

Abstract
Currently, no plague vaccine exists in the United States for human use. The capsular antigen (Caf1 or F1) and two type 3 secretion system (T3SS) components, the low-calcium-response V antigen (LcrV) and the needle protein YscF, represent protective antigens of Yersinia pestis. We used a replication-defective human type 5 adenovirus (Ad5) vector and constructed recombinant monovalent and trivalent vaccines (rAd5-LcrV and rAd5-YFV) that expressed either the codon-optimized lcrV or the fusion gene designated YFV (consisting of ycsF, caf1, and lcrV). Immunization of mice with the trivalent rAd5-YFV vaccine by either the intramuscular (i.m.) or the intranasal (i.n.) route provided protection superior to that with the monovalent rAd5-LcrV vaccine against bubonic and pneumonic plague when animals were challenged with Y. pestis CO92. Preexisting adenoviral immunity did not diminish the protective response, and the protection was always higher when mice were administered one i.n. dose of the trivalent vaccine (priming) followed by a single i.m. booster dose of the purified YFV antigen. Immunization of cynomolgus macaques with the trivalent rAd5-YFV vaccine by the prime-boost strategy provided 100% protection against a stringent aerosol challenge dose of CO92 to animals that had preexisting adenoviral immunity. The vaccinated and challenged macaques had no signs of disease, and the invading pathogen rapidly cleared with no histopathological lesions. This is the first report showing the efficacy of an adenovirus-vectored trivalent vaccine against pneumonic plague in mouse and nonhuman primate (NHP) models.
INTRODUCTION
Yersinia pestis is the causative agent of plague and can be transmitted to humans via an infected flea bite or by direct inhalation of the aerosolized bacilli from an infected person or an animal (1, 2). Plague manifests itself in three major forms in humans, namely, bubonic, septicemic, and pneumonic (2). Pneumonic plague is the most feared form due to its rapid onset and associated high mortality rate (1, 2). Y. pestis has been responsible for at least three pandemics in the past, which killed more than 200 million people (3). Current epidemiological records suggest that there are 4,000 human plague cases annually worldwide (2). The emergence of multi-antibiotic-resistant Y. pestis strains from plague patients and the potential of malicious dissemination of recombinantly engineered bacteria as an airborne bioweapon necessitate the development of an effective preexposure and/or postexposure prophylaxis treatment (1, 2).
Currently, no Food and Drug Administration (FDA)-licensed plague vaccine exists in the United States, and recent efforts have focused on the development of recombinant subunit plague vaccines consisting of two well-characterized Y. pestis antigens, the F1 capsular antigen and the type 3 secretion system (T3SS) component and effector LcrV (4–8). F1, encoded by the caf1 gene, has a polymeric structure and confers bacterial resistance to phagocytosis (9). The F1-V-based vaccines are generally protective against pneumonic plague in rodents and nonhuman primates (NHPs) and are currently undergoing clinical trials (10–17). However, considering the natural existence of fully virulent F1-negative Y. pestis strains (18, 19) or those that have highly diverged LcrV variants (20, 21), such F1-V-based vaccines would most likely not provide optimal protection across all plague-causing Y. pestis strains in humans.
In an effort to search for new immunogenic antigens for the plague subunit vaccines, recent studies have shown that vaccination of mice with recombinant T3SS needle structure protein YscF (rYscF) provided protection to mice against subcutaneous injection of the fully virulent and encapsulated Y. pestis strain CO92 and against an intravenously injected pigmentation locus-negative Y. pestis KIM strain (22, 23). Therefore, the protective antigen YscF could be used in combination with F1 and LcrV to formulate a more effective new-generation trivalent (YFV) vaccine against plague.
In this study, we used a replication-defective human type-5 adenovirus (Ad5) vector to construct recombinant monovalent and trivalent (rAd5-LcrV and rAd5-YFV) vaccines that expressed either the lcrV or the fusion gene designated YFV (ycsF, caf1, and lcrV). We demonstrated that the trivalent rAd5-YFV vaccine provided protection to immunized mice that was superior to that with the monovalent rAd5-LcrV vaccine against both bubonic and pneumonic plague, irrespective of whether or not the preexisting adenoviral immunity was artificially developed in these animals. Most importantly, one dose of the trivalent rAd5-YFV vaccine by the intranasal (i.n.) route in conjunction with a single dose of the purified recombinant fusion protein rYFV by the intramuscular (i.m.) route in a prime-boost strategy provided impressive (up to 100%) protection to both mice and cynomolgus macaques (CMs) against high challenge doses of wild-type (WT) strain CO92 when given by the aerosol route. Vaccinated NHPs rapidly cleared the pathogen, with no signs of disease and histopathological lesions in various organs.
MATERIALS AND METHODS
Bacterial strains and reagents.
Y. pestis strain CO92 (WT CO92) was isolated in 1992 from a fatal human pneumonic plague case and acquired through BEI Resources, Manassas, VA. The bioluminescent WT Y. pestis CO92 luc2 strain (WT CO92 luc2), which contains the luciferase operon (luc or lux), allowing in vivo imaging of mice for bacterial dissemination in real time, was previously constructed in our laboratory (24, 25). Y. pestis strains were grown in heart infusion broth (HIB) medium (Difco, Voigt Global Distribution Inc., Lawrence, KS) at 26 to 28°C with constant agitation (180 rpm) or on either 1.5% HIB agar or 5% sheep blood agar (SBA) plates (Teknova, Hollister, CA). For the aerosol challenge, WT CO92 was grown in HIB enriched with 0.2% xylose (dl-xylose; Sigma-Aldrich, St. Louis, MO) as we previously described (26). Luria-Bertani (LB) medium was used for growing Escherichia coli at 37°C with agitation. Restriction endonucleases and T4 DNA ligase were obtained from Promega (Madison, WI). Advantage cDNA PCR kits were purchased from Clontech (Palo Alto, CA). All digested plasmid DNA or DNA fragments from agarose gels were purified using QIAquick kits (Qiagen, Inc., Valencia, CA).
Production and purification of recombinant proteins.
Genes encoding YscF, Caf1 (F1), and LcrV were amplified from the genome of WT CO92 by PCR with the primer sets YscFHis_F.cln (CACATATGAGTAACTTCTCTGGATTTACGAAAG) and YscFHis_R.cln (CACTCGAGTGGGAACTTCTGTAGGATGCCTT), Caf1His_F.cln (CACATATGAAAAAAATCAGTTCCGTTATCG) and Caf1His_R.cln (CACTCGAGTTGGTTAGATACGGTTACGGTTACAG), and LcrVHis_F.cln (CACATATGATTAGAGCCTACGAACAAAACCC) and LcrVHis_R.cln (CAGTCGACTTTACCAGACGTGTCATCTAGCAGAC), respectively. The underlines denote the restriction enzyme sites in the primers. The amplified genes were individually cloned into the pET20b+ vector at the NdeI and XhoI restriction enzyme sites, which resulted in attaching a histidine (His) tag at the C terminus of each of the gene products. In addition, the yscF, caf1, and lcrV fusion gene (YFV) was synthetically constructed by Epoch Biolabs, Inc. (Houston, TX) after codon optimization for E. coli by using the Blue Heron Biotechnology (Bothell, WA) online service. A flexible linker of 3 GGGGS sequences between the YscF, Caf1 (F1), and LcrV domains was added to facilitate correct folding of the fusion protein. The fusion gene was similarly cloned into the pET20b+ vector with a His tag attached to the C terminus of the YFV protein. Individual genes or the fusion gene was expressed from E. coli BL21(DE3) (New England BioLabs, Ipswich, MA) after induction with 0.5 mM IPTG (isopropyl-beta-d-thiogalactopyranoside) for 4 h at 37°C. The recombinant proteins (rYscF, rF1, rLcrV, and rYFV) were then purified by using Ni2+-charged agarose (27). The recombinant F1 and LcrV fusion protein (rF1-V) was purchased from the BEI Resources and used as a control for some of the experiments.
Construction of recombinant adenoviruses.
The lcrV and the YFV fusion genes were codon optimized for expression in humans by using the Blue Heron Biotechnology online service, which also allowed us to optimize secondary structures of the corresponding RNAs and removal of unwanted sites for the restriction enzymes, except for those used for cloning purposes. The resulting constructs were designed to produce LcrV (37.2 kDa), as well as the YFV fusion protein consisting of YscF (9.5 kDa), the mature form of F1 (15.6 kDa), and LcrV (37.2 kDa), interconnected via a flexible linker, as mentioned above. To improve expression of the corresponding genes, the Kozak consensus sequence was also placed upstream of the start codon. The constructs were then synthesized and verified via DNA sequence analysis by Epoch Biolabs, Inc. Each synthetic construct was cloned into the pShuttleX vector (Clontech Laboratories, Inc., Mountain View, CA) under the control of a cytomegalovirus (CMV) promoter.
To generate recombinant adenoviruses, the above gene constructs with their CMV promoters were removed from the pShuttleX vector and cloned into the replication-defective human type 5 adenovirus plasmid vector Adeno-X (Clonetech Laboratories, Inc.). The adenoviral constructs were created at the Baylor College of Medicine (BCM), Vector Development Laboratory, Houston, TX (https://www.bcm.edu/centers/cell-and-gene-therapy/research/vector-development/vector-services/adenovirus-vectors). The resulting recombinant plasmid vectors, Adeno-XlcrV and Adeno-XYFV, were transfected separately into human embryonic kidney 293 (HEK293) cells, and the plaque formation was monitored. After small-scale expansion, eight plaques from each of the recombinant vector transfections were examined for the production of target proteins by dot blot analysis of the infected whole-cell lysates with a monoclonal antibody to LcrV (MAb-LcrV) (BEI Resources). The positive plaques were selected and designated rAd5-LcrV and rAd5-YFV, respectively. The control adenovirus Ad5-CMV-Empty without recombinant gene insertion was purchased from the BCM Vector Development Laboratory and designated Ad5-Empty.
Ad5-Empty, rAd5-LcrV, and rAd5-YFV were then expanded on a large scale by using HEK293 cells in a chemically defined, protein-free CD-293 medium (Thermo Fisher Scientific, Waltham, MA) and purified at the BCM Vector Development Laboratory under good laboratory practice (GLP) conditions and then were used for the subsequent studies in mice. We also prepared rAd5-YFV as a suspension culture which was bioequivalent to the one produced at the BCM Vector Development Laboratory in a mouse model. This suspension culture was used in all NHP studies. To examine expression of the target-protein-encoding genes in the stocked recombinant viruses, A549 human lung epithelial cells (American Type Culture Collection, Manassas, VA) were infected with Ad5 constructs at 1,000 viral particles (v.p.) per cell. The host cell lysates were harvested after 24 h postinfection (p.i.). An aliquot of the cell lysates was then resolved by SDS-PAGE and subjected to Western blot analysis with MAb-LcrV antibody. The purified rLcrV and rYFV antigens were used as controls. As shown in Fig. S1 in the supplemental material, the size of the major band detected in the A549 cell lysate infected with either rAd5-LcrV (lane 2) or rAd5-YFV (lane 3) corresponded to the size of purified rLcrV (lane 5) or rYFV (lane 6). No band was detected in the A549 cell lysate infected with Ad5-Empty (lane 4). The multiple bands detected in lanes 2, 3, and 6 most likely represented degradation or incomplete synthesis of the target proteins.
Animal studies.
Six- to eight-week-old, female Swiss-Webster mice (17 to 20 g) were purchased from Taconic Laboratories (Germantown, NY). All of the animal studies were performed in the animal biosafety level 3 (ABSL3) facility within the Galveston National Laboratory (GNL) under approved Institutional Animal Care and Use Committee (IACUC) protocols.
(i) Induction of preexisting immunity to adenovirus in mice.
To establish preexisting immunity to adenovirus, animals received a single dose of Ad5-Empty by i.m. injection of 8 × 109 v.p./100 μl into both quadriceps (50 μl each) 30 days prior to vaccination. Mice receiving saline (phosphate-buffered saline [PBS]) served as a control. Blood was collected by the retro-orbital route before and 30 days after the Ad5-Empty injection, and microtiter plates precoated with 0.3 μg/well of Ad5-Empty were used to evaluate antibody titers to adenovirus. Animals with preexisting adenovirus immunity were designated preAd mice.
(ii) Immunization of mice with the recombinant proteins or recombinant Ad5 constructs.
Naive mice (40 per group) were immunized with either the mixture of three recombinant proteins (rYscF, rF1, and rLcrV, 25 μg/each) or 45 μg of the corresponding recombinant fusion protein (rYFV) via the i.m. route. The antigens were emulsified 1:1 in Imject alum adjuvant (Pierce Companies, Dallas, TX). One primary immunization and two identical boosters were given on days 0, 15, and 30. Naive mice receiving adjuvant alone served as a control. For the recombinant Ad5 constructs, naive mice or preAd mice (40 per group) were either i.m. or i.n. immunized with one dose (8 × 109 v.p.) of rAd5-LcrV monovalent or rAd5-YFV trivalent vaccine. Control animals (both naive and preAd mice) received the same dose of Ad5-Empty via the same route as the corresponding immunized mice. In some cases, the dose of Ad5-Empty was split equally into i.m. injection and i.n. instillation for the control naive mice. During i.m. immunizations, the dose in a 100-μl volume was equally split and injected into both quadriceps, while for the i.n. immunizations, the dose in 40 μl was equally distributed into each of the nares of mice, followed by 20 μl of PBS wash.
(iii) Immunization of mice with the combination of rAd5-YFV and rYFV.
PreAd mice (20 per group) were first i.n. immunized with 8 × 109 v.p./40 μl of rAd5-YFV trivalent vaccine and then (two weeks later) by i.m. immunized with 10 μg rYFV (emulsified 1:1 in alum adjuvant). PreAd mice immunized with either 10 μg of rYFV or 8 × 109 v.p./40 μl of rAd5-YFV alone were used for comparison, and preAd mice without further immunizations served as a negative control.
(iv) Evaluation of antibody titers in mice.
Blood was collected by the retro-orbital route from all vaccinated and control mice at day 0 and 12 to 15 days after the last vaccination. Sera were separated, and the antigen-specific antibodies were then evaluated. Briefly, enzyme-linked immunosorbent assay (ELISA) plates were precoated with 200 ng/well of the recombinant proteins (e.g., rLcrV, rF1 or rYscF). Twofold serially diluted serum was then added to the wells of the ELISA microtiter plates, followed by the addition of secondary horseradish peroxidase (HRP)-conjugated anti-mouse specific antibodies to IgG, its isotypes, and/or IgA. The ELISA was performed as we described previously (28).
(v) T-cell responses.
T cells were isolated from splenocytes of preAd mice (n = 5) immunized with rAd5-YFV (i.n., 8 × 109 v.p.) either alone or in a prime-boost combination with rYFV (10 μg, i.m.) on day 15 after the last immunization. The isolated T cells were cocultured with gamma-irradiated splenocytes from naive mice (serving as antigen-presenting cells [APCs]) pulsed or not pulsed with F1-V fusion protein (100 μg/ml). After 72 h of incubation, 1 μCi of [3H]thymidine was added to each well, and the cells were harvested 16 h later using a semiautomated sample harvester (FilterMate Harvester; PerkinElmer, Waltham, MA), followed by the measurement of radioactive counts (TopCount NXT; PerkinElmer) as we previously described (29, 30). To measure gamma interferon (IFN-γ)-producing T cells, the isolated T cells were incubated with the pulsed and unpulsed APCs for 2 days and evaluated by the enzyme-linked immunosorbent spot (ELISpot) assay (R&D Systems Inc., Minneapolis, MN).
(vi) Challenge and rechallenge.
Mice were challenged with WT CO92 on day 14 to 15 after the last vaccination by either the subcutaneous (s.c.), i.n., or aerosol route as we previously described (26, 31). Aerosolization was performed using a 6-jet Collison nebulizer attached to a whole-body mouse aerosol chamber. The challenge doses ranged from 24 to 8,500 50% lethal doses (LD50) for the s.c. route and 21 to 800 LD50 for the i.n. route. The presented dose (Dp) for the aerosol challenge was calculated to be in the range of 3.14 × 105 to 6.34 × 105 CFU. The LD50 of WT CO92 for Swiss-Webster mice is ∼50 CFU for developing bubonic plague (s.c.), ∼500 CFU for inducing pneumonic plague (i.n.), and ∼Dp of 2,100 CFU for the aerosol route (26, 30). For the rechallenge experiment(s), on day 32 after the initial WT CO92 aerosol challenge, the vaccinated mice that survived were infected i.n. with 100 LD50 of the WT CO92 luc2 strain. The age-matched naive mice served as a control. The animals were imaged on day 3 p.i. with the WT CO92 luc2 strain by using an in vivo imaging system (IVIS) 200 bioluminescent and fluorescence whole-body imaging workstation (Caliper Corp. Alameda, CA) in the ABSL3 facility.
NHP study.
Cynomolgus macaques (2.5 to 3.5 kg, males) were purchased from Prelabs, Hines, IL. The nonhuman primates (NHPs) were sedated by the administration of ketamine i.m. during the procedures, and all of the studies were performed in the ABSL3 facility under an approved IACUC protocol.
(i) Induction of preexisting immunity to adenovirus and immunization.
To induce preexisting immunity, four randomly selected NHPs were injected in the left quadriceps muscle with 5 × 1010 v.p./250 μl of Ad5-Empty (day 0). After 30 days, these NHPs were i.n. immunized with 1 × 1011 v.p./500 μl of rAd5-YFV, followed by 50 μg/250 μl of rYFV boost (emulsified 1:1 in alum adjuvant) via the i.m. route on day 42. In the control group, four NHPs received 250 to 500 μl of saline at days 0, 30, and 42 via the same routes as the immunized NHPs and served as controls (Table 1). The nasal administration of rAd5-YFV was performed by using a mucosal atomization device (MAD Nasal; Wolfe Tory Medical, Inc., Salt Lake City, UT) that delivers intranasal medication in a fine mist, thus enhancing the absorption and improving bioavailability.
TABLE 1.
NHP immunization and challenge timeline
| Group (n) | Induction of preexisting antiadenovirus immunity (day 0) | Prime vaccination (day 30) | Boost with rYFV (day 42) | Aerosol challenge (day 85) |
|---|---|---|---|---|
| Immunized (4) | 5 × 1010 v.p./250 μl Ad5-empty, i.m. route | 1 × 1011 v.p./500 μl rAd5-YFV, i.n. route (250 μl per nostril) | 50 μg rYFV mixed with alhydrogel (250 μl), i.m. route | WT CO92 (Dp, 1.32 × 107–8.08 × 107 CFU) |
| Control (4) | Saline (250 μl), i.m. route | Saline (500 μl), i.n. route (250 μl per nostril) | Saline (250 μl), i.m. route |
(ii) Aerosol challenge.
The immunized and control NHPs were challenged with WT CO92 by the aerosol route on day 85 (Table 1). Briefly, WT CO92 was aerosolized with a 6-jet Collison nebulizer. The nebulizer was attached to a head-only NHP aerosol exposure box, and real-time plethysmography was performed on each of the anesthetized NHPs during aerosol challenge. The aerosol/plethysmography system was controlled by a Biaera AeroMP aerosol platform (Biaera Technologies, LLC, Hagerstown, MD) integrated with a respiratory inductive plethysmography (RIP) system (Data Sciences International, St. Paul, MN). Aerosol samples were collected during each animal exposure by using all-glass BioSamplers to ensure accurate aerosol delivery, and the corresponding Dps were calculated (26, 32).
The NHPs were monitored and evaluated closely for developing clinical signs of the disease. Clinical scores were provided after thorough examination of the animals by the veterinarian staff. The NHPs were euthanized when they were found with a clinical score of 8 or above. The parameters examined included, but were not limited to, absence of grooming, decreased breathing, and lack of response to human presence at cage side. All NHP exposures to aerosols of WT CO92 were performed in our ABSL3 facility within the GNL in a specialized aerobiology suite equipped with a class III biosafety glove cabinet.
(iii) Antibody titers, blood cell counts, and bacterial burden.
Blood samples were collected from the femoral veins of NHPs at various time points during the experiment. Antibody titers to Ad5, LcrV, F1, and YscF on days 0, 42, 56, 85, 98, and 112 were evaluated by ELISA as we described above. The last two time points (days 98 and 112) corresponded to days 14 and 28 after WT CO92 challenge. Blood cell counts were analyzed on the day of WT CO92 challenge (day 85) and on days 3 and 6 postchallenge by using a Drew Scientific Hemavet 950 hematology system (Drew Scientific, Inc., Dallas, TX). The bacterial loads were evaluated by plating the blood samples which were drawn when control NHPs were euthanized (on day 3 or 4 after WT CO92 challenge) or at various time points (e.g., days 3, 6, 14, 28, 70, and 82) after WT CO92 challenge in the case of immunized NHPs.
(iv) Necropsy and histopathological analysis.
After euthanasia, necropsies were performed by the certified chief biocontainment veterinarian at UTMB. NHP organs, such as lungs, liver, spleen, and lymph nodes (hilar, submandibular, and mediastinal), were removed and grossly examined. A portion of these organs was homogenized and plated for assessing bacterial load (33), while another portion was fixed in 10% neutral buffered formalin (31, 34) and tissues processed and sectioned at 5 μm. The samples were mounted on slides and stained with hematoxylin and eosin (H&E). Sections from the lungs were also subjected to Gram staining to examine the presence of plague bacilli. Tissue lesions were scored on the basis of a severity scale, which correlated with estimates of lesion distribution and the extent of tissue involvement (minimal, 2 to 10%; mild, >10 to 20%; moderate, >20 to 50%; severe, >50%), as previously described (31, 34). The histopathological evaluation of the tissue sections was performed in a blinded fashion.
Computed tomography (CT) scans.
The CereTom NL 3000 (Neurologica, MA), which is an eight-slice tomography instrument with high-contrast resolution of 0.6 mm (developed for human head imaging in intensive care units [ICU]), was used. The image acquisition settings were as follows: tube voltage, 100 kV; tube current, 5 mA; and axial mode with slice thickness of 1.25 mm. The image resolution was 512 by 512 pixels. The image sharpness was optimized to soft tissue.
Statistical analysis.
Two-way analysis of variance (ANOVA) with Tukey's post hoc test or the multiple Student t test with the Holm-Sidak post hoc test correction was used for data analysis. We used Kaplan-Meier survival estimates for animal studies, and P values of ≤0.05 were considered significant for all of the statistical tests used.
RESULTS
Protective immunity of the recombinant adenoviruses in both bubonic and pneumonic plague mouse models.
Mice were immunized i.m. or i.n. with rAd5-LcrV monovalent or rAd5-YFV trivalent vaccines to evaluate their potential to protect animals from plague. Irrespective of the immunization route, mice that were administered rAd5-YFV trivalent vaccine displayed 100% protection when challenged with 60 LD50 of WT CO92 in a bubonic plague model (Fig. 1A) However, only 50 to 55% of mice receiving the rAd5-LcrV monovalent vaccine were protected, and all control mice died by day 11 p.i. (Fig. 1A). In a more stringent pneumonic plague model (90 LD50 of WT CO92), animals vaccinated by the i.n. route with the rAd5-YFV trivalent vaccine were 60% protected, while the survival rate declined to 10% when immunization occurred by the i.m. route (Fig. 1B). In comparison, either none or 20% of the animals immunized with the Ad5-LcrV monovalent vaccine survived when vaccination occurred by the i.m. versus the i.n. route. Overall, these data indicated vaccines to be more effective when instilled by the i.n. route. The corresponding control mice (receiving Ad5-Empty by the i.m. or the i.n. route) succumbed to infection by day 4 p.i. (Fig. 1B).
FIG 1.
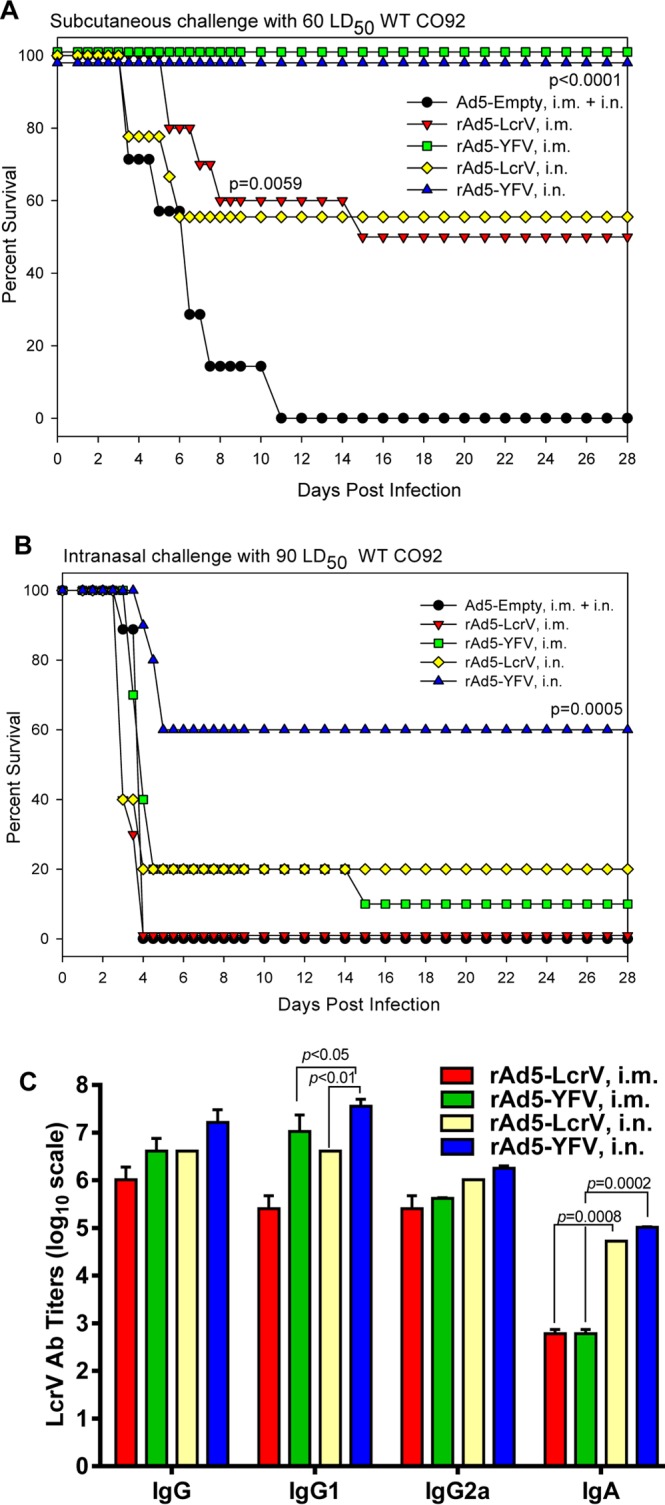
Comparison of immunization routes in mice. Naive mice (n = 40) were either i.m. or i.n. immunized with one dose (8 × 109 v.p.) of rAd5-LcrV or rAd5-YFV vaccine. Animals received the same dose of Ad5-Empty, which was split equally into i.m. injection and i.n. instillation and served as a control. These immunized and control mice were then divided into two sets and challenged on day 15 postimmunization either subcutaneously (s.c.) with 60 LD50 (A) or intranasally (i.n.) with 90 LD50 (B) of WT CO92. The P values are in comparison to the control group and are based on Kaplan-Meier curve analysis. Mice were also bled prior to the challenge to evaluate IgG antibody titers and those of its isotypes to LcrV by ELISA (C). The P values are in comparison to the indicated groups and are based on two-way ANOVA (IgG1 and IgA) with Tukey's post hoc correction.
Higher antibody titers to LcrV were generally observed in mice that received the rAd5-YFV trivalent vaccine than in those that received the rAd5-LcrV monovalent vaccine, reaching statistically significant levels for IgG1 in mice that were immunized by the i.n. route (Fig. 1C). In terms of immunization routes, i.n. vaccinated mice overall had superior antibody titers compared to animals immunized by the i.m. route, reaching statistical significance for the production of IgG1 and IgA (Fig. 1C). Irrespective of the recombinant virus and route of immunization used, all of the vaccinated mice developed more balanced Th1- and Th2-type antibody responses. In comparison, mice intramuscularly immunized with either the mixture of recombinant proteins (rYscF plus rF1 plus rLcrV) or the fusion protein rYFV favored a Th2 response, although both recombinant-protein-immunized animals were 100% protected against the challenges by either the s.c. route (8,500 LD50, to induce bubonic plague) or the i.n. route (800 LD50, to induce pneumonic plague) with WT CO92 (see Fig. S2 in the supplemental material).
Preexisting immunity to adenovirus in mice.
The adenoviral antibody titers on day 30 after injection of Ad5-Empty in mice ranged from 102,400 to 819,200. In a bubonic plague model, at a challenge dose of 24 LD50, a similar level of protection (80 to 90%) was noted in mice immunized with rAd5-YFV trivalent vaccine, irrespective of whether or not preexisting antibodies to adenovirus were developed (Fig. 2A). In contrast, the survival rate was 40% in mice without preexisting Ad5 antibodies and only 10% in preAd mice when immunization was with the rAd5-LcrV monovalent vaccine (Fig. 2A). In a pneumonic plague model (21 LD50), rAd5-YFV-immunized mice with or without preexisting immunity to Ad5 exhibited a similar 55 to 60% survival rate, which was much higher than that in mice immunized with the rAd5-LcrV monovalent vaccine with or without preimmunity to Ad5 (10 to 20% protection) (Fig. 2B). All of the control mice died on the indicated days in a bubonic or pneumonic plague model (Fig. 2A and B).
FIG 2.
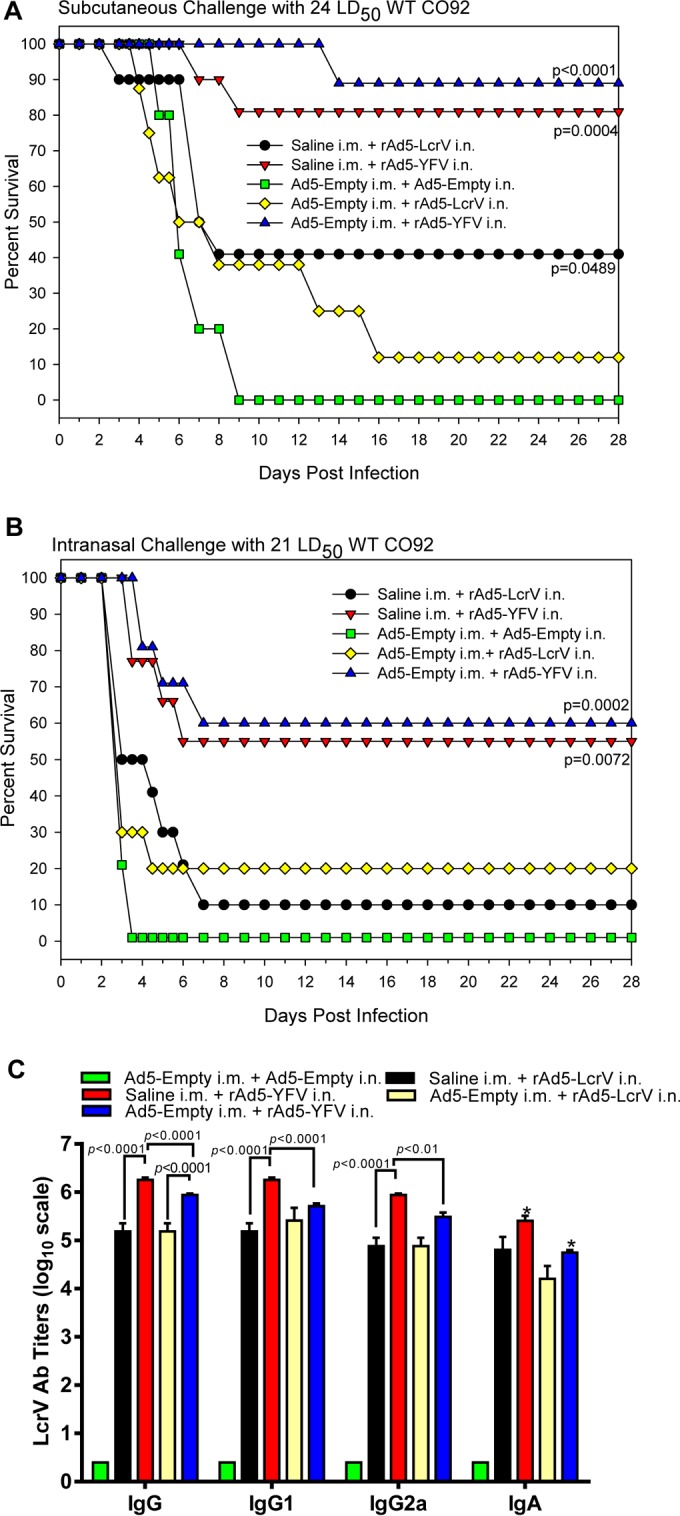
Protection conferred by immunization with the recombinant adenoviruses in mice that had preexisting immunity to adenovirus. To establish preexisting immunity to adenovirus, naive mice (n = 40) received a single dose (8 × 109 v.p./100 μl) in both quadriceps (50 μl each) of Ad5-Empty by i.m. injection at 30 days prior to vaccination. Naive mice receiving saline served as a control. Subsequently, mice were i.n. immunized with one dose (8 × 109 v.p.) of rAd5-LcrV or rAd5-YFV vaccine. Animals received the same dose of Ad5-Empty by i.n. instillation and served as a negative control. These mice were then divided into two sets and challenged on day 15 postimmunization either subcutaneously (s.c.) with 24 LD50 (A) or intranasally (i.n.) with 21 LD50 (B) of the WT CO92. The P values are in comparison to the negative-control group and are based on Kaplan-Meier curve analysis. Mice were also bled prior to the challenge to evaluate IgG antibody titers, titers to its isotypes, and IgA to LcrV by ELISA (C). The P values are in comparison to the indicated groups and are based on two-way ANOVA with Tukey's post hoc correction. The asterisks indicate statistical significance compared to the control (Ad5-Empty) mice for IgA levels by using multiple Student's t test with the Holm-Sidak post hoc test correction.
Balanced Th1- and Th2-type antibody responses with robust titers to LcrV were observed across all immunized mice (Fig. 2C). However, two important observations were drawn from this study: (i) compared to rAd5-LcrV monovalent vaccine-immunized mice, animals that were vaccinated with the rAd5-YFV trivalent vaccine generally developed better antibody titers (both IgG and its isotypes as well as IgA) to LcrV, although some did not reach statistical significance (e.g., IgG1 and IgG2a in preAd mice as well as IgA), and (ii) mice without preexisting adenoviral immunity developed slightly higher IgG and IgA antibody titers to LcrV than preAd mice receiving the trivalent rAd5-YFV vaccination, although only total IgG and its isotopes reached statistical significance (Fig. 2C). As expected, none of the unimmunized control mice developed any detectable level of protective anti-LcrV antibodies, and thus they succumbed to infection (Fig. 2A and B). Importantly, in spite of slight lower antibody titers to LcrV in mice with preexisting Ad5 antibodies, animals were similarly protected when the Ad5-YFV trivalent vaccine was administered by the i.n. route against challenge with WT CO92 in both bubonic and pneumonic plague models (Fig. 2A and B).
Prime-boost and aerosol challenge.
Our data above indicated that the trivalent rAd5-YFV vaccine was better than the monovalent rAd5-LcrV vaccine in providing protection to mice against Y. pestis infection. However, the overall protection rate did not reach 100% in the pneumonic plague model (Fig. 1B and 2B). To enhance protection, a boost with rYFV (10 μg) was administered to mice i.m. 2 weeks later following i.n. instillation of the rAd5-YFV trivalent vaccine. As shown in Fig. 3, mice immunized with only rAd5-YFV had a 70% survival rate after aerosol exposure of WT CO92, irrespective of whether or not preexisting adenoviral immunity was developed. The preAd mice vaccinated with the combination of rAd5-YFV and rYFV displayed a protection rate of 80% with an overall delayed death pattern after WT CO92 aerosol challenge at a Dp of 6.34 × 105 CFU (∼302 LD50). The rYFV-immunized mice alone (single dose, no boosts) had 5% survival, and all unimmunized preAd mice died after aerosol exposure to the pathogen between days 3 and 5 p.i. (Fig. 3).
FIG 3.
Prime-boost immunization provides better protection to mice against lethal WT CO92 aerosol challenge. PreAd mice (groups of 20) were i.n. immunized with 8 × 109 v.p./40 μl of rAd5-YFV either alone or in combination with 10 μg of rYFV (emulsified 1:1 in alum adjuvant) i.m. The immunizations occurred 2 weeks apart. Naive mice immunized with either 10 μg of rYFV (i.m.) or 8 × 109 v.p./40 μl (i.n.) of rAd5-YFV alone were used for comparison, and preAd mice without further immunizations served as a negative control. After 15 days postimmunization, mice were challenged by the aerosol route with WT CO92 at a Dp of 6.34 ×105 CFU. The P values are in comparison to the negative-control group and are based on Kaplan-Meier curve analysis.
To further evaluate the potential of the prime-boost strategy, another set of immunized mice were exposed to a slightly lower WT CO92 aerosol challenge dose (Dp of 4.62 × 105 CFU, ∼220 LD50). As shown in Fig. 4A, the preAd mice first vaccinated with the rAd5-YFV trivalent vaccine and then boosted with rYFV were 100% protected against developing pneumonic plague. On the other hand, preAd mice that were vaccinated with only the rAd5-YFV trivalent vaccine showed a 55% survival rate, with all the unimmunized preAd mice succumbed to infection by day 3 postchallenge.
FIG 4.
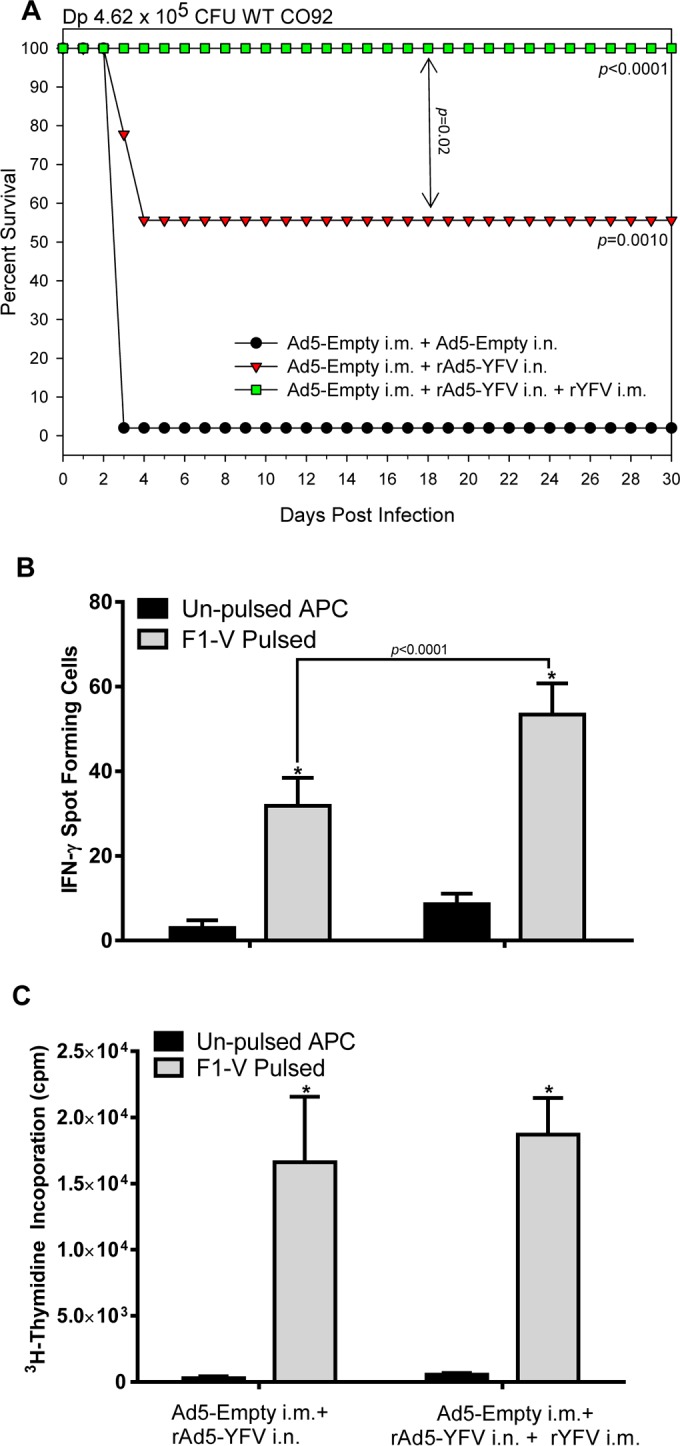
T-cell mediated immune response in mice elicited by immunization with the rAd5-YFV vaccine alone or in combination with rYFV. PreAd mice (n = 10 to 25) were either i.n. immunized with 8 × 109 v.p./40 μl of rAd5-YFV alone or in combination with 10 μg of rYFV (emulsified 1:1 in alum adjuvant) i.m. The immunizations occurred 2 weeks apart. (A) After 15 days postimmunization, 20 mice from each immunized group and 10 from the control group were aerosol challenged with WT CO92 at a Dp of 4.62 ×105 CFU. The P values are in comparison to the negative-control group or between groups (as indicated by the arrow) and are based on Kaplan-Meier curve analysis. (B) On day 15 after the last immunization, T cells were isolated separately from the spleens of the remaining unchallenged 5 mice in each immunized group. The isolated T cells were cocultured with gamma-irradiated APCs pulsed or not with F1-V fusion protein (100 μg/ml). The IFN-γ-producing T cells were measured after 2 days of incubation with the APCs by using the enzyme-linked immunosorbent spot (ELISpot) assay. (C) T-cell proliferation was assessed by measuring incorporation of [3H]thymidine on day 3 of coculture with the APCs. The arithmetic means ± standard deviations were plotted. Data were analyzed by using two-way ANOVA with Tukey's post hoc correction. The statistical significance is indicated by asterisks in comparison of the pulsed and unpulsed T cells within each group or displayed by a horizontal line with the P value.
In addition, 55 to 60% of T cells isolated from the prime-boost group of mice were IFN-γ positive, while this number was only 30% for mice that were immunized with rAd5-YFV trivalent vaccine alone (Fig. 4B). However, there was no difference between the two immunized groups of mice (with or without the prime-boost) in terms of their T-cell proliferative responses upon stimulation with the F1-V antigen (Fig. 4C).
In terms of antibody production, we noted that IgG, its isotypes, and IgA antibody titers to the three antigens (F1, LcrV, and YscF) were generally higher in the prime-boost group of mice than in those animals that received only the rAd5-YFV trivalent vaccine. Further, balanced Th1- and Th2-based antibody responses were observed (Fig. 5A to C).
FIG 5.
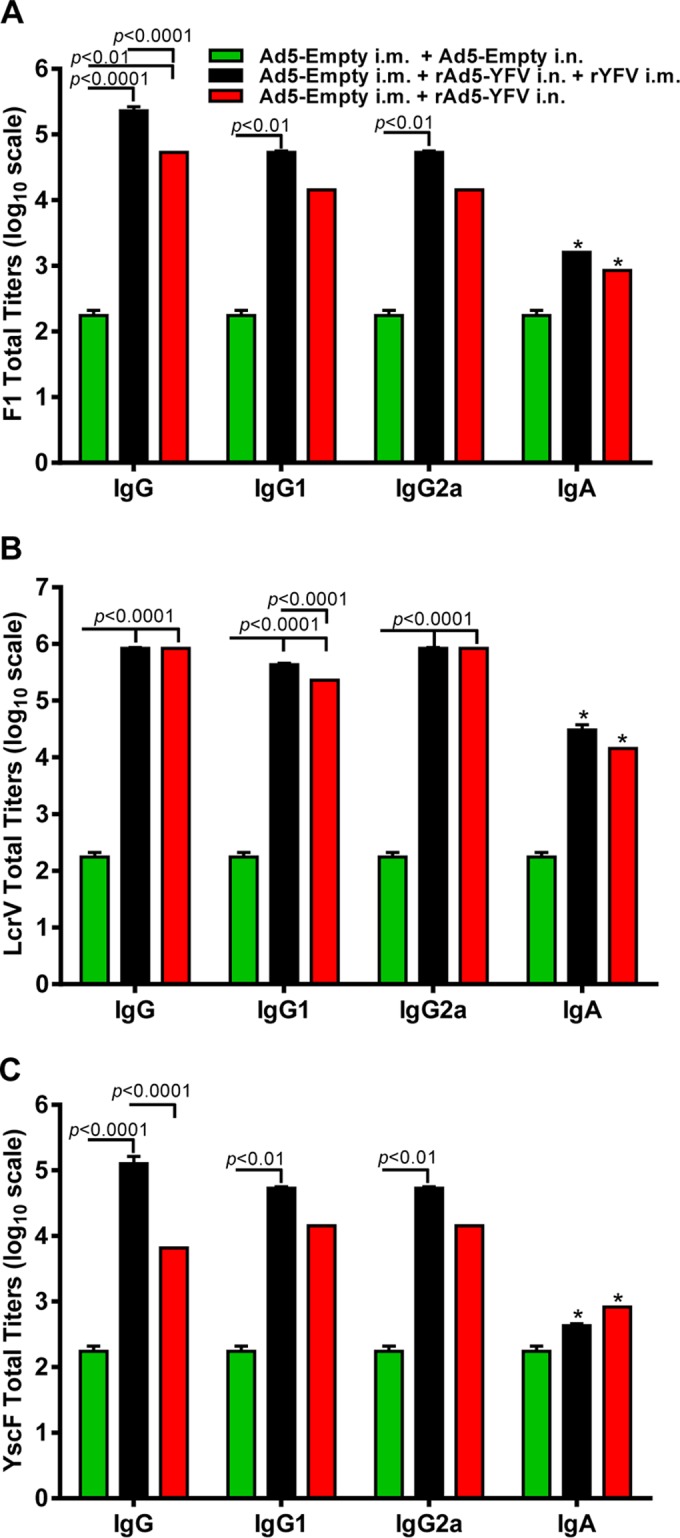
Antibody responses in mice elicited by immunization with the rAd5-YFV vaccine alone or in combination with rYFV. Mice from different groups (Fig. 4) were also bled at 15 days postimmunization, and an ELISA was performed to examine titers of IgG antibody, its isotypes, and IgA to F1 (A), LcrV (B), and YscF (C), respectively. The P values are in comparison to the indicated groups and based on two-way ANOVA with Tukey's post hoc correction. The asterisks indicate statistical significance compared to the control (Ad5-Empty) mice for IgA levels by using multiple Student's t test with the Holm-Sidak post hoc test correction.
Continued protection of mice conferred by the prime-boost vaccination strategy against the initial aerosol challenge and then the subsequent intranasal WT CO92 challenge.
In our subsequent experiment, preAd mice were vaccinated either with the rAd5-YFV trivalent vaccine alone or with an rYFV boost. The preAd mice receiving the Ad5 empty vector alone served as a control. After the vaccination regimen, mice were subjected to WT CO92 aerosol challenge with still a relatively lower Dp (3.14 × 105 CFU, ∼150 LD50) compared to those with the above two aerosol challenges (Fig. 3 and 4A). As noted in Fig. 6A, 100% of the animals survived the initial challenge in all of the immunized groups, while 90% of the control mice died (Fig. 6A). At 32 days after the initial aerosol challenge, the surviving animals from the immunized groups were rechallenged with 100 LD50 of WT strain CO92 luc2 by the i.n. route, and the age-matched uninfected naive mice (n = 5) served as a control. As shown in Fig. 6A, 80% of the mice were protected from developing plague in the rAd5-YFV-immunized group, while this protection was 100% when the prime-boost strategy was used. In contrast, all of the naive rechallenge control mice succumbed to infection within 4 days p.i. The bioluminescent images showed that the plague bacilli disseminated from the initial infection site of lungs to the whole body in all 5 naive control mice after day 3 p.i. (Fig. 6B, panel I). On the other hand, no animals were positive in the group that received vaccination by the prime-boost regimen (Fig. 6B, panel II). However, one mouse from the rAd5-YFV-immunized group was bioluminescence positive, with the organisms confined in the lungs (Fig. 6B, panel III). This bioluminescence-positive animal, along with one other mouse in the same group which did not show bioluminescence at the time of imaging (day 3 p.i.), eventually died, resulting in the overall survival rate of 80% in the rAd5-YFV-immunized group of mice (Fig. 6A).
FIG 6.
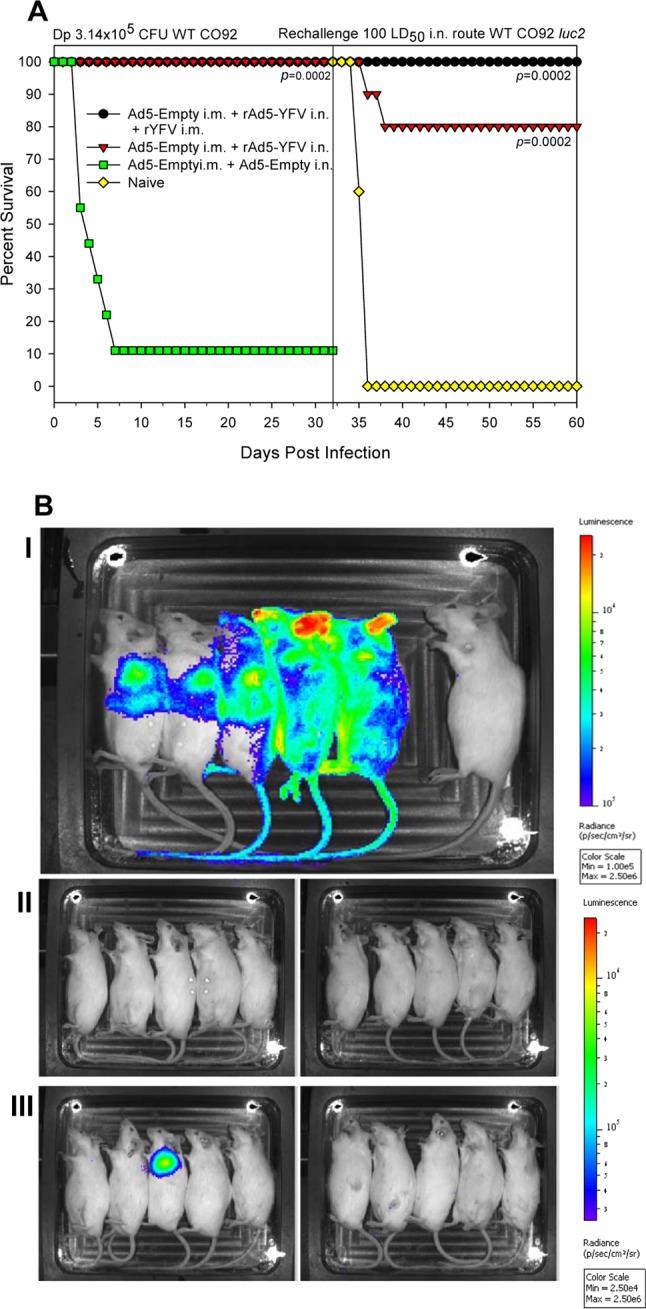
Immunization of mice with the rAd5-YFV vaccine alone or in combination with rYFV provides protection against lethal primary aerosol and subsequent intranasal WT CO92 challenges. PreAd mice (n = 10) were i.n. immunized with 8 × 109 v.p./40 μl of rAd5-YFV either alone or in the combination with 10 μg of rYFV (emulsified 1:1 in alum adjuvant) i.m. The immunizations occurred 2 weeks apart. PreAd mice injected with Ad5-Empty served as a negative control. After 15 days postimmunization, mice were first challenged with aerosolized WT CO92 at a Dp of 4.62 × 105 CFU. At 32 days after the initial aerosol challenge, the surviving animals from the immunized groups along with five age-matched uninfected naive mice were infected with 100 LD50 of WT strain CO92 luc2 by the i.n. route. (A) The deaths were recorded for the initial aerosol challenge and then the subsequent intranasal challenge, and the percentages of survival were plotted. The P values are in comparison to the control group for each challenge and are based on Kaplan-Meier curve analysis. (B) The animals were also imaged by IVIS for bioluminescence on day 3 after WT strain CO92 luc2 i.n. challenge. Panel I, infected naive mice as an i.n. challenge control. The rightmost animal in this panel is an uninfected image control. Panel II, animals immunized with the prime-boost strategy. Panel III, animals immunized with rAd5-YFV vaccine alone. The bioluminescence scale ranges from most intense (red) to least intense (violet).
Evaluation of protection provided by the trivalent rAd5-YFV vaccine in cynomolgus macaques against aerosol challenge with WT CO92.
Four NHPs were initially i.m. injected with Ad5-Empty to generate preexisting adenoviral immunity. This was followed by one dose of rAd5-YFV by i.n. instillation in the form of mist and then one dose of rYFV by the i.m. route. Four unimmunized NHPs served as a control (Table 1). These NHPs were then challenged with the aerosolized WT CO92 at Dp ranging from 1.32 × 107 to 8.08 × 107 CFU (∼13,200 to 80,800 LD90, with 1 LD90 = 864 CFU [35]). No clinic signs were noted in the immunized group of NHPs, and the animals remained healthy and survived the WT CO92 challenge until they were euthanized at the end of the study (Fig. 7). The CT scans of immunized NHPs, which survived the WT CO92 challenge (Fig. 7) and were euthanized on day 82 postchallenge, did not display any abnormalities in the lungs and their surrounding areas compared to the images of the animals before the WT CO92 challenge on day 85 (Table 1; see Fig. S3 in the supplemental material). In contrast, the control NHPs euthanized on day 3 to 4 postchallenge showed consolidation in both the right and left lungs, an indication of severe inflammation (see Fig. S3 in the supplemental material).
FIG 7.
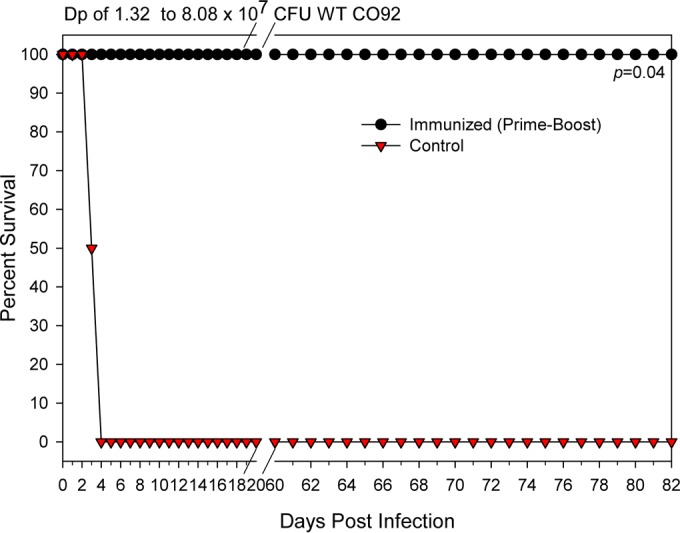
The rAd5-YFV vaccine in combination with rYFV provides protection to NHPs with preexisting adenovirus immunity against lethal aerosol challenge with WT CO92. To induce preexisting adenovirus immunity, four NHPs were injected in the quadriceps muscle with 5 × 1010 v.p. of Ad5-Empty (day 0). On day 30, these NHPs were immunized by the intranasal route with 1 × 1011 v.p. of rAd5-YFV, followed by 50 μg of rYFV boost (emulsified 1:1 in alum adjuvant) via the i.m. route on day 42. Another four NHPs received saline only (without immunization) and served as a control. On day 85, the NHPs were challenged with WT CO92 by the aerosol route with a Dp ranging from 1.32 × 107 to 8.08 × 107 CFU. The animals were euthanized when a clinical score of ≥8 was reached or at the termination of the experiment, and percent survival was plotted. The P values are in comparison to the NHP control group and are based on Kaplan-Meier curve analysis.
Necropsy on immunized NHPs was performed 82 days after the WT CO92 challenge; no gross abnormities were observed, and the internal organs (lungs, liver, spleen, and the lymph nodes) were all free of bacteria (Table 2). In contrast, all unimmunized control NHPs developed clinical signs of the disease as early as 36 h p.i. and reached a clinical score of 8 and higher on day 3 to 4 p.i. The control NHPs had cough, abnormal respiration, lethargy, and a hunched posture. Although we did not notice fever in these animals during the progression of the disease, this could be related to not continuously monitoring these NHPs by using telemetry. Necropsy of these animals revealed serous hemorrhagic fluid in the thorax with respiratory frothy serous discharge. Lungs were hyperinflated with hemorrhagic frothy fluid, and the spleen, liver, and lymph nodes were enlarged. The highest bacterial loads (1.12 × 109 to 1.26 × 109 CFU/node) were noted in the hilar lymph nodes and lungs (2.22 × 107 to 1.06 × 109 CFU/g), followed by the liver (8.16 × 106 to 1.69 × 107 CFU/g), spleen (2.13 × 106 to 4.47 × 106 CFU/g), and submandibular lymph nodes (2.33 × 105 CFU/node). Only one animal showed bacteria in the blood, with a count of 2,500 CFU/ml, and no bacilli were detected in the other control NHPs (Table 2).
TABLE 2.
NHP clinical score, bacterial loads, and necropsy report
| NHP group | Days postinfection | Bacterial loads in blood/organs | Clinical score | Necropsy report |
|---|---|---|---|---|
| Control | 3–4 | Blood, 0–2,500 CFU/ml; lung, 2.22 × 107–1.06 × 109 CFU/g; liver, 8.16 × 106–1.69 × 107 CFU/g; spleen, 2.13 × 106–4.47 × 106 CFU/g; Hilar lymph node, 1.12 × 109–1.26 × 109 CFU/node; submandibular lymph node, 2.0 × 105–2.33 × 105 CFU/node | ≥8 | External, thin, pale, dehydrated, and scruffy coat; respiratory, frothy serous discharge, hyperinflated with hemorrhagic frothy fluid (∼50 ml); lymphatic, enlarged submandibular node; spleen, firm and enlarged; liver, firm, enlarged, and rounded edges; locomotion, lethargic; body cavities, serous hemorrhagic fluid in thorax (∼50 ml) |
| Immunized | 82 | Negative for all organs; blood samples were negative for bacteria as early as day 3 postinfection | 0 | Normal |
NHP blood cell counts and antibody titers.
The changes in the blood cell counts in immunized NHPs versus the controls after WT CO92 challenge are shown in Fig. S4 in the supplemental material. Only the lymphocyte (LY) counts in the control NHPs fell below the normal range by day 3 after WT CO92 challenge before they were euthanized. However, in the immunized NHPs, LY counts remained within the normal range on days 3 and 6 after WT CO92 challenge.
Both immunized and control NHPs showed some level of preexisting Ad5 antibody titers (6,400 to 25,600) on day 0 as a consequence of naturally acquired infection with adenoviruses. The anti-Ad5 titer was increased to 409,600 on day 30 in immunized NHPs after receiving the rAd5-Empty injection, and it continued to climb slightly on days 42 and 56 as a result of immunization with rAd5. The anti-Ad5 antibody titer was maintained at a level similar to that observed on day 0 in the control NHPs (Fig. 8A). No preexisting anti-LcrV, anti-F1, and anti-YscF antibodies were detected in either group of NHPs before immunization (data not shown). However, high antibody titers to three Y. pestis-specific antigens (i.e., F1, LcrV, and YscF) were noticed in all of the immunized NHPs (Fig. 8B to E). Compared to the antibody titers on day 42, the antigen-specific IgG antibodies increased ∼10-fold for LcrV and YscF but nearly 1,000-fold for F1 on day 56 (Fig. 8B to D). Thus, the boost on day 30 with rYFV (Table 1) led to an increase in antibody titers. These antigen-specific antibody titers slightly decreased on day 85 (the day of challenge). A similar trend was observed for the anti-LcrV IgA antibody titers, which were increased ∼10-fold on day 56 after the rYFV boost (Fig. 8E). Compared to all three antigen-specific IgG antibody titers, the anti-LcrV titers were the highest, followed by anti-YscF and anti-F1 across the course of immunization, and the difference could reach up to 1,000-fold (anti-LcrV versus anti-F1 on day 42) (Fig. 8B and C). After WT CO92 aerosol challenge, anti-F1 IgG titers were further boosted, while sustaining IgG titers for LcrV and YscF and IgA LcrV titers up to 28 days after WT CO92 challenge (overall day 112 after initiation of vaccination) (see Fig. S5 in the supplemental material).
FIG 8.
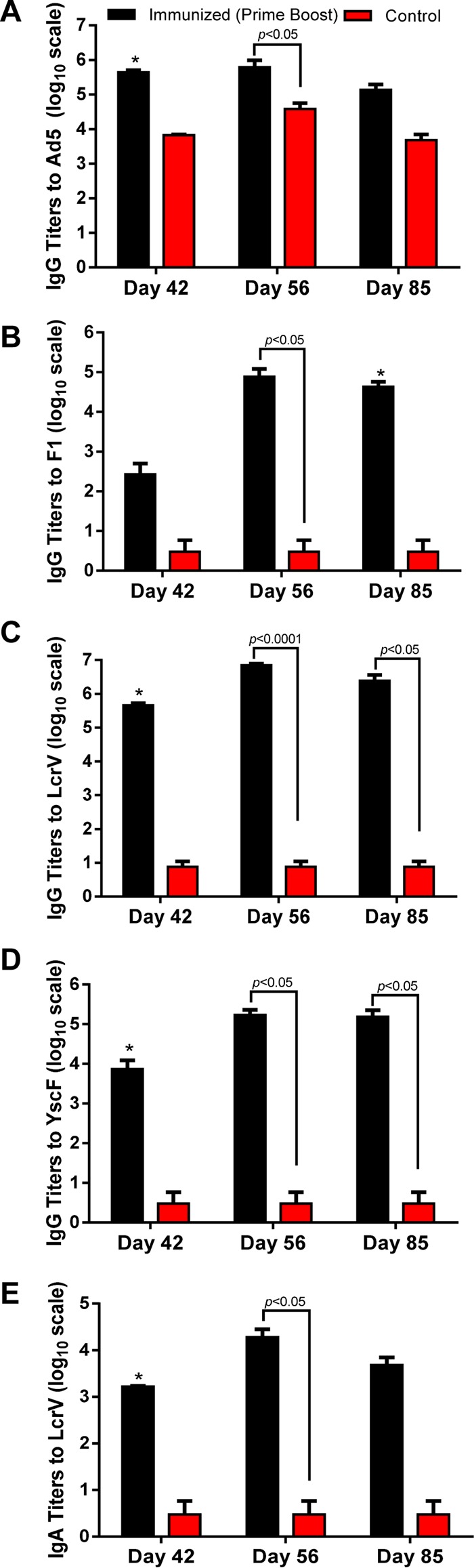
Antibody responses in NHPs immunized with the rAd5-YFV vaccine in combination with rYFV. Four randomly selected NHPs were injected in the quadriceps muscle with 5 × 1010 v.p. of Ad5-Empty to induce preexisting immunity (day 0). On day 30, these NHPs were immunized by the intranasal route with 1 × 1011 v.p. of rAd5-YFV, followed by 50 μg of rYFV boost (emulsified 1:1 in alum adjuvant) via the i.m. route on day 42. Another four NHPs received saline only (without immunization) and served as a control. On day 85, the NHPs were challenged with WT CO92 by the aerosol route. Blood samples were collected from the femoral veins of NHPs at various time points during the experiment. The total IgG titers to Ad5 (A), F1 (B), LcrV (C), and YscF (D) as well as IgA titers to LcrV (E) on days 42, 56, and 85 were evaluated by ELISA. The P values are in comparison to the indicated groups and are based on two-way ANOVA with Tukey's post hoc correction. The asterisks indicate statistical significance compared to the control (Ad5-Empty) mice by using multiple Student's t test with the Holm-Sidak post hoc test correction.
NHP histopathological analysis.
As shown in Fig. 9, the unimmunized control NHPs showed marked acute inflammatory reactions in the lungs, pleurae, and mediastinal lymph nodes. Specifically, multifocal hemorrhage and diffused supportive inflammation were observed in the lungs, with no alveolar spaces. Similar changes were also observed in the pleura and mediastinal lymph nodes of these unimmunized NHPs. Furthermore, tissue sections from the lungs with Gram staining revealed the presence of bacteria, presumptively Y. pestis (Fig. 9, inset). Interestingly, the liver and the spleen tissues of unimmunized NHPs showed normal morphological characteristics in spite of higher bacterial loads (Table 2), indicating that pneumonic changes are the primary cause of death in control groups. In the immunized NHP group, the lungs, pleurae, mediastinal lymph nodes, and liver were normal, and the lungs had alveolar spaces. The only notable and expected changes observed in the prime-boost group was the hyperplasia of lymphoid follicles in mediastinal lymph nodes and the spleen. These changes can mainly be attributed to reaction to vaccination.
FIG 9.

Histopathological analysis of tissues collected from NHPs after WT CO92 aerosol challenge. Lungs, pleurae, mediastinal lymph nodes, liver, and spleen tissues were collected from the control (3 or 4 days after WT CO92 challenge) and immunized (82 days after WT CO92 challenge) NHPs after euthanization and processed for histopathological analysis. The inset from lungs revealed the presence of coccobacilli, presumptively Y. pestis, by Gram staining. The magnifications of the images are indicated.
DISCUSSION
Historically, vaccination has been not only one of the most significant advances in health care but also a cost-effective means of public health intervention. The high mortality rate associated with pneumonic plague, the potential use of Y. pestis as a biological weapon, and the current lack of an FDA-approved plague vaccine highlight the importance of our studies.
Previously, the plague vaccine licensed in the United States (sold under the name USP) was a formaldehyde-killed preparation of the highly virulent 195/P strain of Y. pestis; however, the production of this vaccine was discontinued in 1999. The vaccination regimen included a course of injections over a period of 6 months and then annual boosters (36, 37). The vaccine was effective against bubonic plague, but protection against pneumonic plague was uncertain. The incidence of side effects, such as malaise, headaches, elevated body temperature, and lymphadenopathy, was high-pressure liquid chromatography, and the vaccine was expensive (38). A live-attenuated vaccine based on Y. pestis pigmentation locus-negative EV76 strains is also available in some parts of the world where plague is endemic (1). These types of vaccines have existed since the first half of the 20th century and have proven effective against both subcutaneous and inhalation challenges with Y. pestis. However, the EV76-based vaccines are not genetically uniform and are also highly reactogenic (39), and hence they would not meet the standards for FDA approval.
The major problems encountered in developing live-attenuated vaccines are inadequate attenuation, particularly in immunocompromised individuals, and the potential to revert back to the virulent phenotype. Efforts have been made to generate well-characterized and rationally designed attenuated plague vaccines. For example, mutations that effectively attenuate Salmonella, such as mutations in the aroA, phoP, htrA, and lpp genes, were introduced in Y. pestis, but these mutations had only a limited effect on Y. pestis virulence (31, 40–42). Similarly, a deletion of the Y. pestis global regulator gene rovA significantly attenuated the bacterium during subcutaneous infection, but this mutant was only slightly attenuated when given via an intranasal or intraperitoneal route (43). Recently, a highly attenuated Δlpp ΔmsbB Δail triple mutant, which was deleted for genes encoding Braun lipoprotein (Lpp), an acetyltransferase (MsbB), and the attachment invasion locus (Ail), was constructed (25). Mice immunized with this triple mutant via either the intranasal, subcutaneous, or intramuscular route were protected from lethal WT CO92 challenge, and thus this could be an excellent vaccine candidate (25, 33). However, further evaluation of the efficacy of this triple mutant in higher animal models is warranted.
While the above-described conventional vaccine strategies have focused on live-attenuated or killed bacterial approaches, a new method in the development of vaccines utilizes platform technologies to overcome some of the challenges in vaccine design. The adenoviral vector system has been successfully used as a vaccine platform for a number of pathogens, including Y. pestis (44, 45), with several advantages: (i) the adenoviral genome is well characterized, with the capability of integrating ≥6 kb of the potential insert size for delivering multiple antigens; (ii) the replication-defective Ad5 vector has been developed for gene therapeutic applications at a wide range of doses, with minimal side effects; and (iii) adenoviruses have a broad tropism, infecting a variety of dividing and nondividing cells. Studies have shown that adenoviruses transfer genes effectively to APCs in vivo to promote rapid and robust humoral and cellular immune responses to the transgene products (46–53). In addition, adenoviruses can be grown to high titers in tissue culture cells and can be applied systemically as well as through mucosal surfaces, and they are relative thermostable to facilitate their clinical use.
Our rAd5-YFV trivalent vaccine had an average yield of 1 × 1016 v.p. per batch in a cell suspension culture in CD 293 medium. The vaccine was free of proteins, serum, and animal-derived components, thus making it suitable for a broad range of prophylactic and therapeutic uses. Compared to a favored Th2 response in mice immunized with rYFV or a mixture of rYscF, rLcrV, and rF1 (given with alum, which skews the immune response to Th2) (see Fig. S2A in the supplemental material), a more balanced Th1- and Th2-based antibody response was observed in mice immunized with the rAd5 vaccines (Fig. 1C, 2C, and 5A to C). Indeed, Ad5 has been shown to promote the Th1 response (45). As expected, intranasal administration of rAd5-LcrV monovalent and rAd5-YFV trivalent vaccines elicited IgA production in immunized animals (both mice and NHPs), and most importantly, mice immunized with rAd5-YFV alone or in a prime-boost vaccination strategy exhibited a robust T-cell proliferative responses (Fig. 4C). These features suggest superiority of Ad5-based vaccines over the rF1-V-based subunit vaccines, as the protection by the latter vaccines is largely dependent on systemic antibody responses without mucosal and cellular immune components. Interestingly, although generally a higher IgG antibody titer was observed across all mice immunized intranasally compared to animals immunized intramuscularly with the recombinant adenoviruses, the protection rate was indistinguishable during the development of bubonic plague. However, subtle differences in protection were noted depending upon of the route of immunization of mice in a pneumonic plague model (Fig. 1A and B), which further highlighted the importance of mucosal immunity during the development of pneumonic plague.
Pneumonic plague begins with an anti-inflammatory state (i.e., the first 24 to 36 h after infection), which is characterized by a delay in the inflammatory cell recruitment to the lungs and production of proinflammatory cytokines and chemokines (54). Therefore, a plague vaccine should be able to stimulate a strong mucosal immunity to overcome this initial immune suppression in the host (55). In our future studies, we plan to discern the role of the mucosal immune response (e.g., IgA) that is triggered by the rAd5-YFV vaccine in protection.
Compared to the monovalent rAd5-LcrV vaccine, the trivalent rAd5-YFV vaccine not only mounted higher anti-LcrV antibody titers (both IgG and IgA) (Fig. 1C and 2C) but also generated immune responses to the F1 and YscF (Fig. 5), which correlated with better protection of animals against both bubonic and pneumonic plague (Fig. 1A and B, 2A and B, and 3). In addition, LcrV was more immunogenic than F1 and YscF in both mice and NHPs that were immunized with the trivalent rAd5-YFV vaccine (Fig. 5 and 8). In contrast, the antibody titers to F1 were the lowest among the three examined antigens in the rAd5-YFV-immunized NHPs (Fig. 8). The difference in immunogenicity may be attributed to the nature of each of the antigens; however, the conformation of the antigens in the fusion protein may also play a role, especially as higher anti-LcrV antibody titers were observed in the rAd5-YFV-immunized mice than in rAd5-LcrV vaccinated animals. Alternatively, the presence of the other two antigens could augment antibody production to LcrV.
Previously, an rAd5 (designated rAdsecV) expressing a human Igκ secretion (sec) signal fused to lcrV was reported (44). The rAdsecV produced a secreted form of LcrV and elicited specific T-cell responses as well as high IgG titers in sera, which protected mice from a lethal intranasal challenge of Y. pestis CO92 in a single intramuscular immunization (44). Although there is no direct comparison, the AdsecV provided better protection (80 to 100%) in mice than our monovalent rAd5-LcrV vaccine (∼20%) (Fig. 1B and 2B), indicating that the secreted form of LcrV might be more immunogenic in mice. However, different species of mice (Swiss-Webster versus BALB/c) and challenge doses were used in these studies (44). In our initial study, an rAd5 expressing the Igκ secretion signal fused to YFV was successfully created; however, we found that the secreted YFV (sYFV) was toxic to HEK293 cells, which prevented large-scale expansion of this construct (data not shown).
There are several established plague models using NHPs, such as the langur monkey (56), African green vervet (57, 58), baboon (59, 60), and rhesus macaque (61, 62). However, the current recommendations from FDA and the National Institute of Allergy and Infectious Disease to support plague therapeutic and vaccine studies involve a cynomolgus macaque (Macaca fascicularis) (CM) pneumonic plague model (63). In addition, the lethal dose of Y. pestis has been established for aerosol challenge of CMs with the standard CO92 strain, and this model was utilized in protection studies including F1-V-based subunit vaccines for the past several years as well as in most recent studies (7, 8, 63–68). Importantly, CMs exhibit a clinical course of the disease similar to that described in humans (69).
Indeed, we observed that the unimmunized NHPs after WT CO92 aerosol challenge had cough, respiratory changes, lethargy, and hunched posture, as well as typical pneumonic lesions in the lungs (Fig. 9). However, no fever was observed during the course of infection. This is in contrast to the most recent report that the onset of fever was predominant across all CMs infected with Y. pestis (68). This highlights the importance of using telemetry to observe physiological parameters in a real-time manner. Our study did not employ telemetry, while the other report measured body temperature in real time and a temperature of 1.5°C above the baseline was considered fever (68). One notable finding of our study was that a significant increase in the antibody titer was noted in immunized NHPs, especially to F1, after rYFV boost as well as after WT CO92 challenge (Fig. 8; see Fig. S5 in the supplemental material). These data indicated memory B-cell-evoked recall responses. Similarly, a predominant hyperplasia of lymphoid follicles was observed in the immunized NHPs in the mediastinal lymph nodes and spleen for as long as 82 days after the WT CO92 challenge (Fig. 9), suggesting that a sustained immune response was developed in these NHPs, which could be pivotal in long-term protection of animals against plague. Our studies also indicated that by using the prime-boost strategy in CMs, higher antibody responses were generated than in animals that were immunized with only rAd5-YFV (Fig. 8). Average antibody titers of ∼1.7 × 106 for LcrV, ∼4.3 × 104 for F1, and ∼1.2 × 105 for YscF were mounted when animals were immunized following the prime-boost strategy. These antibodies titers were sufficient for providing complete protection to CMs against high aerosol challenge doses of Y. pestis CO92, although the role of cell-mediated immunity in protection should also be considered.
One of the major concerns involving adenoviral vectors for vaccine development is the preexisting immunity to Ad5 (in ∼95% of the human population) that could lessen the efficacy of the vaccine. Currently, most of the efforts to overcome the concerns regarding neutralizing antibodies have been focused on identifying alternative serotypes of adenovirus (70, 71). While some groups have reported favorable results with this approach, it offers only a short-term solution, as new adenoviral vector adaptation will result in the generation of neutralizing antibodies through widespread use. On the other hand, a number of studies indicated that administration of Ad5-vectored vaccines via the i.n. route might overcome preexisting immunity against the Ad5 vector (72–75). We did observe slightly lower Y. pestis antigen-specific antibody titers in mice with the preexisting adenoviral immunity than in those animals without the preexisting adenoviral immunity when mice were i.n. immunized with either the rAd5-LcrV or the rAd5-YFV vaccine (Fig. 2C). However, the protection conferred in mice against Y. pestis challenge was similar in both groups of mice irrespective of the preexisting adenoviral immunity (Fig. 2A and B). Most importantly, NHPs with preexisting adenoviral immunity and immunized with the rAd5-YFV vaccine plus a boost of rYFV were fully protected from a high aerosol challenge dose of WT CO92 (Fig. 7).
In addition to YscF, other Y. pestis antigens, such as the T3SS components YpkA, YopH, YopE, YopK, and YopN as well as a subunit of pH 6 antigen and purified lipopolysaccharide (LPS), were studied for their immunogenic efficacies against plague infection, but these did not generate promising results (76). The only protection was observed in mice vaccinated with YopD, a protein involved in the delivery of T3SS effectors into the host cell (77). However, YopD vaccination provided protection only against the nonencapsulated bacilli and not against the encapsulated Y. pestis CO92 strain.
As the most promising plague subunit vaccines currently under development are primarily dependent on only two antigens, F1 and LcrV, the incorporation of a new antigen, YscF, may help in formulating a better vaccine against all human plague-causing strains, as we showed using the bacteriophage T4-based platform (78). Furthermore, the adenoviral vector has been demonstrated to have adjuvant activities as well as the ability to promote cellular immunity (49, 79, 80). In this regard, our trivalent rAd5-YFV vaccine has unique advantages as a plague vaccine. Our further studies will include in-depth characterization of cell-mediated immune responses in vaccinated CMs.
Supplementary Material
ACKNOWLEDGMENTS
This research was supported by an NIH/NIAID SBIR grant (5R44AI071634) awarded to E.R. and A.K.C. We also acknowledge a UC7 grant (AI070083), which facilitated our research in the Galveston National Laboratory (GNL), UTMB, Galveston, TX. E.C.F. was supported in part by T32 Biodefense Training Grant AI060549. J.A.A. and B.L.T. were supported in part by the WHO Collaborating Center for Vaccine Development, and the latter also was supported by a Jeane B. Kempner predoctoral fellowship, UTMB. D.P. was supported in part by a James W. McLaughlin postdoctoral fellowship, UTMB.
We appreciate the help of Linsey Yeager during earlier studies with adenovirus and the mouse model of pneumonic plague. We thank members of W. B. Baze's Histopathology Laboratory for help with histopathological examination of tissue specimens. Special thanks go to the Animal Resource Center team (within GNL) for their help during NHP studies.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/CVI.00150-16.
REFERENCES
- 1.Smiley ST. 2008. Current challenges in the development of vaccines for pneumonic plague. Expert Rev Vaccines 7:209–221. doi: 10.1586/14760584.7.2.209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sun W, Roland KL, Curtiss R III. 2011. Developing live vaccines against plague. J Infect Dev Ctries 5:614–627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Perry RD, Fetherston JD. 1997. Yersinia pestis–etiologic agent of plague. Clin Microbiol Rev 10:35–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cornelis GR. 2002. Yersinia type III secretion: send in the effectors. J Cell Biol 158:401–408. doi: 10.1083/jcb.200205077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Powell BS, Andrews GP, Enama JT, Jendrek S, Bolt C, Worsham P, Pullen JK, Ribot W, Hines H, Smith L, Heath DG, Adamovicz JJ. 2005. Design and testing for a nontagged F1-V fusion protein as vaccine antigen against bubonic and pneumonic plague. Biotechnol Prog 21:1490–1510. doi: 10.1021/bp050098r. [DOI] [PubMed] [Google Scholar]
- 6.Alvarez ML, Pinyerd HL, Crisantes JD, Rigano MM, Pinkhasov J, Walmsley AM, Mason HS, Cardineau GA. 2006. Plant-made subunit vaccine against pneumonic and bubonic plague is orally immunogenic in mice. Vaccine 24:2477–2490. doi: 10.1016/j.vaccine.2005.12.057. [DOI] [PubMed] [Google Scholar]
- 7.Williamson ED, Flick-Smith HC, Waters E, Miller J, Hodgson I, Le Butt CS, Hill J. 2007. Immunogenicity of the rF1+rV vaccine for plague with identification of potential immune correlates. Microb Pathog 42:11–21. doi: 10.1016/j.micpath.2006.09.003. [DOI] [PubMed] [Google Scholar]
- 8.Cornelius CA, Quenee LE, Overheim KA, Koster F, Brasel TL, Elli D, Ciletti NA, Schneewind O. 2008. Immunization with recombinant V10 protects cynomolgus macaques from lethal pneumonic plague. Infect Immun 76:5588–5597. doi: 10.1128/IAI.00699-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Baker EE, Somer H, Foster LW, Meyer E, Meyer KF. 1952. Studies on immunization against plague. I. The isolation and characterization of the soluble antigen of Pasteurella pestis. J Immunol 68:131–145. [PubMed] [Google Scholar]
- 10.Rosenzweig JA, Jejelowo O, Sha J, Erova TE, Brackman SM, Kirtley ML, van Lier CJ, Chopra AK. 2011. Progress on plague vaccine development. Appl Microbiol Biotechnol 91:265–286. doi: 10.1007/s00253-011-3380-6. [DOI] [PubMed] [Google Scholar]
- 11.Quenee LE, Ciletti N, Berube B, Krausz T, Elli D, Hermanas T, Schneewind O. 2011. Plague in Guinea pigs and its prevention by subunit vaccines. Am J Pathol 178:1689–1700. doi: 10.1016/j.ajpath.2010.12.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Quenee LE, Ciletti NA, Elli D, Hermanas TM, Schneewind O. 2011. Prevention of pneumonic plague in mice, rats, guinea pigs and non-human primates with clinical grade rV10, rV10-2 or F1-V vaccines. Vaccine 29:6572–6583. doi: 10.1016/j.vaccine.2011.06.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lin JS, Kummer LW, Szaba FM, Smiley ST. 2011. IL-17 contributes to cell-mediated defense against pulmonary Yersinia pestis infection. J Immunol 186:1675–1684. doi: 10.4049/jimmunol.1003303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Smiley ST. 2008. Immune defense against pneumonic plague. Immunol Rev 225:256–271. doi: 10.1111/j.1600-065X.2008.00674.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Agar SL, Sha J, Foltz SM, Erova TE, Walberg KG, Baze WB, Suarez G, Peterson JW, Chopra AK. 2009. Characterization of the rat pneumonic plague model: infection kinetics following aerosolization of Yersinia pestis CO92. Microbes Infect 11:205–214. doi: 10.1016/j.micinf.2008.11.009. [DOI] [PubMed] [Google Scholar]
- 16.Williamson ED, Packer PJ, Waters EL, Simpson AJ, Dyer D, Hartings J, Twenhafel N, Pitt ML. 2011. Recombinant (F1+V) vaccine protects cynomolgus macaques against pneumonic plague. Vaccine 29:4771–4777. doi: 10.1016/j.vaccine.2011.04.084. [DOI] [PubMed] [Google Scholar]
- 17.FDA. 2012. African Green monkey (Chlorocebus aethiops) animal model development to evaluate treatment of pneumonic plague. FDA, Washington, DC. [Google Scholar]
- 18.Sha J, Endsley JJ, Kirtley ML, Foltz SM, Huante MB, Erova TE, Kozlova EV, Popov VL, Yeager LA, Zudina IV, Motin VL, Peterson JW, DeBord KL, Chopra AK. 2011. Characterization of an F1 deletion mutant of Yersinia pestis CO92, pathogenic role of F1 antigen in bubonic and pneumonic plague, and evaluation of sensitivity and specificity of F1 antigen capture-based dipsticks. J Clin Microbiol 49:1708–1715. doi: 10.1128/JCM.00064-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Quenee LE, Cornelius CA, Ciletti NA, Elli D, Schneewind O. 2008. Yersinia pestis caf1 variants and the limits of plague vaccine protection. Infect Immun 76:2025–2036. doi: 10.1128/IAI.00105-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Anisimov AP, Dentovskaya SV, Panfertsev EA, Svetoch TE, Kopylov P, Segelke BW, Zemla A, Telepnev MV, Motin VL. 2010. Amino acid and structural variability of Yersinia pestis LcrV protein. Infect Genet Evol 10:137–145. doi: 10.1016/j.meegid.2009.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Motin VL, Pokrovskaya MS, Telepnev MV, Kutyrev VV, Vidyaeva NA, Filippov AA, Smirnov GB. 1992. The difference in the lcrV sequences between Y. pestis and Y. pseudotuberculosis and its application for characterization of Y. pseudotuberculosis strains. Microb Pathog 12:165–175. doi: 10.1016/0882-4010(92)90050-X. [DOI] [PubMed] [Google Scholar]
- 22.Matson JS, Durick KA, Bradley DS, Nilles ML. 2005. Immunization of mice with YscF provides protection from Yersinia pestis infections. BMC Microbiol 5:38. doi: 10.1186/1471-2180-5-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Swietnicki W, Powell BS, Goodin J. 2005. Yersinia pestis Yop secretion protein F: purification, characterization, and protective efficacy against bubonic plague. Protein Expr Purif 42:166–172. doi: 10.1016/j.pep.2005.02.016. [DOI] [PubMed] [Google Scholar]
- 24.Sha J, Rosenzweig JA, Kirtley ML, van Lier CJ, Fitts EC, Kozlova EV, Erova TE, Tiner BL, Chopra AK. 2013. A non-invasive in vivo imaging system to study dissemination of bioluminescent Yersinia pestis CO92 in a mouse model of pneumonic plague. Microb Pathog 55:39–50. doi: 10.1016/j.micpath.2012.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tiner BL, Sha J, Kirtley ML, Erova TE, Popov VL, Baze WB, van Lier CJ, Ponnusamy D, Andersson JA, Motin VL, Chauhan S, Chopra AK. 2015. Combinational deletion of three membrane protein-encoding genes highly attenuates Yersinia pestis while retaining immunogenicity in a mouse model of pneumonic plague. Infect Immun 83:1318–1338. doi: 10.1128/IAI.02778-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Agar SL, Sha J, Foltz SM, Erova TE, Walberg KG, Parham TE, Baze WB, Suarez G, Peterson JW, Chopra AK. 2008. Characterization of a mouse model of plague after aerosolization of Yersinia pestis CO92. Microbiology 154:1939–1948. doi: 10.1099/mic.0.2008/017335-0. [DOI] [PubMed] [Google Scholar]
- 27.Suarez G, Sierra JC, Kirtley ML, Chopra AK. 2010. Role of Hcp, a type 6 secretion system effector, of Aeromonas hydrophila in modulating activation of host immune cells. Microbiology 156:3678–3688. doi: 10.1099/mic.0.041277-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.van Lier CJ, Tiner BL, Chauhan S, Motin VL, Fitts EC, Huante MB, Endsley JJ, Ponnusamy D, Sha J, Chopra AK. 2015. Further characterization of a highly attenuated Yersinia pestis CO92 mutant deleted for the genes encoding Braun lipoprotein and plasminogen activator protease in murine alveolar and primary human macrophages. Microb Pathog 80:27–38. doi: 10.1016/j.micpath.2015.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sha J, Kirtley ML, van Lier CJ, Wang S, Erova TE, Kozlova EV, Cao A, Cong Y, Fitts EC, Rosenzweig JA, Chopra AK. 2013. Deletion of the Braun lipoprotein-encoding gene and altering the function of lipopolysaccharide attenuate the plague bacterium. Infect Immun 81:815–828. doi: 10.1128/IAI.01067-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.van Lier CJ, Sha J, Kirtley ML, Cao A, Tiner BL, Erova TE, Cong Y, Kozlova EV, Popov VL, Baze WB, Chopra AK. 2014. Deletion of Braun lipoprotein and plasminogen-activating protease-encoding genes attenuates Yersinia pestis in mouse models of bubonic and pneumonic plague. Infect Immun 82:2485–2503. doi: 10.1128/IAI.01595-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sha J, Agar SL, Baze WB, Olano JP, Fadl AA, Erova TE, Wang S, Foltz SM, Suarez G, Motin VL, Chauhan S, Klimpel GR, Peterson JW, Chopra AK. 2008. Braun lipoprotein (Lpp) contributes to virulence of yersiniae: potential role of Lpp in inducing bubonic and pneumonic plague. Infect Immun 76:1390–1409. doi: 10.1128/IAI.01529-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Guyton AC. 1947. Measurement of the respiratory volumes of laboratory animals. Am J Physiol 150:70–77. [DOI] [PubMed] [Google Scholar]
- 33.Tiner BL, Sha J, Ponnusamy D, Baze WB, Fitts EC, Popov VL, van Lier CJ, Erova TE, Chopra AK. 7 October 2015. Intramuscular immunization of mice with a live-attenuated triple mutant of Yersinia pestis CO92 induces robust humoral and cell-mediated immunity to completely protect animals against pneumonic plague. Clin Vaccine Immunol doi: 10.1128/CVI.00499-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Agar SL, Sha J, Baze WB, Erova TE, Foltz SM, Suarez G, Wang S, Chopra AK. 2009. Deletion of Braun lipoprotein gene (lpp) and curing of plasmid pPCP1 dramatically alter the virulence of Yersinia pestis CO92 in a mouse model of pneumonic plague. Microbiology 155:3247–3259. doi: 10.1099/mic.0.029124-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Warren R, Lockman H, Barnewall R, Krile R, Blanco OB, Vasconcelos D, Price J, House RV, Bolanowksi MA, Fellows P. 2011. Cynomolgus macaque model for pneumonic plague. Microb Pathog 50:12–22. doi: 10.1016/j.micpath.2010.10.002. [DOI] [PubMed] [Google Scholar]
- 36.Russell P, Eley SM, Hibbs SE, Manchee RJ, Stagg AJ, Titball RW. 1995. A comparison of plague vaccine, USP and EV76 vaccine induced protection against Yersinia pestis in a murine model. Vaccine 13:1551–1556. doi: 10.1016/0264-410X(95)00090-N. [DOI] [PubMed] [Google Scholar]
- 37.Titball RW, Williamson ED. 2001. Vaccination against bubonic and pneumonic plague. Vaccine 19:4175–4184. doi: 10.1016/S0264-410X(01)00163-3. [DOI] [PubMed] [Google Scholar]
- 38.Titball RW, Williamson ED. 2004. Yersinia pestis (plague) vaccines. Expert Opin Biol Ther 4:965–973. doi: 10.1517/14712598.4.6.965. [DOI] [PubMed] [Google Scholar]
- 39.Cui Y, Yang X, Xiao X, Anisimov AP, Li D, Yan Y, Zhou D, Rajerison M, Carniel E, Achtman M, Yang R, Song Y. 2014. Genetic variations of live attenuated plague vaccine strains (Yersinia pestis EV76 lineage) during laboratory passages in different countries. Infect Genet Evol 26:172–179. doi: 10.1016/j.meegid.2014.05.023. [DOI] [PubMed] [Google Scholar]
- 40.Oyston PC, Dorrell N, Williams K, Li SR, Green M, Titball RW, Wren BW. 2000. The response regulator PhoP is important for survival under conditions of macrophage-induced stress and virulence in Yersinia pestis. Infect Immun 68:3419–3425. doi: 10.1128/IAI.68.6.3419-3425.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Oyston PC, Russell P, Williamson ED, Titball RW. 1996. An aroA mutant of Yersinia pestis is attenuated in guinea-pigs, but virulent in mice. Microbiology 142:1847–1853. doi: 10.1099/13500872-142-7-1847. [DOI] [PubMed] [Google Scholar]
- 42.Williams K, Oyston PC, Dorrell N, Li S, Titball RW, Wren BW. 2000. Investigation into the role of the serine protease HtrA in Yersinia pestis pathogenesis. FEMS Microbiol Lett 186:281–286. doi: 10.1111/j.1574-6968.2000.tb09118.x. [DOI] [PubMed] [Google Scholar]
- 43.Cathelyn JS, Crosby SD, Lathem WW, Goldman WE, Miller VL. 2006. RovA, a global regulator of Yersinia pestis, specifically required for bubonic plague. Proc Natl Acad Sci U S A 103:13514–13519. doi: 10.1073/pnas.0603456103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chiuchiolo MJ, Boyer JL, Krause A, Senina S, Hackett NR, Crystal RG. 2006. Protective immunity against respiratory tract challenge with Yersinia pestis in mice immunized with an adenovirus-based vaccine vector expressing V antigen. J Infect Dis 194:1249–1257. doi: 10.1086/507644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tatsis N, Ertl HC. 2004. Adenoviruses as vaccine vectors. Mol Ther 10:616–629. doi: 10.1016/j.ymthe.2004.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Boyer JL, Kobinger G, Wilson JM, Crystal RG. 2005. Adenovirus-based genetic vaccines for biodefense. Hum Gene Ther 16:157–168. doi: 10.1089/hum.2005.16.157. [DOI] [PubMed] [Google Scholar]
- 47.Barouch DH, Nabel GJ. 2005. Adenovirus vector-based vaccines for human immunodeficiency virus type 1. Hum Gene Ther 16:149–156. doi: 10.1089/hum.2005.16.149. [DOI] [PubMed] [Google Scholar]
- 48.Bessis N, GarciaCozar FJ, Boissier MC. 2004. Immune responses to gene therapy vectors: influence on vector function and effector mechanisms. Gene Ther 11(Suppl 1):S10–S17. doi: 10.1038/sj.gt.3302364. [DOI] [PubMed] [Google Scholar]
- 49.Molinier-Frenkel V, Lengagne R, Gaden F, Hong SS, Choppin J, Gahery-Segard H, Boulanger P, Guillet JG. 2002. Adenovirus hexon protein is a potent adjuvant for activation of a cellular immune response. J Virol 76:127–135. doi: 10.1128/JVI.76.1.127-135.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hackett NR, Kaminsky SM, Sondhi D, Crystal RG. 2000. Antivector and antitransgene host responses in gene therapy. Curr Opin Mol Ther 2:376–382. [PubMed] [Google Scholar]
- 51.Song W, Kong HL, Traktman P, Crystal RG. 1997. Cytotoxic T lymphocyte responses to proteins encoded by heterologous transgenes transferred in vivo by adenoviral vectors. Hum Gene Ther 8:1207–1217. doi: 10.1089/hum.1997.8.10-1207. [DOI] [PubMed] [Google Scholar]
- 52.Wilson JM. 1996. Adenoviruses as gene-delivery vehicles. N Engl J Med 334:1185–1187. doi: 10.1056/NEJM199605023341809. [DOI] [PubMed] [Google Scholar]
- 53.Tripathy SK, Black HB, Goldwasser E, Leiden JM. 1996. Immune responses to transgene-encoded proteins limit the stability of gene expression after injection of replication-defective adenovirus vectors. Nat Med 2:545–550. doi: 10.1038/nm0596-545. [DOI] [PubMed] [Google Scholar]
- 54.Lathem WW, Crosby SD, Miller VL, Goldman WE. 2005. Progression of primary pneumonic plague: a mouse model of infection, pathology, and bacterial transcriptional activity. Proc Natl Acad Sci U S A 102:17786–17791. doi: 10.1073/pnas.0506840102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Do Y, Didierlaurent AM, Ryu S, Koh H, Park CG, Park S, Perlin DS, Powell BS, Steinman RM. 2012. Induction of pulmonary mucosal immune responses with a protein vaccine targeted to the DEC-205/CD205 receptor. Vaccine 30:6359–6367. doi: 10.1016/j.vaccine.2012.08.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chen TH, Meyer KF. 1965. Susceptibility of the langur monkey (Semnopithecus entellus) to experimental plague: pathology and immunity. J Infect Dis 115:456–464. doi: 10.1093/infdis/115.5.456. [DOI] [PubMed] [Google Scholar]
- 57.Hallett AF, Isaacson M, Meyer KF. 1973. Pathogenicity and immunogenic efficacy of a live attentuated plaque vaccine in vervet monkeys. Infect Immun 8:876–881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chen TH, Elbert SS, Eisler DM. 1976. Immunity in plague: protection induced in Cercopithecus aethiops by oral administration of live, attenuated Yersinia pestis. J Infect Dis 133:302–309. doi: 10.1093/infdis/133.3.302. [DOI] [PubMed] [Google Scholar]
- 59.Stacy S, Pasquali A, Sexton VL, Cantwell AM, Kraig E, Dube PH. 2008. An age-old paradigm challenged: old baboons generate vigorous humoral immune responses to LcrV, a plague antigen. J Immunol 181:109–115. doi: 10.4049/jimmunol.181.1.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Byvalov AA, Pautov VN, Chicherin Iu V, Lebedinskii VA, Evtigneev VI. 1984. Effectiveness of revaccinating hamadryas baboons with NISS live dried plague vaccine and fraction I of the plague microbe. Zh Mikrobiol Epidemiol Immunobiol 4:74–76. [PubMed] [Google Scholar]
- 61.Ransom JP, Krueger AP. 1954. Chronic pneumonic plague in Macaca mulatta. Am J Trop Med Hyg 3:1040–1054. [DOI] [PubMed] [Google Scholar]
- 62.Finegold MJ, Petery RF, Berendt RF, Adams HR. 1968. Studies on the pathogenesis of plague: blood coagulation and tissue responses of Macaca mulatta following exposure to aerosols of Pasteurella pestis. Am J Pathol 53:99–114. [PMC free article] [PubMed] [Google Scholar]
- 63.Van Andel R, Sherwood R, Gennings C, Lyons CR, Hutt J, Gigliotti A, Barr E. 2008. Clinical and pathologic features of cynomolgus macaques (Macaca fascicularis) infected with aerosolized Yersinia pestis. Comp Med 58:68–75. [PMC free article] [PubMed] [Google Scholar]
- 64.Mett V, Lyons J, Musiychuk K, Chichester JA, Brasil T, Couch R, Sherwood R, Palmer GA, Streatfield SJ, Yusibov V. 2007. A plant-produced plague vaccine candidate confers protection to monkeys. Vaccine 25:3014–3017. doi: 10.1016/j.vaccine.2007.01.017. [DOI] [PubMed] [Google Scholar]
- 65.Welkos S, Norris S, Adamovicz J. 2008. Modified caspase-3 assay indicates correlation of caspase-3 activity with immunity of nonhuman primates to Yersinia pestis infection. Clin Vaccine Immunol 15:1134–1137. doi: 10.1128/CVI.00091-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Mizel SB, Graff AH, Sriranganathan N, Ervin S, Lees CJ, Lively MO, Hantgan RR, Thomas MJ, Wood J, Bell B. 2009. Flagellin-F1-V fusion protein is an effective plague vaccine in mice and two species of nonhuman primates. Clin Vaccine Immunol 16:21–28. doi: 10.1128/CVI.00333-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Koster F, Perlin DS, Park S, Brasel T, Gigliotti A, Barr E, Myers L, Layton RC, Sherwood R, Lyons CR. 2010. Milestones in progression of primary pneumonic plague in cynomolgus macaques. Infect Immun 78:2946–2955. doi: 10.1128/IAI.01296-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Fellows P, Price J, Martin S, Metcalfe K, Krile R, Barnewall R, Hart MK, Lockman H. 2015. Characterization of a cynomolgus macaque model of pneumonic plague for evaluation of vaccine efficacy. Clin Vaccine Immunol 22:1070–1078. doi: 10.1128/CVI.00290-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Pitt ML. 2004. Non-human primates as a model for pneumonic plague. In Animal Models and Correlates of Protection for Plague Vaccines Workshop. FDA, Washington, DC. [Google Scholar]
- 70.Barouch DH, Pau MG, Custers JH, Koudstaal W, Kostense S, Havenga MJ, Truitt DM, Sumida SM, Kishko MG, Arthur JC, Korioth-Schmitz B, Newberg MH, Gorgone DA, Lifton MA, Panicali DL, Nabel GJ, Letvin NL, Goudsmit J. 2004. Immunogenicity of recombinant adenovirus serotype 35 vaccine in the presence of pre-existing anti-Ad5 immunity. J Immunol 172:6290–6297. doi: 10.4049/jimmunol.172.10.6290. [DOI] [PubMed] [Google Scholar]
- 71.Nanda A, Lynch DM, Goudsmit J, Lemckert AA, Ewald BA, Sumida SM, Truitt DM, Abbink P, Kishko MG, Gorgone DA, Lifton MA, Shen L, Carville A, Mansfield KG, Havenga MJ, Barouch DH. 2005. Immunogenicity of recombinant fiber-chimeric adenovirus serotype 35 vector-based vaccines in mice and rhesus monkeys. J Virol 79:14161–14168. doi: 10.1128/JVI.79.22.14161-14168.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Zhang J, Jex E, Feng T, Sivko GS, Baillie LW, Goldman S, Van Kampen KR, Tang DC. 2013. An adenovirus-vectored nasal vaccine confers rapid and sustained protection against anthrax in a single-dose regimen. Clin Vaccine Immunol 20:1–8. doi: 10.1128/CVI.00280-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Croyle MA, Patel A, Tran KN, Gray M, Zhang Y, Strong JE, Feldmann H, Kobinger GP. 2008. Nasal delivery of an adenovirus-based vaccine bypasses pre-existing immunity to the vaccine carrier and improves the immune response in mice. PLoS One 3:e3548. doi: 10.1371/journal.pone.0003548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Xu Q, Pichichero ME, Simpson LL, Elias M, Smith LA, Zeng M. 2009. An adenoviral vector-based mucosal vaccine is effective in protection against botulism. Gene Ther 16:367–375. doi: 10.1038/gt.2008.181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Yu JR, Kim S, Lee JB, Chang J. 2008. Single intranasal immunization with recombinant adenovirus-based vaccine induces protective immunity against respiratory syncytial virus infection. J Virol 82:2350–2357. doi: 10.1128/JVI.02372-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Benner GE, Andrews GP, Byrne WR, Strachan SD, Sample AK, Heath DG, Friedlander AM. 1999. Immune response to Yersinia outer proteins and other Yersinia pestis antigens after experimental plague infection in mice. Infect Immun 67:1922–1928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Andrews GP, Strachan ST, Benner GE, Sample AK, Anderson GW Jr, Adamovicz JJ, Welkos SL, Pullen JK, Friedlander AM. 1999. Protective efficacy of recombinant Yersinia outer proteins against bubonic plague caused by encapsulated and nonencapsulated Yersinia pestis. Infect Immun 67:1533–1537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Tao P, Mahalingam M, Kirtley ML, van Lier CJ, Sha J, Yeager LA, Chopra AK, Rao VB. 2013. Mutated and bacteriophage T4 nanoparticle arrayed F1-V immunogens from Yersinia pestis as next generation plague vaccines. PLoS Pathog 9:e1003495. doi: 10.1371/journal.ppat.1003495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Jones FR, Gabitzsch ES, Xu Y, Balint JP, Borisevich V, Smith J, Peng BH, Walker A, Salazar M, Paessler S. 2011. Prevention of influenza virus shedding and protection from lethal H1N1 challenge using a consensus 2009 H1N1 HA and NA adenovirus vector vaccine. Vaccine 29:7020–7026. doi: 10.1016/j.vaccine.2011.07.073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Patel A, Zhang Y, Croyle M, Tran K, Gray M, Strong J, Feldmann H, Wilson JM, Kobinger GP. 2007. Mucosal delivery of adenovirus-based vaccine protects against Ebola virus infection in mice. J Infect Dis 196(Suppl 2):S413–S420. doi: 10.1086/520603. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.