

 This work is licensed under a
This work is licensed under a Summary
IgG4-related disease (IgG4-RD) is a rare but increasingly recognised condition, emerging as a clinical entity following the observation of the associations of autoimmune pancreatitis. IgG4-RD is characterised by extensive infiltration of IgG4-positive plasma cells into multiple organs and raised serum IgG4 levels. Clinical manifestations of IgG4 disease classically include autoimmune pancreatitis, lacrimal or salivary gland infiltration (formerly known as Mikulicz disease) and retroperitoneal fibrosis. More rarely, IgG4 disease can cause pituitary hypophysitis. Although most frequently described in middle-aged males, the epidemiology and pathogenesis of the disease remain largely undefined. Nevertheless, an understanding of the wide variety of clinical manifestations of this multi-system condition is undeniably important given the often excellent outcomes following treatment. We describe an unusual presentation of IgG4 disease with isolated diabetes insipidus secondary to pituitary hypophysitis. The patient in question subsequently developed chest pain secondary to mediastinal lymphadenopathy and tubulo-interstitial nephritis leading to renal dysfunction. He was successfully treated with oral steroids and had regular follow-up, and remains well at follow-up 2 years later.
Learning points
IgG4 disease, although rare, is increasing in prevalence largely due to increased recognition of its clinical manifestations, including autoimmune pancreatitis, lacrimal or salivary gland infiltration, retroperitoneal fibrosis and, more rarely, lymphocytic hypophysitis presenting as diabetes insipidus.
IgG4 disease is highly treatable, and symptoms may show complete resolution with administration of steroids, highlighting the importance of correct and timely diagnosis.
Causes of lymphocytic hypophysitis are varied and not distinguishable radiologically. Given the difficulty in biopsying the pituitary, careful attention must be paid to the systemic clinical presentation to provide clues as to the underlying disorder.
Background
We describe a presentation of IgG4 disease with isolated diabetes insipidus. The case demonstrates an unusual presentation of this rare, but increasingly recognised and very treatable, condition.
Case presentation
A 54-year-old Caucasian gentleman presented with a 1-year history of gradually worsening polydipsia with dry mouth and polyuria. He had no significant previous medical history. In particular, he had suffered no head trauma, previous stroke or seizures, although the patient did report a high alcohol intake of approximately 60 units per week.
Random and fasting blood glucose measurements proved to be within range and urine microscopy and cultures were negative and remained so on regular repeat testing. Urine osmolality at that time was 147 and rose to 404 on repeat testing the following year. Monitoring of his fluid balance demonstrated fluid intake and urine output in excess of 6 L of water per day with no glycosuria. The patient was referred to urology regarding his urinary frequency. He was strongly advised to reduce his alcohol intake, and tamsulosin therapy was commenced for presumed prostatism.
Several weeks later, the patient presented directly to hospital with a non-specific history of chest pain. Acute coronary syndrome was excluded, but computed tomography (CT) of the chest revealed sub-carinal lymphadenopathy with para-aortic and para-caval fibrosis (Fig. 1). This raised the suspicion of sarcoidosis, for which the patient underwent further investigations as follows.
Figure 1.

CT chest, abdomen and pelvis showed mediastinal lymph nodes up to 1.3cm, right hilar nodes up to 1.4cm and left hilar nodes of 1.2cm. Mild splenomegaly (14.2cm) was noted. No abdominal lymph nodes or interstitial lung shadowing were present. There was minimal peribronchial thickening.
Investigation
Blood results for urea, electrolytes, calcium, thyroid function tests, cortisol and prolactin were within normal ranges. Magnetic resonance imaging (MRI) of the abdomen showed bilateral renal lesions with an appearance typical of renal sarcoidosis. MRI of the pituitary with contrast showed a thickened pituitary infundibulum and bulky anterior pituitary (Fig. 2). These findings were deemed classical for lymphocytic hypophysitis. The patient’s chest pain resolved with analgesia, and he was discharged for outpatient follow-up.
Figure 2.

MRI pituitary with contrast demonstrated a thickened pituitary infundibulum (3.7mm craniocaudal and 4.9mm transverse) with normal post-contrast diffuse enhancement. The anterior pituitary is also bulky with maximal craniocaudal dimension of 10.4mm. No obvious adenoma was noted. The appearance was deemed to be classical for lymphocytic hypophysitis.
Outpatient water deprivation testing was suggestive of cranial diabetes insipidus (DI) with serum osmolality of 304mmol/kg, urine osmolality of 378mmol/kg and urine osmolality post DDAVP of 404mmol/kg. Desmopressin was prescribed, providing significant symptomatic improvement. Blood tests also demonstrated a degree of possible renal impairment with a creatinine of 120μmol/L (65–111μmol/L) and estimated glomerular filtration rate of 55mL/min (>60mL/min). Subsequent renal biopsy noted the presence of tubulo-interstitial nephritis and provided a definitive diagnosis of IgG4-related disease (Fig. 3). The diagnosis was supported by the finding of raised serum IgG4.
Figure 3.
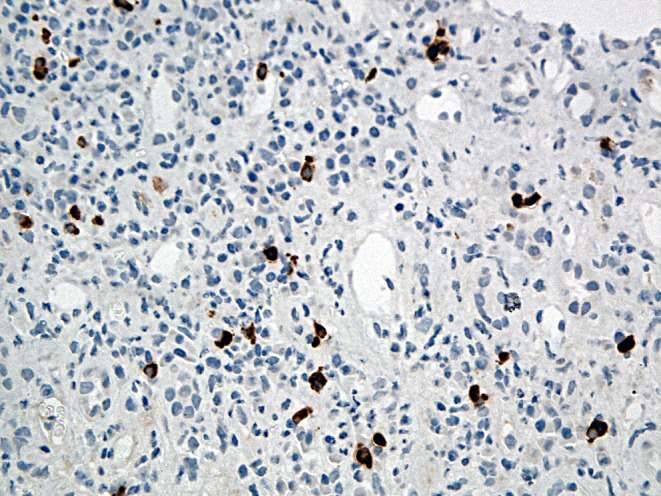
Renal biopsy stain showed a florid and extensive tubulitis with mainly lymphocytes entering tubular epithelium. Staining with IgG4 revealed an increased population of positive plasma cells.
Treatment
Given the diagnosis and progressively deteriorating renal function, the patient was commenced on oral steroids. Renal biochemistry showed an improvement in urinary concentration following presumed renal response to oral steroids. He has responded extremely well to treatment and has been discharged from hospital care. Histological specimens of the pituitary lesion were deemed too invasive and clinically unnecessary, and were, therefore, not obtained. Nevertheless, the clinical response and rise in urinary osmolality with steroid therapy are strongly suggestive of pituitary pathology similar to that observed on renal biopsy.
Outcome and follow-up
Follow-up MRI pituitary after 1 year (Fig. 4) has demonstrated regression of the described radiological findings. Hormonal profiles tested at 1 year post follow-up showed raised testosterone, reduced LH, reduced normal FSH and normal prolactin. The patient remained clinically well 2 years after his initial presentation.
Figure 4.

Follow-up MRI pituitary. The maximum CC dimension of pituitary is 8.4mm vs 10.9mm at diagnosis. Maximum transverse thickness of the infundibulum (stalk) is 3.8mm vs 5.8mm at diagnosis. Maximum CC dimension of the stalk is 3.1mm vs 3.8mm at diagnosis.
Discussion
IgG4-related disease (IgG4-RD) is a rare but increasingly recognised condition, emerging as a clinical entity following the observation of the associations of autoimmune pancreatitis (1). Clinical manifestations classically include autoimmune pancreatitis, lacrimal or salivary gland infiltration (formerly known as Mikulicz disease) and retroperitoneal fibrosis. Although most frequently described in middle-aged males, the epidemiology and pathogenesis of the disease remain largely undefined (2). Further pituitary involvement of IgG4 disease has seldom been documented with only 8 published cases as of 2010 (3). Pituitary hypophysitis, causes of which are radiologically indistinguishable and include IgG4-related adenohypophysitis, occurs at an estimated prevalence of one in nine million individuals per year (4).
IgG4-related hypophysitis usually presents as disturbance of anterior pituitary hormone production including generalised hypopituitarism and isolated central hypogonadism, hypothyroidism and ACTH deficiency. We believe this to be the first case in the published literature of biopsy-confirmed IgG4-RD presenting solely as cranial diabetes insipidus without other features of hypopituitarism and before involvement of other organs. It should be noted that the sensitivity and negative-predictive values of elevated serum IgG4 concentrations for the diagnosis of IgG4-RD are high. However, a number of conditions can be associated with elevated serum IgG4, so specificity and the positive-predictive value of elevated IgG4 levels are low. Although water deprivation testing was only partially suggestive of cranial diabetes insipidus, the significant symptomatic response to desmopressin in addition to pituitary imaging and biopsy confirmation of systemic IgG4-RD provide convincing support for the presenting condition.
The patient developed widespread effects of presumable IgG4-RD relatively rapidly, with pituitary, mediastinal, retroperitoneal and renal involvement over the course of 1 year. As yet, the progression of IgG4-RD has not been well defined. Previous reports of systemic IgG4-RD in the literature have highlighted a clinical course ranging from 1 to 20 years for multi-system involvement (3).
Declaration of interest
The authors declare that there is no conflict of interest that could be perceived as prejudicing the impartiality of the research reported.
Funding
This research did not receive any specific grant from any funding agency in the public, commercial or not-for-profit sector.
Patient consent
Written informed consent has been obtained from the patient for publication of the submitted article and accompanying images.
Author contribution statement
The manuscript was researched and written by R P and W M. Figures and editing of manuscript was done by S K. M S analysed the histological specimen and provided the written description. All authors read and approved the final manuscript.
References
- 1.Kamisawa T, Okamoto A. 2006. Autoimmune pancreatitis: proposal of igg4-related sclerosing disease. Journal of gastroenterology 41 613–625. ( 10.1007/s00535-006-1862-6) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zen Y, Nakanuma Y. 2010. Igg4-related disease: a cross-sectional study of 114 cases. American Journal of Surgical Pathology 34 1812–1819. ( 10.1097/PAS.0b013e3181f7266b) [DOI] [PubMed] [Google Scholar]
- 3.Haraguchi A, Era A, Yasui J, Ando T, Ueki I, Horie I, Imaizumi M, Usa T, Abe K, Origuchi T, et al. 2010. Putative igg4-related pituitary disease with hypopituitarism and/or diabetes insipidus accompanied with elevated serum levels of igg4. Endocrine Journal 57 719–725. ( 10.1507/endocrj.k10e-030) [DOI] [PubMed] [Google Scholar]
- 4.Mittal R, Kalra P, Dharmalingam M, Verma RG, Kulkarni S, Shetty P. 2012. Lymphocytic hypophysitis masquerading as pituitary adenoma. Indian Journal of Endocrinology and Metabolism 16 S304–S306. ( 10.4103/2230-8210.104069) [DOI] [PMC free article] [PubMed] [Google Scholar]