 3T3 assays, was compromised in p53 −/− MEFs expressing TPP1ΔRD (Fig. S6A,B, Supporting information). In contrast, p53 −/− ; p73 −/− MEFs expressing TPP1ΔRD grew robustly. Deletion of p73 did not compromise TIF formation or number of fused chromosomes generated, indicating that p73 status does not impact upon DNA damage signaling or repair of uncapped telomeres (Fig. S6B,C Supporting information). FACS analysis for 7‐ADD and Annexin V staining revealed that p53 −/− MEFs expressing TPP1ΔRD exhibited increased apoptosis (13.8%). In contrast, apoptotic cells were observed in only 1.96% of p53 −/− ; p73 −/− MEFs expressing TPP1ΔRD (Fig. 6D). In support of this result, both Bax and PUMA expression levels, while elevated in p53 −/− MEFs expressing TPP1ΔRD, were reduced to background levels in p53 −/− ; p73 −/− MEFs (Fig. 6E).
To further ascertain the mechanisms underlying this activation of p73, we examined cellular growth in p53 −/− and p53 −/− ; p73 −/− MEFs depleted of TRF2. As shown in Figs 6D,E and S6A,B, depletion of TRF2 did not result in compromised cellular proliferation nor increased apoptosis in either p53 −/− nor p53 −/− ; p73 −/− MEFs. Taken together, these results suggest that removal of Pot1b from telomeres preferentially activates a p53‐independent, p73‐dependent apoptotic program to suppress growth.
p53‐independent apoptosis represses B‐cell, but not T‐cell lymphomagenesis
Previously reports reveal that p73 is able to activate Caspase 3 to induce apoptosis (Cottini et al., 2014). We found that Caspase 3‐mediated cleavage of PARP‐1 was readily detected in Pot1b ∆/∆ ; p53 −/− splenocytes, indicative of robust p53‐independent apoptosis (Fig. 6B). The observation of increased p‐Chk1 levels in Pot1b ∆/∆ ; p53 P/P and Pot1b ∆/∆ ; p53 −/− splenocytes, but not in WT or p53 −/− splenocytes, suggests that apoptosis was induced by dysfunctional telomeres eliciting an ATR‐Chk1‐dependent DDR. To confirm functionally that activation of Caspase 3 is important to limit proliferation of Pot1b ∆/∆ ; p53 −/− splenocytes, we expressed a dominant negative Caspase 3 mutant (MSCV‐Caspase 3C163A‐Myc–GFP) (Huang et al., 2011) in WT, p53 −/− and Pot1b ∆/∆ ; p53 −/− fetal liver stem cells and FACS sorted for cells robustly expressing mutant Caspase 3. The sorted cells were then cultured in media to specifically promote pre‐B‐cell differentiation and proliferation. Western analysis confirmed Caspase 3C163A‐Myc expression after 12 days of culture (Fig. 7A). Expression of Caspase 3C163A significantly improved the ability of Pot1b ∆/∆ ; p53 −/− pre‐B cells to form colonies in vitro, suggesting that p53‐independent activation of Caspase 3 limited B‐cell proliferation (Fig. 7B). In support of this notion, transplantation of Pot1b ∆/∆ ; p53 −/− hematopoietic cells expressing Caspase 3C163A‐Myc into SCID mice resulted in significantly increased induction of B‐cell lymphomas, suggesting that p53‐independent apoptosis normally represses lymphomagenesis through the up‐regulation of Caspase 3 (Fig. 7C). To strengthen this observation, we examined the impact that activation of p53‐independent apoptosis has on spontaneous tumorigenesis in our mouse cohorts. We found that while our cohort of p53−/− mice normally succumbed to B‐cell lymphomas derived from lymph nodes and spleens, a dramatic shift in tumor type to thymic lymphomas was observed in Pot1b ∆/∆ ; p53 −/− mice (Table 3). RT–PCR analysis revealed that in contrast to the high levels of PUMA and Bax transcripts observed in Pot1b ∆/∆ ; p53 −/− splenocytes, both transcripts were significantly reduced in thymocytes and in thymic lymphomas (Figs 6C–E and 7D,E). We also found that p73 expression was markedly reduced in thymic lymphomas (Fig. 7F). Taken together, these results suggest that activation of p53‐independent, p73‐dependent apoptosis in Pot1b ∆/∆ ; p53 −/− mice limited B‐cell proliferation to suppress B‐cell lymphomas but did not suppress the formation of thymic lymphomas.
Figure 7.
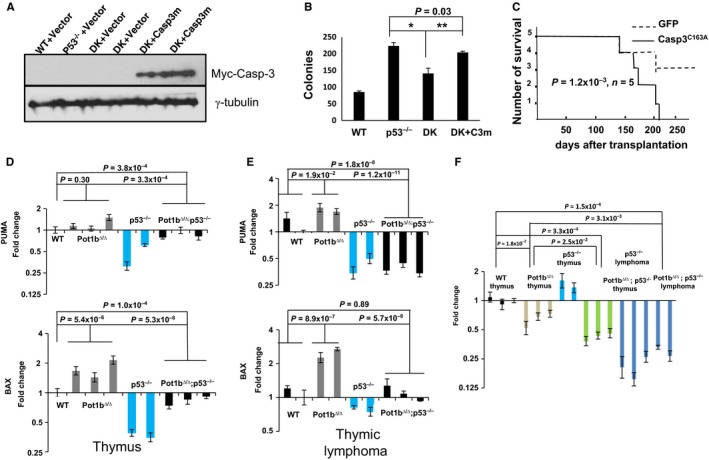
Activation of p53‐independent apoptosis represses B‐cell, but not T‐cell lymphomagenesis. (A) Immunoblotting for Myc‐Caspase‐3C163A expression in FACS sorted GFP+ fetal liver cells of the indicated genotypes. Vector: MSCV‐IRES‐GFP empty vector. Casp3m: MSCV‐Myc‐Caspase‐3C163A‐IRES‐GFP. DK: Pot1b Δ/Δ ; p53 −/− double knockout cells. γ‐tubulin was used as the loading control. (B) Quantification of in vitro colony forming assays for sorted GFP+ fetal liver cells in B‐cell specific M3630 medium. Results are mean from three independent experiments. Error bars represent SEM. A two‐tailed student's t test was used to calculate statistical significance*P = 2.1 × 10−3; **P = 3.1 × 10−3. (C) Kaplan–Meier survival analysis showing the percent survival of SCID mice transplanted with either Pot1b Δ/Δ ; p53 −/− fetal liver cells expressing empty vector or MSCV‐Myc‐Caspase‐3C163A‐IRES‐GFP. (D–G) Real‐time RTPCR analysis of PUMA and BAX expression profiles in thymus (D) and thymic lymphomas (E) from 6 weeks old mice of the indicated genotypes. Each experiment was repeated in triplicate. Error bars represent SEM. A 2‐way ANOVA analysis was performed to calculate statistical significance. (F) Real‐time RT‐PCR analysis of p73 expression profiles in 6 weeks thymus and thymic lymphomas of the indicated genotypes. Each experiment was repeated in triplicate. Error bars represent SEM. A 2‐way ANOVA analysis was performed to calculate statistical significance.
Table 3.
Tumor profile of p53−/ − and Pot1bΔ/ Δ; p53−/ − mice
| p53−/− | Pot1bΔ/Δ; p53−/− | P value | |
|---|---|---|---|
| Lymphoma total | 11 | 13 | 0.80 |
| Thymic lymphomas | 3 | 10 | 0.01 |
| Other tumors | 6 | 4 | 0.35 |
| No gross tumors | 1 | 4 | 0.19 (Chi‐square test) |
| Total | 18 | 20 |
Discussion
The incidence of human cancers is inextricably linked with advancing age. Increased DNA damage and accumulation of somatic mutations is the major driving force behind cancer development in the elderly. Mutations in p53 involve more than half of all human cancers, highlighting p53′s importance in tumor suppression. Understanding mechanistically how induction of p53 suppresses tumor formation is therefore of significant interest in an increasingly aged population. Here, we use an established mouse model of Burkett's B‐cell lymphoma to show that dysfunctional telomeres devoid of the shelterin component Pot1b potently activate a p53‐dependent DDR to almost completely suppress Myc oncogene‐driven B‐cell lymphomagenesis. By generating Eμ‐myc; Pot1b ∆/∆ ; p53 P/+ mice compromised in the induction of p53‐dependent apoptosis and Eμ‐myc; Pot1b ∆/∆ ; p21 −/− mice unable to initiate p53‐dependent cellular senescence, we are able to directly compare the tumor suppressive contributions of p53‐mediated apoptosis vs. cellular senescence. While we and others have previously shown that activation of the p53‐dependent senescence program by dysfunctional telomeres is critical to suppress tumorigenesis (Cosme‐Blanco et al., 2007; Feldser & Greider, 2007), this is the first time that the effects of telomere‐induced senescence and apoptosis are directly compared side by side in mouse littermates. Our results indicate that while cooperation of both programs is required to fully suppress oncogene‐induced lymphomagenesis, dysfunctional telomere‐induced apoptosis plays a more important role than cellular senescence in suppressing tumorigenesis. In addition, our studies uncover a role for the p53‐ortholog p73 in mediating tumor suppression in the absence of p53. Telomeres devoid of Pot1b are able to activate a p53‐independent, p73‐dependent apoptotic program to prevent B‐cell lymphoma development. The data presented here highlight the various p53‐dependent and independent antiproliferative mechanisms employed by mammalian cells to combat dysfunctional telomere‐induced genomic instability and the generation of lymphomas.
The p53 family members p63 and p73 share many features with p53, including transcriptionally regulating a number of genes in response to DNA damage (Yang & McKeon, 2000; Irwin & Kaelin, 2001; Lin et al., 2009). Unlike p53, both p63 and p73 are structurally complex, with multiple isoforms functioning both as oncogenes and tumor suppressors. For example, the p73 gene has two promoters that results in the generation of transactivating (TA) and the ΔN form of p73 (Yang et al., 1998; Grob et al., 2001). Both the TA and ΔN isoforms of p73 can transactivate target genes to induce apoptosis and growth arrest following DNA damage (Irwin et al., 2003; Liu et al., 2004a). Importantly, in the absence of p53, p73 isoforms are able to induce apoptosis and growth arrest (Willis et al., 2003; Lin et al., 2009; Tophkhane et al., 2012). While p53‐dependent apoptosis is highly tumor suppressive in our mouse lymphoma models, we found that highly proliferative tissues from Pot1b ∆/∆ ; p53 −/− mice also displayed robust TUNEL staining and activation of pro‐apoptotic factors, including BAX, PUMA, and Caspase 3. This p53‐independent, p73‐dependent apoptosis was able to inhibit spontaneous B‐cell lymphomagenesis normally observed in p53 −/− mice. Transplantation of Pot1b ∆/∆ ; p53 −/− B cells expressing a dominant negative form of Caspase 3 promotes lymphomagenesis in SCID mice, further supporting the hypothesis that dysfunctional telomeres induced by Pot1b loss activates p53‐independent apoptosis to suppress the formation of B‐cell lymphomas. However, p53‐independent apoptosis induced by Pot1b deletion also compromises organ homeostasis, promoting the onset of premature age‐related phenotypes.
We have previously shown that in the setting of Pot1b deficiency, increased generation of single‐stranded telomeric DNA promotes the activation of the ATR‐Chk1 checkpoint kinases (Guo et al., 2007; He et al., 2009; Flynn et al., 2011; Thanasoula et al., 2012). We now propose a model in which telomeres devoid of Pot1b activates an ATR‐dependent DNA damage response (DDR), activation of Chk1 stabilizes E2F1, and both proteins transcriptionally activate p73 (Sanchez‐Prieto et al., 2002; Urist et al., 2004; Tophkhane et al., 2012) (Fig. S7, Supporting information). Consistent with its p53‐independent tumor suppressive function, the levels of p73 targets including BAX, PUMA, and Caspase 3 are significantly increased in Pot1b ∆/∆ ; p53 −/− spleens Pot1b deficient telomeres induce p73 to activate BAX and PUMA, as the expression of both of these pro‐apoptotic genes is abrogated in p53 −/− ; p73 −/− MEFs. Activation of p73 serves to restrain the progression toward cancer in hematopoietic tissues. However, it also induced undesirable effects, including premature onset of age‐related phenotypes. That an ATR‐Chk1‐dependent DDR may be required to potently activate p53‐independent, p73‐dependent apoptosis is supported by our observation that telomeres devoid of TRF2, which activates an ATM‐Chk2‐dependent DDR (Rai et al., 2010), did not induce apoptosis in either p53 −/− or p53 −/− ; p73 −/− MEFs. p73 has been shown to upregulate several DNA repair genes, including Rad51, BRCA2, and MRE11, and these are also the same ones needed to repair dysfunctional telomeres lacking functional Pot1a/b proteins (Lin et al., 2009; Rai et al., 2010). Taken together, our data suggest that initiation of p73‐dependent apoptosis may be uniquely dependent upon the activation of distinct types of DNA damage responses and repair pathways in precursor cancer cells.
Interestingly, p53‐independent apoptosis was observed to preferentially limit the development of B‐cell but not in thymic lymphomas, resulting in decreased formation of B‐cell lymphomas and an increase in the generation of spontaneous thymic lymphomas in Pot1b ∆/∆ ; p53 −/− mice. This tissue‐specific activation of BAX and PUMA by telomeres devoid of Pot1b in B cells but not in T cells is not currently understood. We speculate that the ATR‐Chk1‐dependent DDR pathway is not activated in Pot1b ∆/∆ ; p53 −/− T cells, resulting in lymphomagenesis.
Expression of mutant forms of the telomerase protein or the shelterin component TIN2 is able to promote cell death in a p53‐independent manner (Hahn et al., 1999; Guiducci et al., 2001; Kim et al., 2008). In addition, telomerase null mice experience increased sterility as their telomeres shorten, and this phenotype is not recused by deleting p53 (Chin et al., 1999). Because perturbation of telomere homeostasis in p53 −/− cells and in late generation mTerc−/−; p53−/− mice results in increased chromosomal fusions, it is generally believed that it is this elevated genomic instability that results in eventual cell death (Begus‐Nahrmann et al., 2009; Sperka et al., 2012b) (Fig. S7). Our data suggest that in addition to elevated chromosomal instability, dysfunctional telomeres lacking Pot1b also activate an ATR‐Chk1 DDR pathway to induce p53‐independent, p73‐dependent apoptosis. Activation of p73 has been recently shown to restrain tumorigenesis in the absence of functional p53, suggesting that this could represent a novel strategy to specifically target p53‐deficient tumors (Venkatanarayan et al., 2015).
Materials and methods
Mice
Eμ‐myc, Pot1b Δ/Δ, Eμ‐myc; Pot1b Δ/Δ, Pot1b Δ/Δ; p21 −/−, Pot1b Δ/Δ; p53 −/−, Pot1b Δ/Δ; p53 P/P mice were generated and maintained according to the IACUC‐approved protocols of Yale University (Harris et al., 1988; Liu et al., 2003; He et al., 2009; Wang et al., 2010; Sperka et al., 2012a). To generate Eμ‐myc; Pot1b Δ/Δ mice, we crossed Eμ‐myc mice with Pot1b Δ/+ mice to get Eμ‐myc; Pot1b Δ/+ mice. We then crossed Eμ‐myc; Pot1b Δ/+ mice with Pot1b Δ/+ mice to get Eμ‐myc; Pot1b Δ/Δ mice. To generate Eμ‐myc; Pot1b Δ/Δ ; p53 −/− mice, we crossed Eμ‐myc; Pot1b Δ/+ mice with Pot1b Δ/+ ; p53 +/− or Pot1b Δ/+ ; p53 −/− mice to get Eμ‐myc; Pot1b Δ/Δ ; p53 +/− mice. We then crossed Eμ‐myc; Pot1b Δ/Δ ; p53 +/− mice with Pot1b Δ/Δ ; p53 +/− mice to get Eμ‐myc; Pot1b Δ/Δ ; p53 −/− mice. To generate Eμ‐myc; Pot1b Δ/Δ ; p53 P/+ mice, we crossed Eμ‐myc; Pot1b Δ/+ mice with Pot1b Δ/+ ; p53 P/P mice to get Eμ‐myc; Pot1b Δ/Δ ; p53 P/+ mice. We then crossed Eμ‐myc; Pot1b Δ/Δ ; p53 P/+ mice with Pot1b Δ/Δ ; p53 P/+ mice to get Eμ‐myc; Pot1b Δ/Δ ; p53 P/+ mice. To generate Eμ‐myc; Pot1b Δ/Δ ; p21 +/− and Eμ‐myc; Pot1b Δ/Δ ; p21 −/− mice, we crossed Eμ‐myc; Pot1b Δ/+ mice with Pot1b Δ/+ ; p21 −/− mice to get Eμ‐myc; Pot1b Δ/Δ ; p21 +/− and Eμ‐myc; Pot1b Δ/+ ; p21 +/− mice. We then crossed Eμ‐myc; Pot1b Δ/+ ; p21 +/− mice with Pot1b Δ/+ ; p21 −/− mice to get Eμ‐myc; Pot1b Δ/Δ ; p21 −/− mice.
Histology and TUNEL assay
Tissues were fixed in 10% formalin, paraffin embedded, sectioned at 5 μm thickness and stained with H&E. The terminal deoxynucleotidyltransferase‐mediated dUTP‐biotin nick end labeling (TUNEL) assay was performed using the ApopTag Plus peroxidase in situ apoptosis detection kit (Chemicon, 28820 Single Oak Drive, Temecula, CA 92590, USA) according to the manufacturer's instructions.
Flow cytometry analysis
To detect lineage markers on the bone marrow (BM) cells, 1 × 107 BM cells were centrifuged and resuspended in 200 μL of HBSS+ (Invitrogen, 542 Flynn Road, Camarillo CA 93012, USA), then stained for 15 min with antibodies. After staining, cells were washed, resuspended in HBSS+, and analyzed by flow cytometry (LSRII; BD, 2350 Qume Drive San Jose, CA 95131, USA). For B‐cell sorting, cells were stained with B220 (RM‐2601; Caltag, 5791 Van Allen Way Carlsbad, CA 92008 USA) and IgM (Caltag). For LSK cell staining, a cocktail of antibodies including nine lineage markers was used as follows: Ter‐119, CD3, CD4, CD8, B220, CD19, IL‐7Rα, GR‐1, and Mac‐1 (eBioscience, 10255 Science Center Dr, San Diego, CA 92121, USA) conjugated with APC‐Cy‐7 (47‐4317; eBioscience). The cocktail also contained anti‐Sca‐1‐PE (12‐598; eBioscience) and anti‐c‐Kit‐APC (17‐1171; eBioscience). LSK populations were selected based on low or negative expression of the mature lineage markers and dual‐positive expression for Sca‐1 and c‐Kit. To determine the proliferative status of LSK cells, BrdU was injected intraperitoneally into mice at a dose of 150 mg/kg body weight 2 h before euthanization. Analysis of BrdU incorporation was performed using the FITC BrdU Flow Kit (BD). To quantitatively determine the percentage of cells that were actively undergoing apoptosis, the Annexin V‐PE Apoptosis Detection Kit (BD Pharmingen, 2350 Qume Drive San Jose, CA 95131, USA) was used.
Isolation of total BM cells and colony‐forming assay
Hindlimb bones were dissected, and the marrow was flushed through a 21‐gauge needle into HBSS+, 2% FBS (Invitrogen), and 10 mM HEPES. The cells were passed through a 25‐gauge needle twice and filtered (40‐μm filter, BD Falcon™, BD Biosciences, 2350 Qume Drive San Jose, CA 95131 ) to ensure a single‐cell suspension. Nucleated cells were counted manually after the lysis of RBCs with 3% acetic acid in methylene blue (STEMCELL Technologies, 570 West Seventh Avenue, Suite 400, Vancouver, BC, V5Z 1B3, Canada). For the colony‐forming assay, 1 × 104 BM mononucleated cells (BM MNCs) or 3 × 104 GFP+ cells were cultured in 35‐mm dishes containing MethylCult 3630 (STEMCELL Technologies) following the manufacturer's protocols. The colonies were counted on day 8.
PNA‐FISH and TIF and immunofluorescence‐FISH assays
Cells were treated with 0.5 μg/mL of colcemid for 2–4 h before harvest. Metaphase chromosomes from BM were prepared 1–2 h after colcemid treatment and subjected to telomere peptide nucleic acid FISH staining to label telomeres. Depending on the quality of metaphase spreads, 20–50 metaphases from each sample were analyzed in detail. Cells were treated with 0.6M KCl, fixed with methanol: acetic acid (3:1), and spread on glass slides. Metaphase spreads were hybridized with 5′‐Tam‐OO‐(CCCTAA) 4‐3′ probe (Panagene, 54, Techno 10‐ro Yuseong‐gu Daejeon, Korea). For the TIF assay, cells were seeded in the 8‐well chambers and immunostained with primary antibody and FITC‐secondary antibody, then hybridized with 5′‐Tam‐OO‐(CCCTAA) 4‐3′ probe. TIF analysis in LSK cells and B220 + cells was performed as previously described (Wang et al., 2011). All microscopy was performed with a Nikon Eclipse 80i and an Andore CCD camera utilizing Metamorph software.
Reverse transcriptase coupled RT–PCR
Total RNA was prepared using QIAGEN's RNeasy Micro kit (QIAGEN, 27220 Turnberry Lane Suite 200 Valencia, CA 91355) according to the manufacturer's instructions. For first‐strand cDNA synthesis, 1 μg of total RNA, 20 pmol of Oligo (dT)12–18, and 200 units of SuperScript II Reverse Transcriptase (Invitrogen) were mixed in a final volume of 20 μL. Synthesized cDNA (1 μL) was added to a 20‐μL PCR mixture containing TaqMan Gene Expression Assay primers (Applied Biosystems, 850 Lincoln Centre Drive Foster City, CA 94404, USA.) and TaqMan Universal PCR Master Mix. Each sample was amplified in triplicate. PCR consisted of 40 cycles of denaturation at 95 °C for 15 s, annealing, and amplification at 60 °C for 60 s in an ABI7900HT Sequence Detection System machine (Applied Biosystems). The specific primers for the TaqMan Gene Expression Assay primers were as follows: PUMA: Mm00519268_m1; BAX: Mm00432050_m1; p21:Mm00432448_ml; FasL: Mm00438864_ml; Caspase 3: Mm01195085_ml. p16: Mm01257348_m1 p73:Mm00660220_ml. Bcl2:Mm00477631_m1. Bcl‐xL:Mm00437783_m1. 18S rRNA primers (4319413E or Mm00519571_ml) were used as internal controls.
Western blot analysis
The antibodies used for Western blot analysis were as follows: phospho‐p53 (Ser15) (Cell Signaling Technology, 3 Trask Lane Danvers, MA 01923, USA; 1:500); phospho‐Chk1 (Cell Signaling Technology; 1:500), anti‐mouse p21 (sc‐397; Santa Cruz Biotechnology Inc., 10410 Finnell Street, Dallas, Texas 75220, USA,1:500), anti‐phospho‐Rb (sc‐56174; Santa Cruz Biotechnology Inc., 1:500), antitotal Rb (sc‐74562; Santa Cruz Biotechnology Inc., 1:500), anti‐E2F1 (sc‐251; Santa Cruz Biotechnology Inc., 1:500), anti‐p38 (sc‐535; Santa Cruz Biotechnology Inc., 1:500), anti‐p73 (sc‐7957; Santa Cruz Biotechnology Inc., 1:500), anti‐Myc (Millipore, 290 Concord Road, Billerica, MA 01821, USA; 1:500), anti‐PARP (Cell signaling 1:500), and anti‐p16 (Abcam, 1 Kendall Square, Suite B2304, Cambridge, MA 02139, USA, 1:500). Anti‐mouse γ‐tubulin (Sigma‐Aldrich, PO Box 14508, St. Louis, MO 63178, USA; 1:10 000) was used as a loading control.
Plasmids
The pcDNA3‐Casp3C163A‐myc plasmid 11814 was purchased from Addgene. The Casp3C163A‐myc insert was subcloned into the BamHI site of MSCV‐IRES‐EGFP (MIG) vector, designated as MSCV‐Caspase 3C163A‐Myc–GFP. Retrovirus production and infection method were performed as described previously (Wang et al., 2011).
MEFs culture and proliferation assays
The 3T3 proliferation assays for MEFs were performed following standard protocols. For colony‐forming assay, 1 × 103 cells were seeded in 6‐well tissue culture plates and cultured for 1 week. Colonies were fixed with ice‐cold methanol and stained with crystal violet (0.5% in 25% methanol).
Statistics
Statistical analyses were performed using GraphPad Prism, version 6 software (GraphPad Software Inc., 7825 Fay Avenue, Suite 230, La Jolla, CA 92037, USA) or Microsoft Excel. Data sets were compared using ANOVA or the Student's t‐test (unpaired, 2‐tailed). Kaplan–Meier survival analyses were performed using the log‐rank test. P values of 0.05 or less were considered significant.
Study approval
The animal care and use program at Yale University maintains full accreditation from the Association for Assessment and Accreditation of Laboratory Animal Care (AAALAC) and complies with U.S. Animal Welfare Regulations, the National Research Council (NRC) Guide for the Care and Use of Laboratory Animals, and Public Health Service Policy on Humane Care and Use of Laboratory Animals. Yale University has an approved Animal Welfare Assurance (#A3230‐01) on file with the NIH Office for Protection from Research Risks. The Assurance was approved May 16, 2014. My mouse personnel have multiple years of experience working with mice. Our veterinarian Dr. Peter Smith is always available should health concerns arise. My animal protocols have been approved by the animal care and use program at Yale University (2010‐11358) for 3 years and renewed annually.
Author contributions
YW and SC conceived the project. YW and XW conducted the experiments, and ERF supplied essential reagents. JY and SC interpreted the data. YW and SC wrote the paper.
Funding
This work was supported by U01DK085570 and ACS RGS‐10‐124‐01‐CCE to JY, R01CA134796, R01CA160394, and CPRIT 140271 to E.R. F., and RO1 CA129037, RO1CA202816, R21CA200506, R21CA202816, and CT 2015‐0901 to S.C.
Conflicts of interest
The authors declare no conflict of interests.
Supporting information
Fig. S1. Representative images of (A) Eμ‐myc; Pot1b ∆/∆; p53 +/− and (B) Eμ‐myc; Pot1b ∆/∆ ; p53 P/+ mice.
Fig. S2. (A) Images of WT, Pot1b∆/∆, Pot1b∆/∆; p53P/P and Pot1b∆/∆; p53−/− mice at the indicated ages. (B) SA‐β‐galactosidase staining and (C) quantification of SA‐β‐galactosidase positive cells in the bone marrows of mice of the indicated genotypes. (D) Western analysis for p21expression in mouse kidney cells. γ‐tubulin was used as the loading control.
Fig. S3. (A) Quantification of total BM nucleated cell counts in 30–35 weeks old mice of the indicated genotypes. (B) Representative FACS analysis of Sca‐1 and C‐kit positive cell populations in 30–35 weeks old mouse BMs of the indicated genotypes.
Fig. S4. (A) Quantification of testicular weights from 20 weeks old mice of the indicated genotypes. (B) Quantification of testicular weights from 30 to 35 weeks old mice of the indicated genotypes. (C) H&E stained testicular sections from mice of the indicated genotypes. (D) Representative photographs of Caspase 3 staining of intestinal sections.
Fig. S5. (A) Representative histograms showing Annexin V profiles of mouse bone marrow cells isolated from 30 to 35 week old mice of the indicated genotypes. (B) Real‐time RT‐PCR analysis of Bcl2 and BCL‐xL expression profiles in spleenocytes from mice of the indicated genotypes.
Fig. S6. (A) Left panel: Representative image of colony forming assays for p53−/− and p53−/−; p73−/− MEFs 72 h after treatment with empty vector (EV), TPP1ΔRD or shTRF2. (B) Left panel: Immunostaining for γ‐H2AX‐positive dysfunctional telomere‐induced DNA damage foci (TIFs) following 72 h expression of TPP1ΔRD in p53−/− or p53−/−; p73−/− MEFs. (C) Left panel: Telomere PNA‐FISH of metaphase spreads showing end‐to‐end chromosome fusions (arrows).
Fig. S7. Model of genetic interactions discussed in the test.
Acknowledgments
We thank members of the Chang laboratory for helpful suggestions.
References
- d'Adda di Fagagna F, Reaper PM, Clay‐Farrace L, Fiegler H, Carr P, Von Zglinicki T , Saretzki G, Carter NP, Jackson SP (2003) A DNA damage checkpoint response in telomere‐initiated senescence. Nature 426, 194–198. [DOI] [PubMed] [Google Scholar]
- Artandi SE, Chang S, Lee SL, Alson S, Gottlieb GJ, Chin L, DePinho RA (2000) Telomere dysfunction promotes non‐reciprocal translocations and epithelial cancers in mice. Nature 406, 641–645. [DOI] [PubMed] [Google Scholar]
- Baumann P, Cech TR (2001) Pot1, the putative telomere end‐binding protein in fission yeast and humans. Science 292, 1171–1175. [DOI] [PubMed] [Google Scholar]
- Begus‐Nahrmann Y, Lechel A, Obenauf AC, Nalapareddy K, Peit E, Hoffmann E, Schlaudraff F, Liss B, Schirmacher P, Kestler H, Danenberg E, Barker N, Clevers H, Speicher MR, Rudolph KL (2009) p53 deletion impairs clearance of chromosomal‐instable stem cells in aging telomere‐dysfunctional mice. Nat. Genet. 41, 1138–1143. [DOI] [PubMed] [Google Scholar]
- Bennardo N, Cheng A, Huang N, Stark JM (2008) Alternative‐NHEJ is a mechanistically distinct pathway of mammalian chromosome break repair. PLoS Genet. 4, e1000110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chin L, Artandi SE, Shen Q, Tam A, Lee SL, Gottlieb GJ, Greider CW, DePinho RA (1999) p53 deficiency rescues the adverse effects of telomere loss and cooperates with telomere dysfunction to accelerate carcinogenesis. Cell 97, 527–538. [DOI] [PubMed] [Google Scholar]
- Cosme‐Blanco W, Shen MF, Lazar AJ, Pathak S, Lozano G, Multani AS, Chang S (2007) Telomere dysfunction suppresses spontaneous tumorigenesis in vivo by initiating p53‐ dependent cellular senescence. EMBO Rep. 8, 497–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cottini F, Hideshima T, Xu C, Sattler M, Dori M, Agnelli L, ten Hacken E, Bertilaccio MT, Antonini E, Neri A, Ponzoni M, Marcatti M, Richardson PG, Carrasco R, Kimmelman AC, Wong KK, Caligaris‐Cappio F, Blandino G, Kuehl WM, Anderson KC, Tonon G (2014) Rescue of Hippo coactivator YAP1 triggers DNA damage‐induced apoptosis in hematological cancers. Nat. Med. 20, 599–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denchi EL, de Lange T (2007) Protection of telomeres through independent control of ATM and ATR by TRF2 and POT1. Nature 448, 1068–1071. [DOI] [PubMed] [Google Scholar]
- Deng Y, Chan SS, Chang S (2008) Telomere dysfunction and tumour suppression: the senescence connection. Nat. Rev. Cancer 8, 450–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng Y, Guo X, Ferguson DO, Chang S (2009) Multiple roles for MRE11 at uncapped telomeres. Nature 460, 914–918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldser DM, Greider CW (2007) Short telomeres limit tumor progression in vivo by inducing senescence. Cancer Cell 11, 461–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flores ER, Tsai KY, Crowley D, Sengupta S, Yang A, McKeon F, Jacks T (2002) p63 and p73 are required for p53‐dependent apoptosis in response to DNA damage. Nature 416, 560–564. [DOI] [PubMed] [Google Scholar]
- Flores ER, Sengupta S, Miller JB, Newman JJ, Bronson R, Crowley D, Yang A, McKeon F, Jacks T (2005) Tumor predisposition in mice mutant for p63 and p73: evidence for broader tumor suppressor functions for the p53 family. Cancer Cell 7, 363–373. [DOI] [PubMed] [Google Scholar]
- Flynn RL, Centore RC, O'Sullivan RJ, Rai R, Tse A, Songyang Z, Chang S, Karlseder J, Zou L (2011) TERRA and hnRNPA1 orchestrate an RPA‐to‐POT1 switch on telomeric single‐ stranded DNA. Nature 471, 532–536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez‐Suarez E, Samper E, Flores JM, Blasco MA (2000) Telomerase‐deficient mice with short telomeres are resistant to skin tumorigenesis. Nat. Genet. 26, 114–117. [DOI] [PubMed] [Google Scholar]
- Greider CW (1996) Telomere length regulation. Annu. Rev. Biochem. 65, 337–365. [DOI] [PubMed] [Google Scholar]
- Grob TJ, Novak U, Maisse C, Barcaroli D, Luthi AU, Pirnia F, Hugli B, Graber HU, De Laurenzi V, Fey MF, Melino G, Tobler A (2001) Human delta Np73 regulates a dominant negative feedback loop for TAp73 and p53. Cell Death Differ. 8, 1213–1223. [DOI] [PubMed] [Google Scholar]
- Guiducci C, Cerone MA, Bacchetti S (2001) Expression of mutant telomerase in immortal telomerase‐negative human cells results in cell cycle deregulation, nuclear and chromosomal abnormalities and rapid loss of viability. Oncogene 20, 714–725. [DOI] [PubMed] [Google Scholar]
- Guo X, Deng Y, Lin Y, Cosme‐Blanco W, Chan S, He H, Yuan G, Brown EJ, Chang S (2007) Dysfunctional telomeres activate an ATM‐ATR‐dependent DNA damage response to suppress tumorigenesis. EMBO J. 26, 4709–4719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hahn WC, Stewart SA, Brooks MW, York SG, Eaton E, Kurachi A, Beijersbergen RL, Knoll JH, Meyerson M, Weinberg RA (1999) Inhibition of telomerase limits the growth of human cancer cells. Nat. Med. 5, 1164–1170. [DOI] [PubMed] [Google Scholar]
- Halazonetis TD, Gorgoulis VG, Bartek J (2008) An oncogene‐induced DNA damage model for cancer development. Science 319, 1352–1355. [DOI] [PubMed] [Google Scholar]
- Harris AW, Pinkert CA, Crawford M, Langdon WY, Brinster RL, Adams JM (1988) The E mu‐ myc transgenic mouse. A model for high‐incidence spontaneous lymphoma and leukemia of early B cells. J. Exp. Med. 167, 353–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He H, Multani AS, Cosme‐Blanco W, Tahara H, Ma J, Pathak S, Deng Y, Chang S (2006) POT1b protects telomeres from end‐to‐end chromosomal fusions and aberrant homologous recombination. EMBO J. 25, 5180–5190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He H, Wang Y, Guo X, Ramchandani S, Ma J, Shen M‐F, Garcia DA, Deng Y, Multani AS, You MJ, Chang S (2009) Pot1b Deletion and Telomerase Haploinsufficiency in Mice Initiate an ATR‐Dependent DNA Damage Response and Elicit Phenotypes Resembling Dyskeratosis Congenita. Mol. Cell. Biol. 29, 229–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hockemeyer D, Daniels JP, Takai H, de Lange T (2006) Recent expansion of the telomeric complex in rodents: two distinct POT1 proteins protect mouse telomeres. Cell 126, 63–77. [DOI] [PubMed] [Google Scholar]
- Hockemeyer D, Palm W, Wang RC, Couto SS, de Lange T (2008) Engineered telomere degradation models dyskeratosis congenita. Genes Dev. 22, 1773–1785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Q, Li F, Liu X, Li W, Shi W, Liu FF, O'Sullivan B, He Z, Peng Y, Tan AC, Zhou L, Shen J, Han G, Wang XJ, Thorburn J, Thorburn A, Jimeno A, Raben D, Bedford JS, Li CY (2011) Caspase 3‐mediated stimulation of tumor cell repopulation during cancer radiotherapy. Nat. Med. 17, 860–866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irwin MS, Kaelin WG (2001) p53 family update: p73 and p63 develop their own identities. Cell Growth Differ. 12, 337–349. [PubMed] [Google Scholar]
- Irwin MS, Kondo K, Marin MC, Cheng LS, Hahn WC, Kaelin WG Jr (2003) Chemosensitivity linked to p73 function. Cancer Cell 3, 403–410. [DOI] [PubMed] [Google Scholar]
- Kim SH, Davalos AR, Heo SJ, Rodier F, Zou Y, Beausejour C, Kaminker P, Yannone SM, Campisi J (2008) Telomere dysfunction and cell survival: roles for distinct TIN2‐ containing complexes. J. Cell. Biol. 181, 447–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirkwood TB (2008) Gerontology: healthy old age. Nature 455, 739–740. [DOI] [PubMed] [Google Scholar]
- Lin YL, Sengupta S, Gurdziel K, Bell GW, Jacks T, Flores ER (2009) p63 and p73 transcriptionally regulate genes involved in DNA repair. PLoS Genet. 5, e1000680. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Liu G, Xia T, Chen X (2003) The activation domains, the proline‐rich domain, and the C‐terminal basic domain in p53 are necessary for acetylation of histones on the proximal p21 promoter and interaction with p300/CREB‐binding protein. J. Biol. Chem. 278, 17557–17565. [DOI] [PubMed] [Google Scholar]
- Liu G, Nozell S, Xiao H, Chen X (2004a) DeltaNp73beta is active in transactivation and growth suppression. Mol. Cell. Biol. 24, 487–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu G, Parant JM, Lang G, Chau P, Chavez‐Reyes A, El‐Naggar AK, Multani A, Chang S, Lozano G (2004b) Chromosome stability, in the absence of apoptosis, is critical for suppression of tumorigenesis in Trp53 mutant mice. Nat. Genet. 36, 63–68. [DOI] [PubMed] [Google Scholar]
- Loayza D, Parsons H, Donigian J, Hoke K, de Lange T (2004) DNA binding features of human POT1: a nonamer 5′‐TAGGGTTAG‐3′ minimal binding site, sequence specificity, and internal binding to multimeric sites. J. Biol. Chem. 279, 13241–13248. [DOI] [PubMed] [Google Scholar]
- Martinez P, Blasco MA (2010) Role of shelterin in cancer and aging. Aging Cell 9, 653–666. [DOI] [PubMed] [Google Scholar]
- Nahle Z, Polakoff J, Davuluri RV, McCurrach ME, Jacobson MD, Narita M, Zhang MQ, Lazebnik Y, Bar‐Sagi D, Lowe SW (2002) Direct coupling of the cell cycle and cell death machinery by E2F. Nat. Cell Biol. 4, 859–864. [DOI] [PubMed] [Google Scholar]
- Nandakumar J, Podell ER, Cech TR (2010) How telomeric protein POT1 avoids RNA to achieve specificity for single‐stranded DNA. Proc. Natl Acad. Sci. USA 107, 651–656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Sullivan RJ, Karlseder J (2010) Telomeres: protecting chromosomes against genome instability. Nat. Rev. Mol. Cell Biol. 11, 171–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palm W, de Lange T (2008) How shelterin protects mammalian telomeres. Annu. Rev. Genet. 42, 301–334. [DOI] [PubMed] [Google Scholar]
- Palm W, Hockemeyer D, Kibe T, de Lange T (2009) Functional dissection of human and mouse POT1 proteins. Mol. Cell. Biol. 29, 471–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polager S, Ginsberg D (2008) E2F – at the crossroads of life and death. Trends Cell Biol. 18, 528–535. [DOI] [PubMed] [Google Scholar]
- Post SM, Quintas‐Cardama A, Pant V, Iwakuma T, Hamir A, Jackson JG, Maccio DR, Bond GL, Johnson DG, Levine AJ, Lozano G (2010) A high‐frequency regulatory polymorphism in the p53 pathway accelerates tumor development. Cancer Cell 18, 220–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quesada V, Conde L, Villamor N, Ordonez GR, Jares P, Bassaganyas L, Ramsay AJ, Bea S, Pinyol M, Martinez‐Trillos A, Lopez‐Guerra M, Colomer D, Navarro A, Baumann T, Aymerich M, Rozman M, Delgado J, Gine E, Hernandez JM, Gonzalez‐Diaz M, Puente DA, Velasco G, Freije JM, Tubio JM, Royo R, Gelpi JL, Orozco M, Pisano DG, Zamora J, Vazquez M, Valencia A, Himmelbauer H, Bayes M, Heath S, Gut M, Gut I, Estivill X, Lopez‐Guillermo A, Puente XS, Campo E, Lopez‐Otin C (2012) Exome sequencing identifies recurrent mutations of the splicing factor SF3B1 gene in chronic lymphocytic leukemia. Nat. Genet. 44, 47–52. [DOI] [PubMed] [Google Scholar]
- Rai R, Zheng H, He H, Luo Y, Multani A, Carpenter PB, Chang S (2010) The function of classical and alternative non‐homologous end‐joining pathways in the fusion of dysfunctional telomeres. EMBO J. 29, 2598–2610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rai R, Li JM, Zheng H, Lok GT, Deng Y, Huen MS, Chen J, Jin J, Chang S (2011) The E3 ubiquitin ligase Rnf8 stabilizes Tpp1 to promote telomere end protection. Nat. Struct. Mol. Biol. 18, 1400–1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramsay AJ, Quesada V, Foronda M, Conde L, Martinez‐Trillos A, Villamor N, Rodriguez D, Kwarciak A, Garabaya C, Gallardo M, Lopez‐Guerra M, Lopez‐Guillermo A, Puente XS, Blasco MA, Campo E, Lopez‐Otin C (2013) POT1 mutations cause telomere dysfunction in chronic lymphocytic leukemia. Nat. Genet. 45, 526–530. [DOI] [PubMed] [Google Scholar]
- Robles‐Espinoza CD, Harland M, Ramsay AJ, Aoude LG, Quesada V, Ding Z, Pooley KA, Pritchard AL, Tiffen JC, Petljak M, Palmer JM, Symmons J, Johansson P, Stark MS, Gartside MG, Snowden H, Montgomery GW, Martin NG, Liu JZ, Choi J, Makowski M, Brown KM, Dunning AM, Keane TM, Lopez‐Otin C, Gruis NA, Hayward NK, Bishop DT, Newton‐Bishop JA, Adams DJ (2014) POT1 loss‐of‐function variants predispose to familial melanoma. Nat. Genet. 46, 478–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudolph KL, Millard M, Bosenberg MW, DePinho RA (2001) Telomere dysfunction and evolution of intestinal carcinoma in mice and humans. Nat. Genet. 28, 155–159. [DOI] [PubMed] [Google Scholar]
- Sanchez‐Prieto R, Sanchez‐Arevalo VJ, Servitja JM, Gutkind JS (2002) Regulation of p73 by c‐ Abl through the p38 MAP kinase pathway. Oncogene 21, 974–979. [DOI] [PubMed] [Google Scholar]
- Sfeir A, de Lange T (2012) Removal of shelterin reveals the telomere end‐protection problem. Science 336, 593–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi J, Yang XR, Ballew B, Rotunno M, Calista D, Fargnoli MC, Ghiorzo P, Bressac‐de Paillerets B, Nagore E, Avril MF, Caporaso NE, McMaster ML, Cullen M, Wang Z, Zhang X, Group NDCSW, Laboratory NDCGR, French Familial Melanoma Study G , Bruno W, Pastorino L, Queirolo P, Banuls‐Roca J, Garcia‐Casado Z, Vaysse A, Mohamdi H, Riazalhosseini Y, Foglio M, Jouenne F, Hua X, Hyland PL, Yin J, Vallabhaneni H, Chai W, Minghetti P, Pellegrini C, Ravichandran S, Eggermont A, Lathrop M, Peris K, Scarra GB, Landi G, Savage SA, Sampson JN, He J, Yeager M, Goldin LR, Demenais F, Chanock SJ, Tucker MA, Goldstein AM, Liu Y, Landi MT (2014). Rare missense variants in POT1 predispose to familial cutaneous malignant melanoma. Nat. Genet. 46, 482–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simsek D, Jasin M (2010) Alternative end‐joining is suppressed by the canonical NHEJ component Xrcc4‐ligase IV during chromosomal translocation formation. Nat. Struct. Mol. Biol. 17, 410–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sperka T, Song Z, Morita Y, Nalapareddy K, Guachalla LM, Lechel A, Begus‐Nahrmann Y, Burkhalter MD, Mach M, Schlaudraff F, Liss B, Ju Z, Speicher MR, Rudolph KL (2012a) Puma and p21 represent cooperating checkpoints limiting self‐renewal and chromosomal instability of somatic stem cells in response to telomere dysfunction. Nat. Cell Biol. 14, 73–79. [DOI] [PubMed] [Google Scholar]
- Sperka T, Wang J, Rudolph KL (2012b) DNA damage checkpoints in stem cells, ageing and cancer. Nat. Rev. Mol. Cell Biol. 13, 579–590. [DOI] [PubMed] [Google Scholar]
- Symington LS, Gautier J (2011) Double‐strand break end resection and repair pathway choice. Annu. Rev. Genet. 45, 247–271. [DOI] [PubMed] [Google Scholar]
- Takai H, Smogorzewska A, de Lange T (2003) DNA damage foci at dysfunctional telomeres. Curr. Biol. 13, 1549–1556. [DOI] [PubMed] [Google Scholar]
- Thanasoula M, Escandell JM, Suwaki N, Tarsounas M (2012) ATM/ATR checkpoint activation downregulates CDC25C to prevent mitotic entry with uncapped telomeres. EMBO J. 31, 3398–3410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tophkhane C, Yang SH, Jiang Y, Ma Z, Subramaniam D, Anant S, Yogosawa S, Sakai T, Liu WG, Edgerton S, Thor A, Yang X (2012) p53 inactivation upregulates p73 expression through E2F‐1 mediated transcription. PLoS One 7, e43564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urist M, Tanaka T, Poyurovsky MV, Prives C (2004) p73 induction after DNA damage is regulated by checkpoint kinases Chk1 and Chk2. Genes Dev. 18, 3041–3054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkatanarayan A, Raulji P, Norton W, Chakravarti D, Coarfa C, Su X, Sandur SK, Ramirez MS, Lee J, Kingsley CV, Sananikone EF, Rajapakshe K, Naff K, Parker‐Thornburg J, Bankson JA, Tsai KY, Gunaratne PH, Flores ER (2015) IAPP‐driven metabolic reprogramming induces regression of p53‐deficient tumours in vivo . Nature 517, 626–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Klumpp S, Amin HM, Liang H, Li J, Estrov Z, Zweidler‐McKay P, Brandt SJ, Agulnick A, Nagarajan L (2010) SSBP2 is an in vivo tumor suppressor and regulator of LDB1 stability. Oncogene 29, 3044–3053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Shen MF, Chang S (2011) Essential roles for Pot1b in HSC self‐renewal and survival. Blood 118, 6068–6077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Sharpless N, Chang S (2013) p16(INK4a) protects against dysfunctional telomere‐ induced ATR‐dependent DNA damage responses. J. Clin. Investig. 123, 4489–4501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willis AC, Pipes T, Zhu J, Chen X (2003) p73 can suppress the proliferation of cells that express mutant p53. Oncogene 22, 5481–5495. [DOI] [PubMed] [Google Scholar]
- Wu L, Multani AS, He H, Cosme‐Blanco W, Deng Y, Deng JM, Bachilo O, Pathak S, Tahara H, Bailey SM, Behringer RR, Chang S (2006) Pot1 deficiency initiates DNA damage checkpoint activation and aberrant homologous recombination at telomeres. Cell 126, 49–62. [DOI] [PubMed] [Google Scholar]
- Yang A, McKeon F (2000) P63 and P73: P53 mimics, menaces and more. Nat. Rev. Mol. Cell Biol. 1, 199–207. [DOI] [PubMed] [Google Scholar]
- Yang A, Kaghad M, Wang Y, Gillett E, Fleming MD, Dotsch V, Andrews NC, Caput D, McKeon F (1998) p63, a p53 homolog at 3q27‐29, encodes multiple products with transactivating, death‐inducing, and dominant‐negative activities. Mol. Cell 2, 305–316. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Representative images of (A) Eμ‐myc; Pot1b ∆/∆; p53 +/− and (B) Eμ‐myc; Pot1b ∆/∆ ; p53 P/+ mice.
Fig. S2. (A) Images of WT, Pot1b∆/∆, Pot1b∆/∆; p53P/P and Pot1b∆/∆; p53−/− mice at the indicated ages. (B) SA‐β‐galactosidase staining and (C) quantification of SA‐β‐galactosidase positive cells in the bone marrows of mice of the indicated genotypes. (D) Western analysis for p21expression in mouse kidney cells. γ‐tubulin was used as the loading control.
Fig. S3. (A) Quantification of total BM nucleated cell counts in 30–35 weeks old mice of the indicated genotypes. (B) Representative FACS analysis of Sca‐1 and C‐kit positive cell populations in 30–35 weeks old mouse BMs of the indicated genotypes.
Fig. S4. (A) Quantification of testicular weights from 20 weeks old mice of the indicated genotypes. (B) Quantification of testicular weights from 30 to 35 weeks old mice of the indicated genotypes. (C) H&E stained testicular sections from mice of the indicated genotypes. (D) Representative photographs of Caspase 3 staining of intestinal sections.
Fig. S5. (A) Representative histograms showing Annexin V profiles of mouse bone marrow cells isolated from 30 to 35 week old mice of the indicated genotypes. (B) Real‐time RT‐PCR analysis of Bcl2 and BCL‐xL expression profiles in spleenocytes from mice of the indicated genotypes.
Fig. S6. (A) Left panel: Representative image of colony forming assays for p53−/− and p53−/−; p73−/− MEFs 72 h after treatment with empty vector (EV), TPP1ΔRD or shTRF2. (B) Left panel: Immunostaining for γ‐H2AX‐positive dysfunctional telomere‐induced DNA damage foci (TIFs) following 72 h expression of TPP1ΔRD in p53−/− or p53−/−; p73−/− MEFs. (C) Left panel: Telomere PNA‐FISH of metaphase spreads showing end‐to‐end chromosome fusions (arrows).
Fig. S7. Model of genetic interactions discussed in the test.