

Abstract
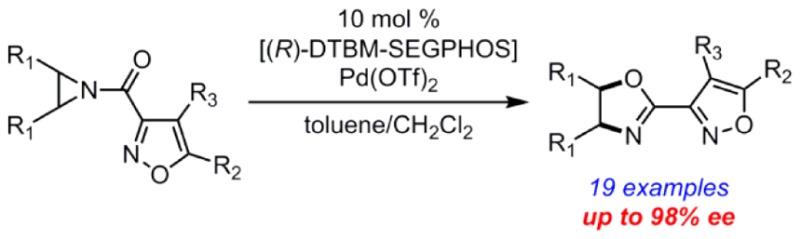
Aziridines are important synthetic intermediates for the generation of nitrogen-containing molecules. N-Acylaziridines undergo rearrangement by ring expansion to produce oxazolines, a process known as the Heine reaction. The first catalytic, enantioselective Heine reaction is reported for meso-N-acylaziridines where a palladium(II)–diphosphine complex is employed. The highly enantioenriched oxazoline products are valuable organic synthons and potential ligands for transition-metal catalysis.
Keywords: enantioselective, aziridine, palladium, heterocycles, desymmetrization
Nonracemic oxazolines are highly valued structures in organic synthesis.1−4 The five-membered heterocycles are employed as functionalized building blocks for the production of biologically active molecules and ligands for enantioselective catalysis.5−8 Typically enantioenriched oxazolines are synthesized from 1,2-amino alcohols available from the chiral pool.2,9−14 The enantioselective synthesis of oxazolines from alkenes can be performed with stoichiometric chiral modifiers;15−17 however, a catalytic, enantioselective synthesis of oxazolines from achiral starting materials is not available.18−23 Herein we report the first example of an enantioselective Heine reaction, catalyzed by palladium(II)–diphosphine complexes,24,25 as a new strategy for generating enantioenriched oxazolines that might be otherwise difficult to access.
Heine and co-workers were the first to utilize N-acylaziridines as synthetic precursors to oxazolines under Lewis acid catalysis.26 Subsequently, Heine’s group also explored mild Lewis base catalysts for the rearrangement reaction that now bears his name.27−30 Chiral N-acylaziridines can undergo stereospecific rearrangement to enantioenriched oxazolines under Lewis acid catalysis,19,31−34 and a mechanism for this process has been proposed.35,36meso-N-Acylaziridines offer the opportunity to control the absolute stereochemistry during oxazoline formation by enantioselective catalysis; however, only racemic oxazolines have been produced by a direct Lewis acid-catalyzed Heine reaction.19 Two methods for enantioenriched oxazoline formation from aziridine desymmetrization intermediates are known (Scheme 1).37,38 In both examples, the ring-opened intermediate (1) was isolated with varying levels of enantiomeric excess, and only one substrate was cyclized to the oxazoline 2. The current work accomplishes a direct catalytic, highly enantioselective aziridine rearrangement to oxazoline (3 → 4, Scheme 1).
Scheme 1. Enantioselective Synthesis of Oxazolines by N-Acylaziridine Desymmetrization.

Our research group has been interested in novel synthetic methods that incorporate N-acylaziridines.34,39 The majority of enantioselective processes incorporating N-acylaziridines have been Lewis acid-catalyzed desymmetrization of meso-aziridines by ring opening reactions with nucleophiles.37,38,40−54 Benzoyl-protected aziridines have proven to be the most successful in these desymmetrization reactions; however, early efforts in our group to develop an enantioselective Heine reaction using these aziridines were completely nonselective despite evidence for metal catalysis. We considered the strategy of transition-state organization by 2-point binding as a solution. While N-(2-picolinoyl)-aziridines have been successfully employed for ring opening desymmetrization,48−54 we identified 3-acylisoxazoles as a novel directing group (3, Scheme 1). The isoxazole structure is highly modular at the C4 and C5 positions which enables tuning of reaction yield and enantioselectivity.
Aziridine 5 was produced from the corresponding N-tosylaziridine for a catalyst survey (Table 1). Commercially available metal triflate salts and ligands (Figure 1) were complexed in dichloromethane with 4 Å molecular sieves prior to the addition of aziridine 5. A large variety of catalysts produced oxazoline 6 as a single diastereomer, with a relevant sample shown in Table 1. Interestingly, oxazoline-based ligands gave the best initial hits with lanthanide and copper triflates (entries 1–4). The copper(II)–BOX complexes were promising with high conversion and good enantioselectivity, but adjustment of reaction parameters could not improve selectivity above 82% ee (entry 4). While considering other late transition metals with BOX ligands (entries 5–6), we discovered that palladium(II)–BINAP derived catalysts produced very good ee with reduced conversion (entries 7–8). The μ-hydroxy dimeric palladium(II) catalyst55,56 (7) was inactive (entry 9). Further investigation of diphosphine ligands revealed DTBM-SEGPHOS as the optimal choice following a solvent switch to toluene (entries 10–11).57−60 Alternative noncoordinating anions were inferior to triflate (entry 12–13), and lowering the catalyst loading to 5 mol % was possible, albeit with diminished enantioselectivity (entry 14).
Table 1. Catalyst Discovery for an Enantioselective Heine Reactiona.

| entry | metal | ligand | conversion (%)b | ee (%)b |
|---|---|---|---|---|
| 1 | Cu(OTf)2 | L1 | 12 | 28 |
| 2 | La(OTf)3 | L1 | 24 | –33 |
| 3 | Cu(OTf)2 | L2 | 95 | 39 |
| 4 | Cu(OTf)2 | L3 | 93 | 82 |
| 5 | Ni(OTf)2 | L3 | 23 | –35 |
| 6 | Pd(OTf)2 | L3 | 41 | 60 |
| 7 | Pd(OTf)2 | L4 | 29 | 85 |
| 8 | Pd(OTf)2 | L5 | 27 | 95 |
| 9 | 7 | — | 0 | — |
| 10 | Pd(OTf)2 | L6 | 33 | 92 |
| 11c | Pd(OTf)2 | L6 | 79 | 94 |
| 12c | Pd(ClO4)2 | L6 | 60 | 76 |
| 13 | Pd(BF4)2 | L6 | 0 | — |
| 14c,d | Pd(OTf)2 | L6 | 89 | 88 |
Reaction conditions: aziridine 5 (0.1 mmol) and 50 mg of 4 Å powdered molecular sieves were added to the preformed catalyst (0.01 mmol of metal and 0.012 mmol of ligand) in CH2Cl2 (0.2 M) at room temperature for 24 h.
Determined by HPLC peak area ratios of 5 and 6 at 250 nm using the CHIRALCEL OJ column.
Reaction solvent was toluene.
Reaction performed with 5 mol % catalyst.
Figure 1.

Ligand structures surveyed for the catalytic, enantioselective Heine reaction.
The effect of isoxazole structure on the reaction outcome was investigated (8, Table 2). Slight modifications to reaction conditions were employed with 7 mol % of [DTBM-SEGPHOS]Pd(OTf)2 producing adequate yield and enantioselectivity after 40 h at room temperature. Adjusting the steric size of the 5-isoxazole substituent (R2) had only a slight effect on yield and enantioselectivity (entries 1–6). The trend was not linear, with methyl (entry 2) and tert-butyl (entry 6) producing the highest enantioselectivities. A 5-phenyl substituent suppressed the yield but generated high levels of enantioselectivity (entry 7). Aziridines containing a 4-isoxazole substituent resulted in similar yields and enantioselectivities (entries 8–9). The 4-iodo derivative is an interesting compound for further functionalization.
Table 2. Isoxazole Substituent Effects on the Enantioselective Heine Reactiona.

| entry | R1 | R2 | yield (%)b | ee (%)c |
|---|---|---|---|---|
| 1 | H | H | 61 | 86 |
| 2 | H | methyl | 81 | 92 |
| 3 | H | ethyl | 82 | 89 |
| 4 | H | isopropyl | 74 | 90 |
| 5 | H | cyclopropyl | 69 | 83 |
| 6 | H | tert-butyl | 75 | 95 |
| 7 | H | phenyl | 62 | 91 |
| 8 | I | methyl | 68 | 85 |
| 9 | –CH2CH2CH2CH2– | 63 | 89 | |
Reaction conditions: aziridine (0.2 mmol), [(R)-DTBM-SEGPHOS]Pd(OTf)2 (0.014 mmol), and 100 mg of 4 Å powdered molecular sieves in 9:1 toluene/CH2Cl2 (0.2 M) at room temperature for 40 h.
Isolated yield; average of two or more runs.
Determined by HPLC.
meso-Aziridines with structurally diverse backbones were synthesized as both the 5-methyl and 5-tert-butyl-3-acylisoxazole derivatives (10, Table 3). The changes in aziridine structure led to significant decreases in turnover for most substrates at room temperature. To combat slower reactions, catalyst loading was increased to 10 mol %, and reaction temperature was optimized for yield and enantioselectivity of oxazoline 11. Under the modified standard conditions, substrates containing a 6-membered ring carbocyclic aziridine backbone gave high yields and excellent ee values (entries 1–2, Table 3). Decreasing the ring size gave comparable results (entry 3) where incorporation of the 5-tert-butyl isoxazole produced a decrease in yield, but the highest ee observed in the study (entry 4). A 7-membered carbocyclic backbone resulted in significant issues with conversion though enantioselectivity remained high (entries 5–6). After 7 days at 30 °C, only 40% and 44% yields could be isolated for the two isoxazole variations. When accounting for recovered starting material, high yield was observed for the tert-butyl derivative (entry 6). Incorporation of oxygen in the aziridine backbone was possible utilizing a dioxepine ring (entry 7) or tetrahydrofuran ring (entry 8), albeit with reduced enantioselectivity and yield. An acyclic meso-aziridine was a very good substrate when performed at room temperature where enantioselectivities were highest (entries 9–10).
Table 3. Substrate Scope for meso-Aziridine Desymmetrization by Enantioselective Rearrangementa.


Reaction conditions: aziridine (0.2 mmol), [(R)-DTBM-SEGPHOS]Pd(OTf)2 (0.02 mmol), and 100 mg of 4 Å powdered molecular sieves in 2:1 toluene/CH2Cl2 (0.2 M).
Isolated yield for an average of two or more runs. Yield in parentheses is based on recovered starting material (bmrs).
Determined by HPLC.
Reaction solvent was 1:1 toluene/CH2Cl2.
Reaction solvent was CH2Cl2.
Aqueous HCl in ethyl acetate rapidly converts oxazoline 12 to 1,2-hydroxyamide 13 by ring opening (Scheme 2). Such rapid hydrolysis of 12 was not surprising as certain purified oxazolines produced by rearrangement hydrolyzed during storage under ambient conditions. Independent synthesis of amide 13 proved the absolute configuration of oxazoline 12 to be 4S,5R when the R catalyst was employed.
Scheme 2. Hydrolysis of Enantioenriched Oxazoline 12.

The enantioselective rearrangement of aziridine 14 was scaled to 1 mmol with reduced yield, but an increase in ee (Scheme 3). Following the reaction, the catalyst mixed with 4 Å mol sieves was collected by filtration before purification of oxazoline 12 by column chromatography. The recovered catalyst was a single diastereomer by 1H NMR and has a structure consistent with 15 by HRMS (Figure 2). Confirmation of 15 was obtained following treatment with pyridine to free the oxazoline portion for analysis. A crossover experiment was performed by subjecting aziridine 5 to rearrangement conditions with 7 mol % of 15 (Scheme 3). Oxazoline 6 was produced in nearly identical yield and enantioselectivity as standard conditions (entry 2, Table 2). This experiment suggests that the active catalyst is formed in situ from 15.61
Scheme 3. Enantioselective Heine Reaction Scale-Up.

Figure 2.

Structure of recovered catalyst from reaction scale-up.
A proposed reaction mechanism is provided in Figure 3. The anhydrous, premade catalyst58 (16) coordinates the aziridine (17) by 2-point binding. We favor binding to both nitrogen atoms to form intermediate 18 based on the azaphilic nature of palladium. Bond reorganization directly to the product bound catalyst 19 occurs in the enantiodetermining step. We propose that the cis-oxazoline product is generated exclusively by C–O bond construction via an SNi mechanism leading to retention of stereochemistry.36 The orientation of 18 would project substituents on the aziridine backbone toward the chiral ligand. The requirement of a triflate counterion may suggest a double inversion mechanism involving triflate, which has not been ruled out. Intermediate 19 releases the product, likely by dissociative substitution, to turn over the catalytic cycle. Slow release of product (20) by intermediate 19 may explain the requirement for higher catalyst loading. The recovered catalyst 15 appears to be a low energy intermediate that can be formed directly from 18, but re-enter the catalytic cycle.
Figure 3.

Proposed mechanism for the palladium-catalyzed, enantioselective Heine reaction.
The first example of a catalytic, enantioselective Heine reaction has been described. A cationic palladium(II)–diphosphine catalyst produces highly enantioenriched oxazoline products with good to excellent yields across a series of structural modifications. The application of the isoxazole-based oxazoline products as synthetic intermediates and ligands for catalysis is currently being explored. Efforts are underway to discern the reaction mechanism and apply this catalyst system to new aziridine ring-opening reactions.
Acknowledgments
The authors are grateful to the National Institute of General Medical Sciences of the National Institutes of Health (R15GM114766) for financial support. The HRMS data was collected from a Bruker MicrOTOF-Q II instrument purchased with funds from the National Science Foundation (CHE-1039784) and University of North Carolina Wilmington. The 600 MHz NMR data was obtained using a Bruker instrument purchased with funds from the National Science Foundation (CHE-0821552).
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acscatal.6b01400.
The authors declare no competing financial interest.
Supplementary Material
References
- Frump J. A. Chem. Rev. 1971, 71, 483. 10.1021/cr60273a003. [DOI] [Google Scholar]
- Gant T. G.; Meyers A. I. Tetrahedron 1994, 50, 2297. 10.1016/S0040-4020(01)86953-2. [DOI] [Google Scholar]
- Meyers A. I. J. Org. Chem. 2005, 70, 6137. 10.1021/jo050470h. [DOI] [PubMed] [Google Scholar]
- Hoogenboom R. Angew. Chem., Int. Ed. 2009, 48, 7978. 10.1002/anie.200901607. [DOI] [PubMed] [Google Scholar]
- Johnson J. S.; Evans D. A. Acc. Chem. Res. 2000, 33, 325. 10.1021/ar960062n. [DOI] [PubMed] [Google Scholar]
- McManus H. A.; Guiry P. J. Chem. Rev. 2004, 104, 4151. 10.1021/cr040642v. [DOI] [PubMed] [Google Scholar]
- Desimoni G.; Faita G.; Jorgensen K. A. Chem. Rev. 2006, 106, 3561. 10.1021/cr0505324. [DOI] [PubMed] [Google Scholar]
- Desimoni G.; Faita G.; Jorgensen K. A. Chem. Rev. 2011, 111, PR284. 10.1021/cr100339a. [DOI] [PubMed] [Google Scholar]
- Bansal S.; Halve A. K. Int. J. Pharm. Sci. Res. 2014, 5, 4601. [Google Scholar]
- For additional methods not covered in ref (9), see refs (11−14).
- Winstein S.; Boschan R. J. Am. Chem. Soc. 1950, 72, 4669. 10.1021/ja01166a090. [DOI] [Google Scholar]
- Wipf P.; Miller C. P. Tetrahedron Lett. 1992, 33, 907. 10.1016/S0040-4039(00)91572-7. [DOI] [Google Scholar]
- Phillips A. J.; Uto Y.; Wipf P.; Reno M. J.; Williams D. R. Org. Lett. 2000, 2, 1165. 10.1021/ol005777b. [DOI] [PubMed] [Google Scholar]
- Crosignani S.; Young A. C.; Linclau B. Tetrahedron Lett. 2004, 45, 9611. 10.1016/j.tetlet.2004.10.143. [DOI] [Google Scholar]
- Tiecco M.; Testaferri L.; Santi C.; Tomassini C.; Marini F.; Bagnoli L.; Temperini A. Eur. J. Org. Chem. 2000, 2000, 3451.. [DOI] [Google Scholar]
- Minakata S.; Nishimura M.; Takahashi T.; Oderaotoshi Y.; Komatsu M. Tetrahedron Lett. 2001, 42, 9019. 10.1016/S0040-4039(01)01963-3. [DOI] [Google Scholar]
- Nishimura M.; Minakata S.; Takahashi T.; Oderaotoshi Y.; Komatsu M. J. Org. Chem. 2002, 67, 2101. 10.1021/jo016146d. [DOI] [PubMed] [Google Scholar]
- For racemic examples, see refs (19−23).
- Ferraris D.; Drury W. J.; Cox C.; Lectka T. J. Org. Chem. 1998, 63, 4568. 10.1021/jo980558d. [DOI] [PubMed] [Google Scholar]
- Yeung Y. Y.; Gao X. R.; Corey E. J. J. Am. Chem. Soc. 2006, 128, 9644. 10.1021/ja063675w. [DOI] [PubMed] [Google Scholar]
- Minakata S.; Morino Y.; Ide T.; Oderaotoshi Y.; Komatsu M. Chem. Commun. 2007, 3279. 10.1039/b706572h. [DOI] [PubMed] [Google Scholar]
- Hajra S.; Bar S.; Sinha D.; Maji B. J. Org. Chem. 2008, 73, 4320. 10.1021/jo8003937. [DOI] [PubMed] [Google Scholar]
- Scholz S. O.; Farney E. P.; Kim S.; Bates D. M.; Yoon T. P. Angew. Chem., Int. Ed. 2016, 55, 2239. 10.1002/anie.201510868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sodeoka M.; Hamashima Y. Chem. Commun. 2009, 5787. 10.1039/b911015a. [DOI] [PubMed] [Google Scholar]
- Sheshenev A. E.; Smith A. M. R.; Hii K. K. Nat. Protoc. 2012, 7, 1765. 10.1038/nprot.2012.095. [DOI] [PubMed] [Google Scholar]
- Heine H. W.; Proctor Z. J. Org. Chem. 1958, 23, 1554. 10.1021/jo01104a605. [DOI] [Google Scholar]
- Heine H. W.; Fetter M. E.; Nicholson E. M. J. Am. Chem. Soc. 1959, 81, 2202. 10.1021/ja01518a048. [DOI] [Google Scholar]
- Heine H. W. Angew. Chem., Int. Ed. Engl. 1962, 1, 528. 10.1002/anie.196205281. [DOI] [Google Scholar]
- Heine H. W. Mech. Mol. Migr. 1971, 3, 145. [Google Scholar]
- Kurti L.; Czako B.. Strategic Applications of Named Reactions in Organic Synthesis; Elsevier Academic Press: San Diego, 2005. [Google Scholar]
- Nishiguchi T.; Tochio H.; Nabeya A.; Iwakura Y. J. Am. Chem. Soc. 1969, 91, 5841. 10.1021/ja01049a024. [DOI] [Google Scholar]
- Nabeya A.; Shigemoto T.; Iwakura Y. J. Org. Chem. 1975, 40, 3536. 10.1021/jo00912a014. [DOI] [Google Scholar]
- Cardillo G.; Gentilucci L.; Mohr G. P. Eur. J. Org. Chem. 2001, 2001, 3545.. [DOI] [Google Scholar]
- Cockrell J.; Wilhelmsen C.; Rubin H.; Martin A.; Morgan J. B. Angew. Chem., Int. Ed. 2012, 51, 9842. 10.1002/anie.201204224. [DOI] [PubMed] [Google Scholar]
- Nishiguchi T.; Tochio H.; Nabeya A.; Iwakura Y. J. Am. Chem. Soc. 1969, 91, 5835. 10.1021/ja01049a023. [DOI] [Google Scholar]
- Hori K.; Nishiguchi T.; Nabeya A. J. Org. Chem. 1997, 62, 3081. 10.1021/jo961089n. [DOI] [PubMed] [Google Scholar]
- Mita T.; Jacobsen E. N. Synlett 2009, 2009, 1680. 10.1055/s-0029-1217344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senatore M.; Lattanzi A.; Santoro S.; Santi C.; Della Sala G. Org. Biomol. Chem. 2011, 9, 6205. 10.1039/c1ob05837a. [DOI] [PubMed] [Google Scholar]
- Martin A.; Casto K.; Morris W.; Morgan J. B. Org. Lett. 2011, 13, 5444. 10.1021/ol202410v. [DOI] [PubMed] [Google Scholar]
- Mita T.; Fujimori I.; Wada R.; Wen J.; Kanai M.; Shibasaki M. J. Am. Chem. Soc. 2005, 127, 11252. 10.1021/ja053486y. [DOI] [PubMed] [Google Scholar]
- Fujimori I.; Mita T.; Maki K.; Shiro M.; Sato A.; Furusho S.; Kanai M.; Shibasaki M. J. Am. Chem. Soc. 2006, 128, 16438. 10.1021/ja067003h. [DOI] [PubMed] [Google Scholar]
- Fukuta Y.; Mita T.; Fukuda N.; Kanai M.; Shibasaki M. J. Am. Chem. Soc. 2006, 128, 6312. 10.1021/ja061696k. [DOI] [PubMed] [Google Scholar]
- Rowland E. B.; Rowland G. B.; Rivera-Otero E.; Antilla J. C. J. Am. Chem. Soc. 2007, 129, 12084. 10.1021/ja0751779. [DOI] [PubMed] [Google Scholar]
- Della Sala G.; Lattanzi A. Org. Lett. 2009, 11, 3330. 10.1021/ol901209n. [DOI] [PubMed] [Google Scholar]
- Larson S. E.; Baso J. C.; Li G. L.; Antilla J. C. Org. Lett. 2009, 11, 5186. 10.1021/ol902123h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu B.; Gallucci J. C.; Parquette J. R.; RajanBabu T. V. Angew. Chem., Int. Ed. 2009, 48, 1126. 10.1002/anie.200804415. [DOI] [PubMed] [Google Scholar]
- Xu Y. J.; Lin L. Q.; Kanai M.; Matsunaga S.; Shibasaki M. J. Am. Chem. Soc. 2011, 133, 5791. 10.1021/ja201492x. [DOI] [PubMed] [Google Scholar]
- Hayashi M.; Shiomi N.; Funahashi Y.; Nakamura S. J. Am. Chem. Soc. 2012, 134, 19366. 10.1021/ja309963w. [DOI] [PubMed] [Google Scholar]
- Kalow J. A.; Doyle A. G. Tetrahedron 2013, 69, 5702. 10.1016/j.tet.2013.01.062. [DOI] [Google Scholar]
- Li J.; Liao Y. T.; Zhang Y. L.; Liu X. H.; Lin L. L.; Feng X. M. Chem. Commun. 2014, 50, 6672. 10.1039/c4cc02206h. [DOI] [PubMed] [Google Scholar]
- Yang D. X.; Wang L. Q.; Han F. X.; Li D.; Zhao D. P.; Cao Y. M.; Ma Y. X.; Kong W. D.; Sun Q. T.; Wang R. Chem. - Eur. J. 2014, 20, 16478. 10.1002/chem.201404354. [DOI] [PubMed] [Google Scholar]
- Li D.; Wang L. Q.; Yang D. X.; Zhang B. Z.; Wang R. ACS Catal. 2015, 5, 7432. 10.1021/acscatal.5b02177. [DOI] [Google Scholar]
- Wang L. Q.; Yang D. X.; Han F. X.; Li D.; Zhao D. P.; Wang R. Org. Lett. 2015, 17, 176. 10.1021/ol503455r. [DOI] [PubMed] [Google Scholar]
- Wang L. Q.; Yang D. X.; Li D.; Wang R. Org. Lett. 2015, 17, 3004. 10.1021/acs.orglett.5b01291. [DOI] [PubMed] [Google Scholar]
- Fujii A.; Hagiwara E.; Sodeoka M. J. Am. Chem. Soc. 1999, 121, 5450. 10.1021/ja9902827. [DOI] [Google Scholar]
- Hagiwara E.; Fujii A.; Sodeoka M. J. Am. Chem. Soc. 1998, 120, 2474. 10.1021/ja973962n. [DOI] [Google Scholar]
- Hamashima Y.; Yagi K.; Takano H.; Tamas L.; Sodeoka M. J. Am. Chem. Soc. 2002, 124, 14530. 10.1021/ja028464f. [DOI] [PubMed] [Google Scholar]
- Corkey B. K.; Toste F. D. J. Am. Chem. Soc. 2005, 127, 17168. 10.1021/ja055059q. [DOI] [PubMed] [Google Scholar]
- Corkey B. K.; Toste F. D. J. Am. Chem. Soc. 2007, 129, 2764. 10.1021/ja068723r. [DOI] [PubMed] [Google Scholar]
- Brazeau J. F.; Zhang S. Y.; Colomer I.; Corkey B. K.; Toste F. D. J. Am. Chem. Soc. 2012, 134, 2742. 10.1021/ja210388g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The active catalyst likely contains a palladium(II)−diphosphine core structure. NMR of the reaction over time shows multiple 31P signals, but no indication of the precatalyst or free diphosphine ligand. Also, the rearrangement of 5 with a product-derived catalyst, (6)Pd(OTf2), produced 6 in 40% yield and 17% ee.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.