

Abstract

In bottom-up mass spectrometry (MS)-based proteomics, peptide isotopic and chromatographic traces (features) are frequently used for label-free quantification in data-dependent acquisition MS but can also be used for the improved identification of chimeric spectra or sample complexity characterization. Feature detection is difficult because of the high complexity of MS proteomics data from biological samples, which frequently causes features to intermingle. In addition, existing feature detection algorithms commonly suffer from compatibility issues, long computation times, or poor performance on high-resolution data. Because of these limitations, we developed a new tool, Dinosaur, with increased speed and versatility. Dinosaur has the functionality to sample algorithm computations through quality-control plots, which we call a plot trail. From the evaluation of this plot trail, we introduce several algorithmic improvements to further improve the robustness and performance of Dinosaur, with the detection of features for 98% of MS/MS identifications in a benchmark data set, and no other algorithm tested in this study passed 96% feature detection. We finally used Dinosaur to reimplement a published workflow for peptide identification in chimeric spectra, increasing chimeric identification from 26% to 32% over the standard workflow. Dinosaur is operating-system-independent and is freely available as open source on https://github.com/fickludd/dinosaur.
Keywords: proteomics, mass spectrometry, electrospray ionization, feature detection, chimeric spectra, algorithm, software
Introduction
Mass-spectrometry-based proteomics, fueled by seemingly ever-increasing instrument performance, has gained considerable traction as a high-throughput method for biomarker discovery and systems biology applications. New and flexible workflows have extended the range of mass spectrometry (MS) applications in life science research from biomarker discovery to complex interaction mapping,1,2 whole-proteome assay libraries,3,4 mRNA and protein dynamics,5 or full structural characterization of a protein complex.6 In bottom-up MS proteomics, mass-spectrometry analysis of peptides, derived from intact proteins using proteolytic processing, allows the measurement of thousands of isotope envelopes of individual peptide ions. These isotope envelopes appear as ion intensity patterns in the retention time and m/z dimensions and are often referred to as features. Accurate feature detection is an important step in many mass-spectrometry-based proteomics workflows7−9 to quantify identified peptides in shotgun mass spectrometry by using the integrated or apex feature intensity. This holds in particular for label-free10 and SILAC11 workflows. Accurately determined features are also used to increase the accuracy of estimated precursor masses, to determine chromatography performance and optimize chromatographic gradients,12 and to determine the total number of detectable peptides by analyzing feature charge states, masses, and retention times. Features can also be used to perform untargeted analysis of data-independent acquisition (DIA) experiments.13 Lastly, recent work has shown that feature information can increase the identification rates of chimeric MS/MS spectra, providing a cheap possibility to increase the number of identified spectra and the quality of the data analysis.14
Feature detection in proteomics and metabolomics mass spectrometry has historically received considerable attention. Although Listgarten and Emili thoroughly summarize early work on the subject,7 label-free quantification of shotgun proteomics experiments has resulted in several additional published feature-detection tools like, for example, msInspect,15 SuperHirn,16 OpenMS,17 centWave,18 Hardklör,19 and MZmine.20 In 2008, MaxQuant introduced a graph model to improve feature detection in dense proteomics data derived from complex biological samples.21 Later work has been driven by changing data quality; modern Fourier transform high-resolution instruments have reduced the need for spectral noise reduction and are not suited for m/z range binning, which were both prominent parts of the early algorithms.8 Kalman filters have also been employed for feature detection.22,23 The large number of available feature detection algorithms calls for strategies to evaluate the outcome of feature detection. A straightforward method for such evaluation is to ensure that the feature list returned by the feature detection algorithm contains the peaks that were confidently identified by data-dependent MS/MS spectra.24
A feature-detection tool should ideally have several properties. First, the tool should detect and distinguish all features for the detectable peptide ions in an LC–MS experiment. Second, it should provide accurate measurements of feature retention times and monoisotopic masses in addition to providing accurate quantitative values. Third, it should preferably compute as fast as possible, be robust against irregularities in input data, and be flexible in terms of input data formats, output, and computational environment. Finally, the tool should provide parameters for optimization and means for the users to evaluate the effects of any modified parameter. Although MS1 feature detection is a well-studied problem,7−9,25 we believe further progress can be made toward these ideal properties. Problems with existing algorithms are, for example, operating system limitations, lack of support for standard file formats, slow algorithm execution, limited control output, and closed or outdated source code. We also experience difficulties in understanding how modification of algorithm parameters influences performance, which prevents adoption of a given tool to new instruments or acquisition conditions.
In the work presented here, we describe a new MS1 feature-detection tool called Dinosaur, based on concepts adapted from the feature detection algorithm in the MaxQuant21 software, which is widely used for analysis of shotgun mass-spectrometry data. Importantly, with Dinosaur, we introduce a new strategy of creating a set of quality-control plots, referred to as a plot trail, to support the monitoring of computation performance. On the basis of an extensive evaluation of the plot trail, we introduce several modifications to the original feature-detection algorithm to improve performance and robustness. The Dinosaur implementation is heavily optimized and parallelized and handles data files from modern mass spectrometers in minutes on any major operating system.
We evaluated the Dinosaur performance by benchmarking against four previously published feature-detection algorithms on a 5 orders of magnitude dilution series of synthetic peptides in a complex biological background.26 Dinosaur achieved an equally high level of linearity compared to the other tools while matching features to a larger percentage of identified peptides. We also demonstrate the utility of Dinosaur by implementing it in a chimeric spectrum identification workflow14 to increase the number of detected unique peptides by 32% at a 1% false discovery rate (FDR).
Experimental Methods
Data Acquisition
The synthetic peptide dilution series (PXD001091) and HeLa data sets (PXD000999) were downloaded from ProteomeXchange. The eight sample-type representative samples were prepared in-lab using standard protocols and have been deposited to the ProteomeXchange Consortium27 via the PRIDE partner repository with the data set identifier PXD003405. Acquisition was performed on a Q Exactive Plus mass spectrometer (Thermo Scientific) in top-15 mode, coupled to an EASY-nLC 1000 ultrahigh-pressure liquid chromatography system (Thermo Scientific). Linear gradients of between 5% and 35% acetonitrile in aqueous 0.1% formic acid were run for 30, 60, 90, or 120 min at a flow rate of 300 nL/min. The “purified protein” sample is a His-tagged Streptococcus pyogenes M-protein expressed in Escherichia coli; “synthetic peptides” refers to 20 synthetic peptides of 80% purity (Proteogenix); “AP-MS” is an affinity purification experiment using His-tagged S. pyogenes M-protein as a bait in mouse plasma; “plasma” and “depl. plasma” are nondepleted and, respectively, depleted mouse plasma; “bacteria” refers to blood agar grown S. pyogenes; “yeast” is MS-compatible yeast protein extract digest (Promega, V7461); and “tissue lysate” is a complete mouse liver tissue lysate. Mouse-derived data are from Malmström et al.28 All raw data files were converted to gzipped and MS-Numpressed mzML29 using the command-line version of the Proteowizard (3.0.5930) tool msconvert.30
MS Data Analysis
When we analyzed the synthetic peptide dilution series, MaxQuant 1.3.0.5 and msInspect (build 599) settings were as previously published.26 MaxQuant 1.5 has become available since we setup our analysis pipeline, but feature results from v1.3 and v1.5 are very similar, and thus, we have kept our results from v1.3. MaxQuant 1.1.0.25 was used to represent the original feature detection algorithm using default settings. Identification of MS/MS spectra was performed and combined within the Proteios Software Environment,31 using X!Tandem TORNADO 2008.12.01.1 (www.thegpm.org/tandem) and Mascot 2.3.01 (www.matrixscience.com) with 7 ppm precursor tolerance and 0.5 Da fragment tolerance, using fixed carbamidomethylation of cysteins and variable oxidation of methionines as described previously. Features were matched to MS/MS identifications passing a 1% peptide-spectrum-match level FDR using a 0.005 Da and 0.2 min threshold. When we mapped multiple identifications to a feature, no control was made for identification consistency, an effect with an estimated magnitude of <0.5%. Advanced feature-identification matching is available as a Proteios plugin, but for our purposes, we reimplemented a simplified version of this functionality to be able to count the mock matches. This code is available at https://github.com/fickludd/dinosaur. Dinosaur 1.1.0 was run using default parameter settings on all files. OpenMS (1.11.1, May 2014) settings for the DeMix workflow were as originally described; in short, a default PeakPickerHiRes configuration was used for centroiding, followed by a FeatureFinderCentroided configuration that allowed precursor charges of 2–7. The used TOPPAS32 workflow is accessible along with the Dinosaur source code. For the DeMix pipeline, searches were performed with MS-GF+,33 with trypsin set as the enzyme, an instrument setting of 3, an initial precursor tolerance of 10 ppm, a minimum peptide length of 7, and fixed carbamidomethylation of cysteins and variable oxidation of methionines as modifications, as in the DeMix publication.14
Runtime Measurement
Program run times were measured by the reported total time for Dinosaur (enabled using the −profiling flag) and as the total feature finder execution time for OpenMS, excluding centroiding. For the OpenMS–Dinosaur comparison, both programs were run with concurrency of eight on a 12 core computation server running Ubuntu Server 14.04 LTS. MaxQuant-ref was timed by starting the analysis and measuring the time taken until MS2-related files were first written. For MaxQuant, the total time for running steps 1–4 is reported as “MaxQuant full”, and the time taken by the “feature detection” subprocess until the appearance of MS2-related files is reported as “MaxQuant part”. Dinosaur–MaxQuant comparisons were performed with concurrency of one on a SSD-equipped desktop computer running Windows 7. All time measurements refer to wall time.
Software Implementation
Dinosaur is implemented in Scala 2.10.0 and compiled for Java Virtual Machine (JVM) using reactive programing techniques and Akka actors to enable parallelization. Scala is a high-level language that gracefully blends object-oriented programming with functional programming, and actors are a program construct that send immutable messages to one another asynchronously and receives and processes each message atomically. These choices of language and parallelization library do not affect the appearance of the final program, which appears as a regular Java program. Dependencies are managed using Maven, and in-house libraries (https://github.com/fickludd/proteomicore) were used for mzML parsing, supporting all mzML versions including zlib, gzip and MS-Numpress34 compression, centroid or profile, indexed or not. All Dinosaur source code and a self-contained standalone executable can be downloaded or compiled from https://github.com/fickludd/dinosaur along with modified scripts for running DeMix with Dinosaur under the Apache2 open-source license. The label-free shotgun quantification workflow using Dinosaur is freely available as part of Proteios at www.proteios.org.
Results
Implementation of Dinosaur and Algorithm Improvements
The implemented Dinosaur feature detection algorithm consists schematically of four computational steps, as shown in Figure 1. In the first step, profile spectra are centroided (Figure 1a). Because the most common MS methods perform repeated MS measurements during the elution of an analyte, the second step assembles centroid peaks with similar m/z in consecutive spectra into hills, meaning that one hill represents the full chromatographic trace of one analyte ion isotope (Figure 1b). In the third step, the hills are clustered according to plausible m/z differences based on carbon and sulfur natural isotopes and considered charge states (Figure 1c). Because these clusters are not consistent in charge state or expected isotopic prevalence, the fourth and last step is hill cluster isotopic deconvolution into individual features with consistent charge (Figure 1d).
Figure 1.

Overview of the Dinosaur feature-finding algorithm. Features are detected by (a) centroiding MS1 spectra; (b) assembling centroid peaks into single isotope traces, also referred to as hills; (c) clustering of hills by theoretically possible m/z differences; and (d) deconvolution of clusters into charge-state-consistent features. The plot trail plots randomly selected parts of the intermediary data to support the tuning of parameters and increase transparency of the computational steps. Created plot types are (e) a line graph of a peak centroid, (f) a heatmap and histogram of hill construction, (g) an isotopic profile compared to an averagine, and (h) a complete heatmap of a data section with annotated detected features.
To improve the understanding and transparency of the algorithm and its parameters, Dinosaur randomly selects and visualizes parts of the data at each computation step. We call this a plot trail (Figure 1e–h). The plot trail facilitates intelligent optimization of the 50 Dinosaur parameters and supports the users to minimize the risk of major computational mistakes or acquisition errors. The Dinosaur plot trail consists of four types of plots; a line graph of a centroided peak (Figure 1e), a heatmap and histogram of a constructed hill (Figure 1f), a line graph of a feature isotopic profile (Figure 1g), and a heatmap of a whole section of data with annotated features (Figure 1h). Plot trail samples are selected uniformly over the considered dimensions. The centroid peak plots consist of the least, most, and median intense centroided peak from one spectrum. The hill plots consist of the shortest, longest, and median length hills with their apex at the same spectrum. Isotopic profile plots are of one feature each, sampled uniformly from all features. Heatmaps are 15 m/z times 150 spectra by default. All sampling in the m/z and spectrum (rt) dimensions is done uniformly. Additionally, Dinosaur supports a targeted plot trail, where a list of m/z and retention time coordinates creates corresponding annotated heatmaps. The targeted plot trail is practical to evaluate the algorithm performance for features of particular interest.
The implementation of the plot trail supports the detailed analysis of individual features in conjunction with MS/MS identifications. Dinosaur was initially a reimplementation of the MaxQuant feature-finding algorithm, as described in the original manuscript21 (hereafter called MaxQuant-ref to distinguish it from later MaxQuant versions with unpublished algorithms). However, using the plot trail during implementation motivated several algorithm modifications to improve performance and robustness. These improvements are documented below and made the final algorithm to distinctly deviate from its ancestor. The complete algorithm and implementation is described within the source code.
First, several changes were introduced in the hill-building step to reduce the occasional intermingling of traces from separate features, which happens as multiple analytes frequently share almost identical retention time and m/z. The centWave18 algorithm, used for hill construction in MaxQuant-ref, relies on a simple maximal offset in ppm between centroid peaks. We reduce the feature intermingling by instead comparing new centroid peaks to the sliding average of the last three peaks in a hill to reduce the effect of single-spectrum m/z fluctuations. Further, this matching is performed in a greedy fashion in centWave, meaning that the first found match is selected, but this also leads to intermingling in crowded or noisy parts of the data because of the relatively large 7 ppm maximal offset. In contrast, Dinosaur temporarily stores all matching hills and centroid peaks in a list until the next pair is outside the maximal offset. At this point, the hills and centroid peaks are matched from the list in the order of the minimal m/z difference until no more matches are within the maximal offset. Finally, hill-profile smoothing in Dinosaur uses only three-point sliding windows versus a five-point window in MaxQuant-ref. After the removal of the artifacts introduced by intermingling, the improvements of the hill construction are expected to reduce the misassignment of features in the later steps and, therefore, increase the algorithm’s sensitivity.
A second improvement relates to the hill cluster deconvolution of large clusters. This phenomenon is more frequent in highly complex samples and at the end of a chromatographic run, when remnants on the column are washed off by a sudden increase in organic solvent. During deconvolution, all of the hills in a cluster are compared against each other, giving a computation of at least quadratic complexity, which becomes prohibitively slow for clusters with thousands of hills. This was solved in MaxQuant-ref by arbitrarily splitting clusters above 100 hills into two halves and recursively running the deconvolution on each half instead. This solution may split the hills of a feature into separate subclusters, resulting in failure to detect such a feature. Dinosaur instead sorts the hills by summed intensity and only seeds isotope patterns from the 100 most intense hills, after which the longest pattern is selected and the process repeated on the remaining hills until no further patterns are found. This heuristic limits computation complexity while avoiding arbitrary splitting of clusters and thus potentially increases algorithm sensitivity.
Third, the trimming of isotope patterns is not well-documented in the original MaxQuant-ref algorithm. From the original source code, it can be seen that the algorithm relies on a few heuristic rules based on the seed mass. Instead, Dinosaur compares the suggested isotope pattern intensity profile to the shifted versions of an averagine peptide35 of the same mass. The shifted averagine pattern is scored by the cosine correlation to the feature isotope profile, multiplied by the explanation percentage of the matched averagine peaks. The highest-scoring shift is selected to determine the monoisotopic mass of the isotope pattern and to discard features that are not similar enough (by default, cosine corr of ≥0.6) to the averagine isotopic pattern. This change is expected to increase the correct assignment of the feature monoisotopic mass, which is crucial for matching features to MS/MS identifications and for label-free alignment in any high-resolution MS workflow.
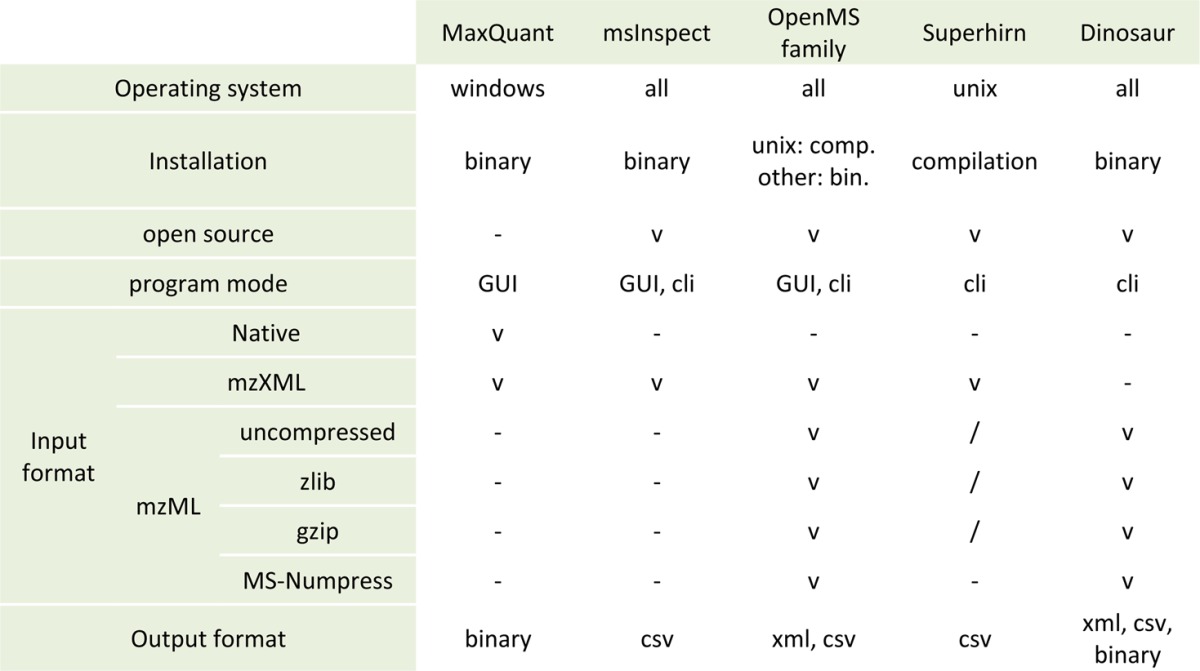
Apart from direct algorithm improvements, Dinosaur aims to be a flexible tool. It runs on all major operating systems and accepts all configurations of the MS data standard format mzML,29 including gzipped and MS-Numpressed files.34 Parameters are controlled directly via the command line or through a configuration file. Dinosaur is parallelized with a configurable degree of concurrency. In addition, Dinosaur is provided in a self-contained executable jar file without any other requirement than the Java Runtime Environment 1.6 or higher. The complete Scala source code for compiling Dinosaur is freely available from http://github.com/fickludd/dinosaur. These properties are summarized for Dinosaur and other common feature detection tools in Table 1. Finally, the plot trail allows users to correctly configure Dinosaur and transparently evaluate its performance on their samples (see the Supplementary Discussion section).
Table 1. Compatibility and Metadata of Dinosaur and Common-Feature-Detection Tools.
Dinosaur Comparative Performance and Usability
Dinosaur was evaluated on three data sets to test different aspects of the tool. First, we compare the raw algorithm performance to four other feature-detection tools on a dilution series of synthetic peptides. Second, we investigate the computation speed and usability of feature data on eight samples that were representative of different mass-spectrometry-based experiments. Third, we demonstrate an application of Dinosaur in a workflow using three HeLa cell lysate injections in a chimeric spectrum identification workflow.
We evaluated the performance of Dinosaur by comparing Dinosaur results to those obtained with msInspect,15 MaxQuant 1.1 (representing MaxQuant-ref), MaxQuant 1.3, and the OpenMS FeatureFinderCentroided17 using a dilution series of synthetic peptides in a bacterial background.26 The dilution series consisted of 273 crude synthetic peptides, log–linearly cross-diluted at 12 dilution levels ranging from 20 pmol/μL to 200 amol/μL. Each dilution point was analyzed in four or seven replicates for a total of 57 separate injections. Data was collected using a standard data-dependent acquisition method on an Orbitrap XL mass spectrometer.
In our hands, Dinosaur compares favorably to the other tested algorithms (Figure 2). We used the same set of 350 551 synthetic and bacterial peptide spectrum matches at 1% FDR to evaluate the performance of the different feature detection tools. The percentage of feature-matched MS/MS identifications was higher for Dinosaur (97.8 ± 0.5%) than MaxQuant (94.5 ± 0.9%), Open MS (95.7% ± 0.7%), MaxQuant-ref (89.0% ± 1.4%), and msInspect (81.3 ± 0.9%) (Figure 2a). These numbers of matched MS/MS identifications were compensated for spurious pairings by control-matching with shuffled MS2 events (see the Supplementary Material section) and therefore represents a quality measure of the detected features because identified peptides were filtered at 1% FDR and should in almost all cases have corresponding MS1 features. The tests with matching of shuffled MS2 events also indicated that the high number of MS/MS matches seen for Dinosaur was not an effect of matches due to high numbers of randomly detected features, as the algorithms matched 2–7% of mock MS/MS events (Figure S-2).
Figure 2.

Dinosaur, msInspect, MaxQuant, MaxQuant-ref, and Open MS feature detection performance based on 57 LC–MS injections of a dilution series of synthetic peptides in bacterial background. (a) The proportion of MS/MS identifications at 1% FDR that were matched to a feature. (b) Log–log coefficients of correlation of feature summed intensities vs theoretical synthetic peptide concentration.
In terms of quantitative linearity versus theoretical concentrations of the synthetic peptides, results seem limited by instrumentation rather than algorithm, as all tested algorithms have similar median coefficients of correlation (r2) around 0.95 (Figure 2b). In total, all algorithms detected features for a median 17% of the synthetic peptides across all of the analyzed LC–MS injections. The low coverage is expected because of the sensitivity limitations of shotgun MS/MS identifications and the very diluted synthetic peptides in the sample and because no identity propagation between samples was performed.26 We conclude that Dinosaur achieves higher sensitivity of matched MS/MS identifications without sacrificing quantitative accuracy compared to the other algorithms.
To deeper characterize the observed increased coverage of MS/MS identifications, we normalized the reported features from all tools by the median intensity of each tool on identifications that were matched by all, and we compared feature intensity distributions (Figure 3). In absolute numbers, the feature-intensity distributions of the five tools are highly similar, presumably reflecting the overall composition of detectable peptide ions in the sample (Figure 3a). If we, however, account for this by analyzing the relative number of features in each intensity bin, differences in the tools become apparent (Figure 3b). The MaxQuant tools have a relatively strong performance on the lower end of feature intensities, and the OpenMS feature detector performs very well in the high-intensity end. Dinosaur, however, achieves top numbers of matched features across the normalized intensity scale, explaining the observed high degree of matched MS/MS identifications. We thus conclude that Dinosaur is capable of full-dynamic-range feature detection and appears to match the differential strength of two previous top-performing tools.
Figure 3.
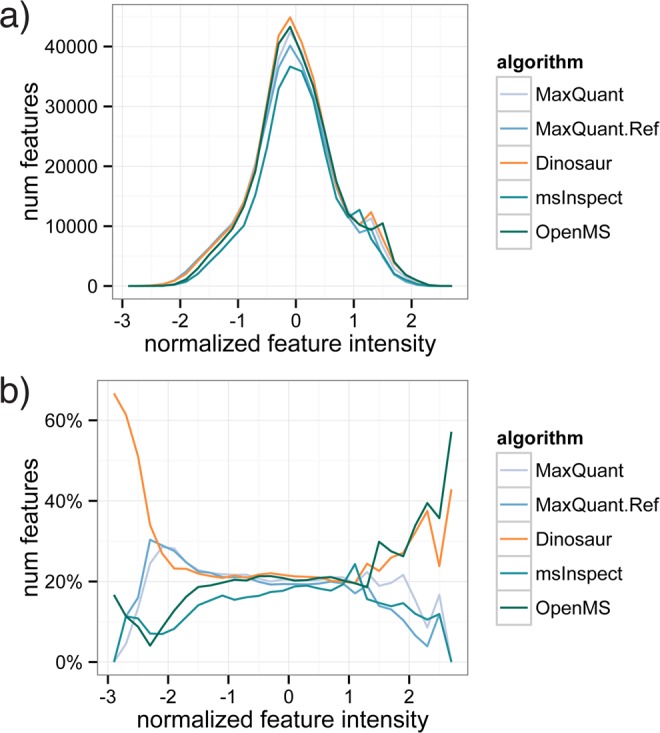
Intensity distribution of ID-matched features for compared feature-detection tools. Feature intensities from each tool were normalized by the division by the median intensity of features linked to IDs that all tools matched. (a) Absolute distribution of features over the normalized intensity range. (b) The relative number of features in each intensity bin. MaxQuant tools are relatively strong at low-intensity features, and the OpenMS tool is relatively strong at high-intensity features. Dinosaur shares both these strengths.
Apart from hard performance metrics, a mature software tool should be applicable in a variety of contexts and be reasonably fast. We challenged Dinosaur by analyzing MS data from eight different samples ranging from a purified protein to full tissue lysates of thousands of proteins. The eight different samples represent typical samples analyzed in proteomics studies with a representative range of complexities (Figure 4a–d). In total, Dinosaur detected 412 331 features of charge 2 or higher in the eight files. These features were used to plot the empirical distributions of feature intensities and retention times and to characterize each sample’s complexity and chromatographic performance. The number of features and their intensity was clearly dependent on the expected sample complexity (Figure 4a), with the highest number of features being detected in a murine liver tissue lysate and the least in the injection of a purified protein. When we analyzed feature retention times, the different gradient length became evident. The highest rate of detected features per minute was found in a 60 min gradient of a yeast sample (Figure 4b), where Dinosaur detected roughly 1000 features per min. These results are in accordance with a previously reported rate of 1500 features per min.36 Finally, we profiled the speed of Dinosaur by measuring the computation time of feature detection in the eight representative files. Dinosaur’s speed was compared to MaxQuant-, MaxQuant-ref-, and the OpenMS-based feature-finding workflow. In our results, no tool displayed a strong correlation between computation time and sample complexity, but Dinosaur often achieved the shortest runtime, reducing runtimes by 5–50% over the MaxQuant versions and by 70% over OpenMS (Figure 4c,d). In summary, the analysis of the eight representative samples show the robustness, speed, and usability of Dinosaur and how descriptive information regarding sample complexity and chromatography can be obtained from Dinosaur-derived features.
Figure 4.

Usability of Dinosaur. (a–d) Typical proteomics samples were represented by eight samples of different complexity. In total, 583 793 features were detected, of which the 412 331 of charge two or higher are included here. (a) Distributions of detected feature intensities for the eight samples. (b) Distributions of feature retention times for the eight samples. (c) Computation times of the eight samples for Dinosaur compared to MaxQuant and MaxQuant-ref. Because of difficulties with timing the feature-detection part of MaxQuant, two alternative measures are reported. (d) Computation times as in (c) but for Dinosaur compared to an OpenMS feature finder. The missing measurement of the synthetic peptide OpenMS sample is likely due to some corner-case implementation issue. (e) The number of unique peptides identified in three HeLa cell replicates using a new Dinosaur-based implementation of the DeMix workflow compared to the original workflow and analysis without DeMixing.
An important parameter for a feature-detection tool is the applicability of the tool to different workflows. To illustrate the ability to integrate Dinosaur in downstream workflows, we first incorporated it into the label-free quantification workflow100 in the Proteios software environment31 as one of the selectable feature detectors. We further used Dinosaur to implement the recently presented DeMix workflow for searching chimeric spectra.14 We exchanged the original feature finder to Dinosaur with some minor modifications in the original workflow script. For an unbiased comparison with the original workflow, we used the three HeLa lysate injections from the DeMix publication. The DeMix workflow with Dinosaur increased the number of unique identified peptides (1% peptide-spectrum match FDR) by 32%, compared to a standard search on the total 151 260 MS/MS spectra in the three replicate injections, and the OpenMS feature finder of the original publication produced a 26% increase on the same files (Figure 4e). The increase in DeMix effectiveness should be directly related to the increased MS/MS matching of Dinosaur over Open MS, as seen in Figure 2a, and is likely a general property that is transferrable to other samples. With the successful incorporation into a label-free quantification workflow and a chimeric spectrum workflow, we conclude that Dinosaur is well-adapted for general-purpose usage.
Discussion
In this work, we have devised an improved MS1 feature detection tool called Dinosaur. This tool includes a plot trail, a new strategy for performance monitoring, which was used to improve the algorithm in most major computational steps. From these improvements, we demonstrate an increased percentage of feature-matched 1% FDR MS/MS identifications and equal levels of quantitative accuracy compared to other feature-detection tools. We further demonstrate runtime decreases of 5–70% over alternative tools and the applicability of Dinosaur, both by itself on eight representative sample injections and as a part of a label-free quantification workflow and a chimeric-spectrum-identification workflow. Collectively, the results imply that Dinosaur is robust and mature.
Given the high data-generation rate in MS-based proteomics, a natural focus is the post-acquisition data analysis. The past years have resulted in numerous developed and published feature detection algorithms in which some are heavily used and others are not. Although most algorithms are useful for specific purposes, underdeveloped and under-tested software, possibly related to difficulties in publishing refined software,37 tends to limit common usage. It cannot be ruled out that many of the best available algorithms remain unused due to implementation problems like noncompatible input or output formats, no support for the used operating system, or simply undocumented and unexplainable crashes on apparently valid input. In this paper, we have attempted to address the need for an accurate and powerful MS1 feature detection program by consolidating and further developing a successful existing algorithm.
Although the ideal feature detection tool might not be achievable, we believe Dinosaur closes the gap a little, given the observed increase in computation speed and percentage of feature-matched MS/MS identifications. It should be noted that acquiring 100% MS/MS identification coverage is a game of diminishing returns, as small improvements in absolute numbers result in increasingly complex algorithms with associated increasing computation time. For the achievement of high coverage in complex proteomes, a DIA-based workflow is possibly a better alternative38−40 because these workflows avoid the stochastic sampling of shotgun MS and rely on quantification at the MS/MS level.
The algorithm improvements that differentiate Dinosaur from MaxQuant-ref increased the percentage of feature-matched MS/MS identifications in the synthetic peptide dilution data set from 89% to 98%, due to decreased feature intermingling, avoided feature-splitting during cluster deconvolution, and improved averagine matching and monoisotopic assignment. We used the Dinosaur targeted plot trail to manually assess 158 coordinates mapping to the ∼2% of nonmatched identifications in a representative file. For most nonmatched identifications, the corresponding features are manually detectable in the MS data. Missed features are often caused by mistakes in cluster deconvolution, but often, features are entangled with other features in such a way that it would not be possible to separate them without considering combinations of isotope patterns, which is not readily possible in the current algorithm. This could potentially be solved by using the Harklör19 decharging algorithm or a two-dimensional template-based feature detector,15−17 although using such a technique on high-resolution data might be computationally prohibitive and would warrant further investigation. We were also unable to devise a way to correctly evaluate the precision of the reported features as the absence of an associated identification does not necessarily indicate that the feature is incorrect because of the limited sequencing speed of the instrument. After the completion of the Dinosaur project, we have become aware of a small manually annotated feature data set that could be used for this purpose in future studies.23
Apart from the missing 2% of the identifications, it should be noted that Dinosaur has been developed and optimized for Orbitrap data. An initial test of application on high-resolution Q-TOF data was, for example, not satisfactory according to the plot trail, and no data from other mass analyzers such as low-resolution ion traps has been analyzed. Here is a clear avenue for further study and algorithm tuning. Finally, one might consider reproducing the final parts of the MaxQuant algorithm for SILAC data analysis to enable operating-system-neutral and open-source analysis of SILAC data using Dinosaur.
Dinosaur exposes more than 50 different configuration parameters (Table S-1), which are configurable directly on the command line or in a parameter file. To guide the user in tuning and evaluating these parameters, we have implemented a plot trail to stochastically sample the results of the major computation steps and to create plots describing computation effect on real data. The plot trail was used in this paper to improve the Dinosaur algorithm, but the main purposes are computational quality control and tuning Dinosaur parameters for application on data from other MS instruments. Plot trail plots are stored in a quality control folder or zip folder and can quickly be opened to check analysis performance and evaluate the effects of particular parameter settings (see the Supplemental Discussion section). The plot trail is distinctly different from the visualizations offered by programs like TOPPView (OpenMS), MaxQuant, and Skyline. In these programs, plots need to be manually generated by loading the corresponding MS data and result files and the plot space manually selected, leaving room for human bias. Also, all metadata present in the Dinosaur plot trail plots is not available in the final feature results. In summary, we believe the plot trail is a novel concept that is useful for explaining the software function to users and to enable the earlier detection of errors in data or feature-detection computation with minimal user effort.
Multiple groups have recently reported alternative techniques for identifying41 and analyzing chimeric MS/MS spectra using either MS1 features,14,42 MS/MS-based precursor mass guessing,43 or iterative searching with fragment attenuation.44 These recent studies consistently report up to 30% gains in the number of unique peptides identified and examples of coisolation of four or more peptides. In this way, peptide information in the available measured data that was previously ignored becomes accessible. Our results using Dinosaur concur with previous reports, contributing to a growing body of evidence that suggests that chimeric spectral searching should be a part of standard workflows.
The intention of Dinosaur is to provide a flexible, fast, and robust feature finding algorithm. Even though the devised improvements give some small increase in feature sensitivity, we emphasize that the main benefit of Dinosaur lies in its usability and fast incorporation in various workflows (for example, as demonstrated in the implemented label-free quantification and chimeric-spectral-search workflows).
Acknowledgments
This project was supported by the Swedish Research Council (projects 2008:3356 and 621-2012-3559), the Swedish Foundation for Strategic Research (grant FFL4), the Crafoord Foundation (grant 20100892), Stiftelsen Olle Engkvist Byggmästare, the Wallenberg Academy Fellow KAW (2012.0178), the European Research Council (starting grant ERC-2012-StG-309831), and the Swedish Foundation for Strategic Environmental Research (Mistra Biotech)
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jproteome.6b00016.
A full list and description of the Dinosaur parameters (Table S-1), a description of the estimation of true MS2 identification-feature-matches (Supplemental Material and Figures S-1 and S-2), and a discussion and exemplification on how to interpret the Dinosaur plot trail (Supplemental Discussion and Figure S-3 to S-14). (PDF)
Author Contributions
∥ F.L. and J.M. contributed equally to this paper.
The authors declare no competing financial interest.
Supplementary Material
References
- Hauri S.; Wepf A.; van Drogen A.; Varjosalo M.; Tapon N.; Aebersold R.; Gstaiger M. Interaction proteome of human Hippo signaling: modular control of the co-activator YAP1. Mol. Syst. Biol. 2013, 9, 713. 10.1002/msb.201304750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hein M. Y.; Hubner N. C.; Poser I.; Cox J.; Nagaraj N.; Toyoda Y.; Gak I. A.; Weisswange I.; Mansfeld J.; Buchholz F.; et al. A Human Interactome in Three Quantitative Dimensions Organized by Stoichiometries and Abundances. Cell 2015, 163 (3), 712–723. 10.1016/j.cell.2015.09.053. [DOI] [PubMed] [Google Scholar]
- Karlsson C.; Malmström L.; Aebersold R.; Malmström J. Proteome-wide selected reaction monitoring assays for the human pathogen Streptococcus pyogenes. Nat. Commun. 2012, 3, 1301. 10.1038/ncomms2297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberger G.; Koh C. C.; Guo T.; Röst H. L.; Kouvonen P.; Collins B. C.; Heusel M.; Liu Y.; Caron E.; Vichalkovski A.; et al. A repository of assays to quantify 10,000 human proteins by SWATH-MS. Sci. Data 2014, 1, 140031. 10.1038/sdata.2014.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng Z.; Teo G.; Krueger S.; Rock T. M.; Koh H. W.; Choi H.; Vogel C. Differential dynamics of the mammalian mRNA and protein expression response to misfolding stress. Mol. Syst. Biol. 2016, 12 (1), 855–855. 10.15252/msb.20156423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greber B. J.; Bieri P.; Leibundgut M.; Leitner A.; Aebersold R.; Boehringer D.; Ban N. Ribosome. The complete structure of the 55S mammalian mitochondrial ribosome. Science 2015, 348 (6232), 303–308. 10.1126/science.aaa3872. [DOI] [PubMed] [Google Scholar]
- Listgarten J.; Emili A. Statistical and Computational Methods for Comparative Proteomic Profiling Using Liquid Chromatography-Tandem Mass Spectrometry. Mol. Cell. Proteomics 2005, 4 (4), 419–434. 10.1074/mcp.R500005-MCP200. [DOI] [PubMed] [Google Scholar]
- Cappadona S.; Baker P. R.; Cutillas P. R.; Heck A. J. R.; van Breukelen B. Current challenges in software solutions for mass spectrometry-based quantitative proteomics. Amino Acids 2012, 43 (3), 1087–1108. 10.1007/s00726-012-1289-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandin M.; Teleman J.; Malmström J.; Levander F. Data processing methods and quality control strategies for label-free LC-MS protein quantification. Biochim. Biophys. Acta, Proteins Proteomics 2014, 1844 (1A), 29–41. 10.1016/j.bbapap.2013.03.026. [DOI] [PubMed] [Google Scholar]
- Ono M.; Shitashige M.; Honda K.; Isobe T.; Kuwabara H.; Matsuzuki H.; Hirohashi S.; Yamada T. Label-free quantitative proteomics using large peptide data sets generated by nanoflow liquid chromatography and mass spectrometry. Mol. Cell. Proteomics 2006, 5 (7), 1338–1347. 10.1074/mcp.T500039-MCP200. [DOI] [PubMed] [Google Scholar]
- Ong S.-E.; Blagoev B.; Kratchmarova I.; Kristensen D. B.; Steen H.; Pandey A.; Mann M. Stable isotope labeling by amino acids in cell culture, SILAC, as a simple and accurate approach to expression proteomics. Mol. Cell. Proteomics 2002, 1 (5), 376–386. 10.1074/mcp.M200025-MCP200. [DOI] [PubMed] [Google Scholar]
- Moruz L.; Pichler P.; Stranzl T.; Mechtler K.; Käll L. Optimized nonlinear gradients for reversed-phase liquid chromatography in shotgun proteomics. Anal. Chem. 2013, 85 (16), 7777–7785. 10.1021/ac401145q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsou C.-C.; Avtonomov D.; Larsen B.; Tucholska M.; Choi H.; Gingras A.-C.; Nesvizhskii A. I.. DIA-Umpire: comprehensive computational framework for data-independent acquisition proteomics. Nat. Methods; 2015, 12, (3), , 258–264; 10.1038/nmeth.3255; 7 p following 264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B.; Pirmoradian M.; Chernobrovkin A.; Zubarev R. A. DeMix workflow for efficient identification of cofragmented peptides in high resolution data-dependent tandem mass spectrometry. Mol. Cell. Proteomics 2014, 13 (11), 3211–3223. 10.1074/mcp.O114.038877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellew M.; Coram M.; Fitzgibbon M.; Igra M.; Randolph T.; Wang P.; May D.; Eng J.; Fang R.; Lin C.; et al. A suite of algorithms for the comprehensive analysis of complex protein mixtures using high-resolution LC-MS. Bioinformatics 2006, 22 (15), 1902–1909. 10.1093/bioinformatics/btl276. [DOI] [PubMed] [Google Scholar]
- Mueller L. N.; Rinner O.; Schmidt A.; Letarte S.; Bodenmiller B.; Brusniak M.-Y.; Vitek O.; Aebersold R.; Müller M. SuperHirn – a novel tool for high resolution LC-MS-based peptide/protein profiling. Proteomics 2007, 7 (19), 3470–3480. 10.1002/pmic.200700057. [DOI] [PubMed] [Google Scholar]
- Sturm M.; Bertsch A.; Gröpl C.; Hildebrandt A.; Hussong R.; Lange E.; Pfeifer N.; Schulz-Trieglaff O.; Zerck A.; Reinert K.; et al. OpenMS – An open-source software framework for mass spectrometry. BMC Bioinf. 2008, 9 (1), 163. 10.1186/1471-2105-9-163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tautenhahn R.; Bottcher C.; Neumann S. Highly sensitive feature detection for high resolution LC/MS. BMC Bioinf. 2008, 9 (1), 504. 10.1186/1471-2105-9-504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoopmann M. R.; Finney G. L.; MacCoss M. J. High-speed data reduction, feature detection, and MS/MS spectrum quality assessment of shotgun proteomics data sets using high-resolution mass spectrometry. Anal. Chem. 2007, 79 (15), 5620–5632. 10.1021/ac0700833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pluskal T.; Castillo S.; Villar-Briones A.; Orešič M. MZmine 2: Modular framework for processing, visualizing, and analyzing mass spectrometry-based molecular profile data. BMC Bioinf. 2010, 11 (1), 395. 10.1186/1471-2105-11-395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox J.; Mann M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 2008, 26 (12), 1367–1372. 10.1038/nbt.1511. [DOI] [PubMed] [Google Scholar]
- Åberg K. M.; Torgrip R. J. O.; Kolmert J.; Schuppe-Koistinen I.; Lindberg J. Feature detection and alignment of hyphenated chromatographic–mass spectrometric data. J. Chromatogr. A 2008, 1192 (1), 139–146. 10.1016/j.chroma.2008.03.033. [DOI] [PubMed] [Google Scholar]
- Conley C. J.; Smith R.; Torgrip R. J. O.; Taylor R. M.; Tautenhahn R.; Prince J. T. Massifquant: open-source Kalman filter-based XC-MS isotope trace feature detection. Bioinformatics 2014, 30 (18), 2636–2643. 10.1093/bioinformatics/btu359. [DOI] [PubMed] [Google Scholar]
- Sandin M.; Krogh M.; Hansson K.; Levander F. Generic workflow for quality assessment of quantitative label-free LC-MS analysis. Proteomics 2011, 11 (6), 1114–1124. 10.1002/pmic.201000493. [DOI] [PubMed] [Google Scholar]
- Katajamaa M.; Orešič M. Data processing for mass spectrometry-based metabolomics. J. Chromatogr. A 2007, 1158 (1–2), 318–328. 10.1016/j.chroma.2007.04.021. [DOI] [PubMed] [Google Scholar]
- Chawade A.; Sandin M.; Teleman J.; Malmström J.; Levander F. Data processing has major impact on the outcome of quantitative label-free LC-MS analysis. J. Proteome Res. 2015, 14 (2), 676–687. 10.1021/pr500665j. [DOI] [PubMed] [Google Scholar]
- Vizcaíno J. A.; Deutsch E. W.; Wang R.; Csordas A.; Reisinger F.; Ríos D.; Dianes J. A.; Sun Z.; Farrah T.; Bandeira N.; et al. ProteomeXchange provides globally coordinated proteomics data submission and dissemination. Nat. Biotechnol. 2014, 32 (3), 223–226. 10.1038/nbt.2839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malmström E.; Kilsgård O.; Hauri S.; Smeds E.; Herwald H.; Malmström L.; Malmström J. Large-scale inference of protein tissue origin in gram-positive sepsis plasma using quantitative targeted proteomics. Nat. Commun. 2016, 7, 10261. 10.1038/ncomms10261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martens L.; Chambers M.; Sturm M.; Kessner D.; Levander F.; Shofstahl J.; Tang W. H.; Römpp A.; Neumann S.; Pizarro A. D.; et al. mzML--a community standard for mass spectrometry data. Mol. Cell. Proteomics 2011, 10 (1), R110000133. 10.1074/mcp.R110.000133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kessner D.; Chambers M.; Burke R.; Agus D.; Mallick P. ProteoWizard: open source software for rapid proteomics tools development. Bioinformatics 2008, 24 (21), 2534–2536. 10.1093/bioinformatics/btn323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Häkkinen J.; Vincic G.; Månsson O.; Wårell K.; Levander F. The proteios software environment: an extensible multiuser platform for management and analysis of proteomics data. J. Proteome Res. 2009, 8 (6), 3037–3043. 10.1021/pr900189c. [DOI] [PubMed] [Google Scholar]
- Junker J.; Bielow C.; Bertsch A.; Sturm M.; Reinert K.; Kohlbacher O. TOPPAS: a graphical workflow editor for the analysis of high-throughput proteomics data. J. Proteome Res. 2012, 11 (7), 3914–3920. 10.1021/pr300187f. [DOI] [PubMed] [Google Scholar]
- Kim S.; Pevzner P. A. MS-GF+ makes progress towards a universal database search tool for proteomics. Nat. Commun. 2014, 5, 5277. 10.1038/ncomms6277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teleman J.; Dowsey A. W.; Gonzalez-Galarza F. F.; Perkins S.; Pratt B.; Röst H. L.; Malmström L.; Malmström J.; Jones A. R.; Deutsch E. W.; et al. Numerical compression schemes for proteomics mass spectrometry data. Mol. Cell. Proteomics 2014, 13 (6), 1537–1542. 10.1074/mcp.O114.037879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senko M. W.; Beu S. C.; McLaffertycor F. W. Determination of monoisotopic masses and ion populations for large biomolecules from resolved isotopic distributions. J. Am. Soc. Mass Spectrom. 1995, 6 (4), 229–233. 10.1016/1044-0305(95)00017-8. [DOI] [PubMed] [Google Scholar]
- Michalski A.; Cox J.; Mann M. More than 100,000 detectable peptide species elute in single shotgun proteomics runs but the majority is inaccessible to data-dependent LC-MS/MS. J. Proteome Res. 2011, 10 (4), 1785–1793. 10.1021/pr101060v. [DOI] [PubMed] [Google Scholar]
- Sandin M.; Ali A.; Hansson K.; Månsson O.; Andreasson E.; Resjö S.; Levander F. An adaptive alignment algorithm for quality-controlled label-free LC-MS. Mol. Cell. Proteomics 2013, 12 (5), 1407–1420. 10.1021/pr501318d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martens L.; Kohlbacher O.; Weintraub S. T. Managing expectations when publishing tools and methods for computational proteomics. J. Proteome Res. 2015, 14 (5), 2002–2004. 10.1021/pr501318d. [DOI] [PubMed] [Google Scholar]
- Gillet L. C.; Navarro P.; Tate S.; Röst H.; Selevsek N.; Reiter L.; Bonner R.; Aebersold R. Targeted data extraction of the MS/MS spectra generated by data-independent acquisition: a new concept for consistent and accurate proteome analysis. Mol. Cell. Proteomics 2012, 11 (6), O111016717. 10.1074/mcp.O111.016717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Röst H. L.; Rosenberger G.; Navarro P.; Gillet L.; Miladinović S. M.; Schubert O. T.; Wolski W.; Collins B. C.; Malmström J.; Malmström L.; et al. OpenSWATH enables automated, targeted analysis of data-independent acquisition MS data. Nat. Biotechnol. 2014, 32 (3), 219–223. 10.1038/nbt.2841. [DOI] [PubMed] [Google Scholar]
- Teleman J.; Röst H. L.; Rosenberger G.; Schmitt U.; Malmström L.; Malmström J.; Levander F. DIANA--algorithmic improvements for analysis of data-independent acquisition MS data. Bioinformatics 2015, 31 (4), 555–562. 10.1093/bioinformatics/btu686. [DOI] [PubMed] [Google Scholar]
- Luethy R.; Kessner D. E.; Katz J. E.; MacLean B.; Grothe R.; Kani K.; Faça V.; Pitteri S.; Hanash S.; Agus D. B.; et al. Precursor-Ion Mass Re-Estimation Improves Peptide Identification on Hybrid Instruments. J. Proteome Res. 2008, 7 (9), 4031–4039. 10.1021/pr800307m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niu M.; Mao X.; Ying W.; Qin W.; Zhang Y.; Qian X. Determination of monoisotopic masses of chimera spectra from high-resolution mass spectrometric data by use of isotopic peak intensity ratio modeling. Rapid Commun. Mass Spectrom. 2012, 26 (16), 1875–1886. 10.1002/rcm.6293. [DOI] [PubMed] [Google Scholar]
- Gorshkov V.; Verano-Braga T.; Kjeldsen F. SuperQuant: A Data Processing Approach to Increase Quantitative Proteome Coverage. Anal. Chem. 2015, 87 (12), 6319–6327. 10.1021/acs.analchem.5b01166. [DOI] [PubMed] [Google Scholar]
- Shteynberg D.; Mendoza L.; Hoopmann M. R.; Sun Z.; Schmidt F.; Deutsch E. W.; Moritz R. L. reSpect: Software for Identification of High and Low Abundance Ion Species in Chimeric Tandem Mass Spectra. J. Am. Soc. Mass Spectrom. 2015, 26, 1837. 10.1007/s13361-015-1252-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.