

ABSTRACT
The classification of a gene as an oncogene or a tumor suppressor has been a staple of cancer biology for decades. However, as we delve deeper into the biology of these genes, this simple classification has become increasingly difficult for some. In the case of heterogeneous nuclear ribonuclear protein K (hnRNP K), its role as a tumor suppressor has recently been described in acute myeloid leukemia and demonstrated in a haploinsufficient mouse model. In contrast, data from other clinical correlation studies suggest that hnRNP K may be more fittingly described as an oncogene, due to its increased levels in a variety of malignancies. hnRNP K is a multifunctional protein that can regulate both oncogenic and tumor suppressive pathways through a bevy of chromatin-, DNA-, RNA-, and protein-mediated activates, suggesting its aberrant expression may have broad-reaching cellular impacts. In this review, we highlight our current understanding of hnRNP K, with particular emphasis on its apparently dichotomous roles in tumorigenesis.
KEYWORDS: 9q21.32, acute myeloid leukemia, c-Myc, C/EBP, hnRNP K, haploinsufficiency, mouse models, p53, p21
Introduction
Over the past several decades, clinical and basic science studies have attempted to determine the critical genetic alterations that directly influence tumorigenesis. In doing so, it has been useful to categorize genes as either tumor suppressors (resulting from gene loss or inactivating mutations) or oncogenes (when gene products are overexpressed). This has allowed researchers and clinicians to delineate the functional and clinical consequences of many genetic alterations. However, these classifications do not always accurately reflect the reality of aberrant gene expression. As an example, the protein p53 was first thought to be an oncogene when it was originally discovered due to its increased stability when mutated.1,2 However, once its cellular function was better understood, it was proven to be a potent tumor suppressor.3 Recently, its role as an oncogene (or at least having oncogenic potential) has been reassessed following the discovery that stabilized mutant p53 does in fact confer gain-of-function phenotypes.4,5 Such blurring of traditional oncogene/tumor suppressor roles is now becoming more accepted, as evidenced by similar observations in the TGF-β pathway.6,7
More recently, evidence has emerged that heterogeneous nuclear ribonucleoprotein K (hnRNP K) may be even harder to classify as a tumor suppressor or an oncogene. This is due to the fact that biochemical and in vitro studies have shown that hnRNP K has the capacity to regulate both tumor suppressive and oncogenic pathways, and both its overexpression and knockdown results in cell proliferation and apoptotic defects.8-13 In support of its potential oncogenic functions, clinical association studies suggest that hnRNP K overexpression correlates with poor clinical outcomes and advanced disease status in a variety of malignancies, including melanoma, prostate, breast, lung, colorectal, hepatocellular, and esophageal cancers.14-18 In contrast, other clinical studies suggest reduced hnRNP K expression, due to deletion of or mutations in the HNRNPK gene, may underpin the pathogenesis of acute myeloid leukemia (AML).19-23 In the case of mutation, there are further questions concerning whether specific mutations inactivate the protein or potentially stabilize or confer gain-of-function phenotypes, reminiscent of mutations observed in the c-Myc protein and mutant p53 proteins, respectively.
Given the lack of consensus between the biochemical, in vitro, and clinical data, it is apparent that hnRNP K has more to its story than just a simple classification as either an oncogene or tumor suppressor. This review will highlight aspects of our current knowledge of hnRNP K's role in cancer biology, describe studies that have evaluated hnRNP K-dependent transcriptional and translational activities, examine the molecular consequence of Hnrnpk haploinsufficiency in vivo, and provide insight into future investigations using Hnrnpk mouse models.
HnRNP K-mediated transcriptional and translational activities regulate pathways involved in tumorigenesis
hnRNP K is an extremely versatile and multifunctional protein that influences transcription, translation, splicing, RNA stability, and chromatin remodeling through its capacity to stringently bind RNA and single-stranded (ss) DNA via its KH domains.24 The roles that hnRNP K plays in these diverse cellular functions are well documented and have been reviewed elsewhere.25,26 Here; however, we will highlight key points to emphasize its potential role in both oncogenic and tumor suppressive pathways.
At the transcriptional level, hnRNP K has been implicated in directly and indirectly regulating gene expression. With respect to cancer biology, hnRNP K directly mediates the expression of both oncogenes and tumor suppressors, such as SRC, MYC, CDKN1A (p21), HDM2 (Mdm2 in the mouse), and EIF4E through its direct binding to C-rich regions in the promoters of these genes.9-12,27 In addition to this direct transcriptional gene regulation, hnRNP K is implicated in positively influencing gene expression through direct interaction with the TATA-binding protein (TBP) of the RNA polymerase machinery,28,29 as well as negatively regulating gene expression through its interactions with lincRNA-21.30,31 Furthermore, hnRNP K also plays a critical role in chromatin remodeling by acting as a scaffold protein that binds to DNA matrix attachment regions (MARs) and thus stabilizes the chromatin.32 While this finding suggests that hnRNP K positively influences global transcription through its chromatin interactions, hnRNP K also interacts with and regulates the localization of the Polycomb repressive complex through its interaction with a core component, Eed.33 Taken as a whole, hnRNP K appears to have conflicting activating and repressive influences on gene expression. On the one hand, hnRNP K has the ability to directly enhance or repress transcription of specific genes, while on the other hand, it can simultaneously enhance or diminish global gene expression. Thus, given its proposed role in regulating critical cellular pathways, any change in its expression may result in significant consequences.
In addition to transcriptionally regulating gene expression through its direct interaction with DNA, hnRNP K also binds to mRNA transcripts and translationally regulates protein expression. Similar to its conflicting roles in both positively and negatively regulating transcription, hnRNP K has the capacity to activate or inhibit the translation of some mRNA transcripts. For example, hnRNP K has been shown to positively stimulate MYC translation,8,34,35 while it inhibits the translation of 15-LOX, a key regulator of erythroid differentiation.36,37 Collectively, these duplicitous and opposing mechanisms of gene regulation present a complex situation, whereby hnRNP K may play a critical balancing act between its roles in directly and globally activating and repressing gene expression, and simultaneously controlling translation of mRNA transcripts. Thus, it is easy to envision scenarios where small changes in hnRNP K expression (either increased or decreased) could result in drastic cellular defects that impact oncogenic or tumor suppressor pathways and directly impact tumorigenesis.
Biochemical and in vitro evidence for a direct role of HnRNP K in tumorigenesis
Two of the classic tumor suppressor and oncogenic pathways directly impacted by hnRNP K-mediated transcriptional and translational activities are the p53/p21 and c-Myc pathways, respectively.9-11 Biochemical experiments performed nearly a decade ago provided the first evidence that hnRNP K may play a pivotal role in tumor suppression. In these studies, hnRNP K was shown to be phosphorylated by ATM and ATR following DNA damage, which directly resulted in activation of p21.10,11 Building on these findings, hnRNP K was later shown to be sumoylated in an ATR-dependent manner following UV-irradiation, resulting in enhanced p53-dependent transcriptional activation of p21.38,39 These results indicate that hnRNP K may act as a putative tumor suppressor and suggest that its loss may directly impact tumor formation. Interestingly, hnRNP K has recently been shown to also negatively impact the translation of the p21 transcript by binding to its 3′ UTR, although this effect was demonstrated in the context of neuronal differentiation.40 These observations suggest that within tissue-specific contexts, hnRNP K may have the capacity to either suppress or promote tumorigenesis through a single pathway; however, detailed in vivo studies are needed to test the veracity of such a notion.
In addition to controlling tumor suppressive programs, hnRNP K has also been shown to influence the expression of oncogenic pathways.26 As a classic example, hnRNP K directly interacts with C-rich regions in the MYC promoter, leading to increased c-Myc expression.9 In addition, hnRNP K is also thought to translationally regulate c-Myc expression by binding to these same C-rich regions in the 5′ UTR of the MYC transcript to promote ribosomal loading and further drive c-Myc expression.8 The transcriptional and translational hnRNP K-mediated regulation of c-Myc suggest that hnRNP K overexpression may result in oncogenic phenotypes, and perhaps most importantly, implicates hnRNP K as a potential driver of c-Myc-dependent malignancies when the MYC gene is not amplified or translocated.
Clinical evidence for a role of aberrant HnRNP K in tumorigenesis
Much of the clinical data regarding hnRNP K's role in tumorigenesis originates from pathologic and immunohistochemical analyses of archived patient samples. These studies revealed that increased hnRNP K expression associated with poor clinical status in melanoma, prostate, breast, lung, colorectal, hepatocellular, and esophageal cancers.14-18 Given the myriad of tumor types that overexpress hnRNP K in these studies and its correlation with disease prognosis, these data suggest that hnRNP K may have oncogenic functions when overexpressed.
However, in contrast to hnRNP K's putative oncogenic roles, HNRNPK is one of 6 genes that maps to the minimally deleted region of the 9q21.32 locus, which is thought to harbor a haploinsufficient tumor suppressor in patients with acute myeloid leukemia (AML).19-21,23 Furthermore, TCGA recently demonstrated that HNRNPK mutations have the capacity to act as driving events in AML.22 However, it is currently unknown if these HNRNPK mutations confer gain-of-function or haploinsufficient phenotypes.
Therefore, to more fully evaluate how hnRNP K expression contributes to the pathogenesis of AML, we examined HNRNPK expression in AML patients with diploid karyotypes that carried a 9q21.32 deletion. To this end, we developed a dual-color fluorescence in situ hybridization (FISH) assay using bacterial artificial chromosomes containing the entire HNRNPK gene (RP11- 101L4) and a control probe to the 9p region (RP11-19G1). Using this FISH assay, we observed that HNRNPK is specifically lost in a subset of malignant haematopoietic cells isolated from the bone marrows of de novo AML patients but not in healthy controls (Fig. 1). Additionally, qRT-PCR analyses revealed that deletion of the 9q21.32 locus significantly reduced HNRNPK expression in patients with AML.41 Critically, 8 of 12 patients had deletion of this 9q21.32 region as their sole karyotypic abnormality, suggesting reduced HNRNPK expression directly contributes to the pathogenesis of AML.
Figure 1.
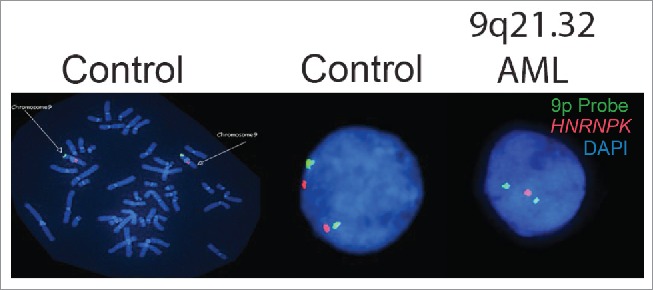
The HNRNPK gene is lost in a subset of de novo AML patients. (Left) Functional validation of the control probe (RP11-19G1, green) to the distal arm of chromosome 9p and HNRNPK probe (RP11-101L4, red) to the 9q21.32 locus using metaphase FISH on haematopoietic cells from healthy donors. DAPI is used as a counterstain to denote the DNA (chromosomes). (Middle). Nuclei of an interphase haematopoietic cell isolated from the bone marrow of a healthy donor indicating two HNRNPK alleles on chromosome 9 (two red/2 green). DAPI is used as a counterstain to denote the nucleus. (Right). Nuclei of an interphase haematopoietic cell isolated from the bone marrow of a de novo AML patient with haploinsufficient loss of the HNRNPK gene (one red/2 green). DAPI is used as a counterstain to denote the nucleus.
An in vivo model of Hnrnpk haploinsufficiency
To directly examine if HNRNPK is, in fact, the haploinsufficient tumor suppressor residing at the 9q21.32 locus, our laboratory generated an Hnrnpk haploinsufficient mouse model and recently published a manuscript that examines the role of Hnrnpk haploinsufficiency in vivo.41 Using mouse embryo fibroblasts (MEFs) and haematopoietic stem progenitor cells (HSPCs) in cytokine-dependent colony formation assays, we determined that reduced hnRNP K expression directly resulted in an increase in cellular proliferation. Molecularly, this increased proliferation was directly attributed to hnRNP K's inability to transcriptionally activate p21 when its levels were reduced, supporting the notion that hnRNP K has tumor suppressive functions.
In addition to the increased proliferation observed in colony formation assays and in the bone marrow of these mice, Hnrnpk haploinsufficiency also contributed to defects in myeloid differentiation. Similar to the hnRNP K-mediated effects on p21, reduced hnRNP K expression also directly impacted the expression of critical myeloid differentiation factors C/EBP-α and -β. Molecular analyses revealed that reduced hnRNP K levels dampened its ability to transcriptionally regulate the expression of these genes. Interestingly, only the C/EBPα p42 isoform, but not p30, was significantly downregulated in Hnrnpk+/− mice. This lack of expression of the p42 isoform partially explains the myeloproliferative phenotype developed by Hnrnpk+/− mice, as previous studies have identified the p42 isoform as a critical myeloid tumor suppressor that significantly alters myelopoiesis. Given hnRNP K's role in translation and the fact that both the p30 and p42 isoforms are translated from the same mRNA, we hypothesized that hnRNP K may influence the expression of specific C/EBPα isoforms. This notion is supported by our finding that hnRNP K directly interacts with the C/EBPα transcript in human leukemic cell lines and in wild type mice. Importantly, these interactions were significantly reduced in bone marrow cells isolated from Hnrnpk haploinsufficient mice. Together, these clinical and animal model data suggest that reduced hnRNP K contributes to these leukemic phenotypes through its direct regulation of the p53/p21- and C/EBP- pathways and support the hypothesis that HNRNPK is the haploinsufficient tumor suppressor at the 9q21.32 locus (Fig. 2A and B).
Figure 2.
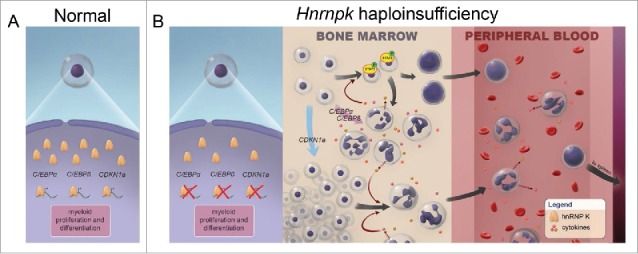
Hnrnpk haploinsufficiency results in proliferative and differentiation defects in the haematopoietic compartment. A. Under normal conditions, hnRNP K is required to maintain a homeostatic balance in haematopoietic development through its regulation of p21, C/EBP-αand β. B. (Left) Hnrnpk haploinsufficiency results in reduced hnRNP K expression and diminished p21, C/EBP-α and β levels. (Middle) In the bone marrow, reduced hnRNP K allows for expansion of the haematopoietic compartment, increased cytokine expression, and activation of Stat-3 signaling. (Right) Cells from the hyperproliferative bone marrow are mobilized and extravasate from the marrow into the periphery.
Reduced hnRNP K expression impacts developmental processes
With the multitude of pathways and cellular programs influenced by hnRNP K, it is easy to envision how alterations in its expression influence tumorigenesis. However, other striking phenotypes observed in haploinsufficient Hnrnpk mice were not related to increased cancer risk, but rather developmental defects.41 Hnrnpk haploinsufficiency resulted in mice being born at a sub-mendelian ratio, extreme runtedness, a propensity for facial malformations, and a significant neonatal lethal phenotype. Analogous to our observations in mice, pediatric patients with germline HNRNPK mutations (c.953 + 1dup and c.257G > A; respectively) have been recently described.42 These mutations result in haploinsufficient loss of wild type hnRNP K expression, and have been identified as causal events in the development of a syndrome consisting of craniofacial malformations, intellectual disability, and skeletal and connective tissue abnormalities. Furthermore, studies in Xenopus laevis have shown that antisense morpholino-mediated loss of hnRNP K expression also results in neuronal development defects.43 However, perhaps the most striking evidence that hnRNP K is required for development stems from our observation that bi-allelic loss of Hnrnpk results in an embryo lethal phenotype prior to day 13.5 (E13.5).41 Given that hnRNP K has been previously shown to transcriptionally regulate Mdm2 expression,10,11 we postulated that bi-allelic Hnrnpk loss may potentially result in an embryo-lethal phenotype that is similar to loss of Mdm2 (i.e; an inability to negatively regulate p53-dependent apoptosis in utero).44 However, when we generated Hnrnpk+/−;Trp53+/− mice and then performed sibling matings, we did not rescue the Hnrnpk-null embryo-lethal phenotype. These initial observations suggest that hnRNP K's influence on embryonic development primarily resides in its impact on cellular programs outside of the p53 pathway.
Summary and future directions
Based on recent studies, there is now accumulating evidence that hnRNP K plays a critical role in regulating many fundamental cellular process that directly impact human diseases, such as tumorigenesis and congenital defects. In the context of malignancies, the impact that aberrant hnRNP K expression has on disease progression is complex, as there is evidence that hnRNP K has both oncogenic and tumor suppressive functions. However, determining its precise impact on cancer risk is complicated by the fact that hnRNP K is capable of both strengthening and attenuating diametrically opposed oncogenic and tumor suppressive pathways. With the development of an Hnrnpk haploinsufficient mouse model, we have made initial strides in examining the tumor suppressive functions of hnRNP K and delineated the importance of reduced hnRNP K expression in cancer development through the p53/p21- and C/EBP-pathways.41 As a result, this model system could become an effective pre-clinical platform to test therapies for patients with AML who harbor a 9q21.32 deletion. In these studies, standard frontline therapies could be used in combination with agents that antagonize the Mdm2-dependent degradation of p53, such as Nutlin-3, DS-3032b, or RG7112 in order to “re-engage” the p53 pathway.
In addition to its tumor suppressive functions, there are also clinical association studies, as well as biochemical and in vitro studies that strongly suggest hnRNP K may also serve as an oncogene when overexpressed.14-18 However, to directly test this notion, there is an absolute need for the development of transgenic animal models that overexpress wild-type hnRNP K. We have currently undertaken this task by generating tissue-specific Hnrnpk transgenic mice. These model systems will directly test whether hnRNP K is a bona fide oncogene and will be useful in identifying the genes and cellular programs critical for tumorigenesis when hnRNP K is overexpressed. Similar to the Hnrnpk haploinsufficient mouse model, these mice will be instrumental in testing and developing hnRNP K overexpression-dependent therapies.
The challenge that remains (and will be central in our functional evaluation of HNRNPK mutations) is the generation of mutant knock-in Hnrnpk mouse models. This endeavor may prove a difficult task, as unlike the hotspot mutations observed in genes like Tp53, IDH1, and KRAS, the HNRNPK mutation spectrum (in cancers and in pediatric patients) is significantly more heterogeneous.45 Thus, there is currently no clear indication as to which alteration is “the” critical mutation to initially examine. As such, there is a need for stringent biochemical and in vitro studies using mutant hnRNP K in order to guide future in vivo studies of mutant hnRNP K.
Even though numerous studies have examined the functional importance of hnRNP K since its discovery nearly a generation ago,46 there remains much work to be done in order to fully understand the role of hnRNP K in human malignancies.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Author contributions
M.G., M.J.H, X.Z, and S.M.P. wrote the manuscript. X.Z. generated artwork. C.B.-R. and P.H. performed and analyzed pathologic and karyotype analyses. All authors reviewed and accepted the manuscript.
Funding
We thank members of the Genetically Engineered Mouse Facility who were supported by an NCI Cancer Center Support Grant CA16672. These studies have been supported by an NIH Career Development Award (P50 CA1000632-09), Leukemia Research Foundation Award, Center for Genetics and Genomics Award, Ladies Leukemia League, MDACC MDS/AML Moonshot, and MDACC start-up funds to (S.M.P).
References
- [1].Lane DP, Crawford LV. T antigen is bound to a host protein in SY40-transformed cells. Nature 1979; 278:261-3; PMID:218111; http://dx.doi.org/ 10.1038/278261a0 [DOI] [PubMed] [Google Scholar]
- [2].Linzer DIH, Levine AJ. Characterization of a 54 K Dalton cellular SV40 tumor antigen present in SV40-transformed cells and uninfected embryonal carcinoma cells. Cell 1979; 17:43-52; PMID:222475; http://dx.doi.org/ 10.1016/0092-8674(79)90293-9 [DOI] [PubMed] [Google Scholar]
- [3].Finlay CA, Hinds PW, Levine AJ. The p53 proto-oncogene can act as a suppressor of transformation. Cell 1989; 57:1083-93; PMID:2525423; http://dx.doi.org/ 10.1016/0092-8674(89)90045-7 [DOI] [PubMed] [Google Scholar]
- [4].Lang GA, Iwakuma T, Suh YA, Liu G, Rao VA, Parant JM, Valentin-Vega YA, Terzian T, Caldwell LC, Strong LC, et al.. Gain of Function of a p53 Hot Spot Mutation in a Mouse Model of Li-Fraumeni Syndrome. Cell 2004; 119:861-72; PMID:15607981; http://dx.doi.org/ 10.1016/j.cell.2004.11.006 [DOI] [PubMed] [Google Scholar]
- [5].Olive KP, Tuveson DA, Ruhe ZC, Yin B, Willis NA, Bronson RT, Crowley D, Jacks T. Mutant p53 gain of function in two mouse models of Li-Fraumeni Syndrome. Cell 2004; 119:847-60; PMID:15607980; http://dx.doi.org/ 10.1016/j.cell.2004.11.004 [DOI] [PubMed] [Google Scholar]
- [6].Massague J. TGF[β] signalling in context. Nat Rev Mol Cell Biol 2012; 13:616-30; PMID:22992590; http://dx.doi.org/ 10.1038/nrm3434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Bierie B, Moses HL. Tumour microenvironment: TGF[β]: the molecular Jekyll and Hyde of cancer. Nat Rev Cancer 2006; 6:506-20; PMID:16794634; http://dx.doi.org/ 10.1038/nrc1926 [DOI] [PubMed] [Google Scholar]
- [8].Notari M, Neviani P, Santhanam R, Blaser BW, Chang JS, Galietta A, Willis AE, Roy DC, Caligiuri MA, Marcucci G, et al.. A MAPK/HNRPK pathway controls BCR/ABL oncogenic potential by regulating MYC mRNA translation. Blood 2006; 107:2507-16; http://dx.doi.org/ 10.1182/blood-2005-09-3732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Tomonaga T, Levens D. Heterogeneous Nuclear Ribonucleoprotein K Is a DNA-binding Transactivator. J Biol Chem 1995; 270:4875-81; PMID:7876260; http://dx.doi.org/ 10.1074/jbc.270.9.4875 [DOI] [PubMed] [Google Scholar]
- [10].Enge M, Bao W, Hedström E, Jackson SP, Moumen A, Selivanova G. MDM2-dependent downregulation of p21 and hnRNP K provides a switch between apoptosis and growth arrest induced by pharmacologically activated p53. Cancer Cell 2009; 15:171-83; PMID:19249676; http://dx.doi.org/ 10.1016/j.ccr.2009.01.019 [DOI] [PubMed] [Google Scholar]
- [11].Moumen A, Masterson P, O'Connor MJ, Jackson SP. hnRNP K: An HDM2 Target and Transcriptional Coactivator of p53 in Response to DNA Damage. Cell 2005; 123:1065-78; PMID:16360036; http://dx.doi.org/ 10.1016/j.cell.2005.09.032 [DOI] [PubMed] [Google Scholar]
- [12].Lynch M, Chen L, Ravitz MJ, Mehtani S, Korenblat K, Pazin MJ, Schmidt EV. hnRNP K binds a core polypyrimidine element in the eukaryotic translation initiation factor 4 E (eIF4E) promoter, and its regulation of eIF4E contributes to neoplastic transformation. Mol Cell Biol 2005; 25:6436-53; PMID:16024782; http://dx.doi.org/ 10.1128/MCB.25.15.6436-6453.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Bao X, Wu H, Zhu X, Guo X, Hutchins AP, Luo Z, Song H, Chen Y, Lai K, Yin M, et al.. The p53-induced lincRNA-p21 derails somatic cell reprogramming by sustaining H3K9me3 and CpG methylation at pluripotency gene promoters. Cell Res 2015; 25:80-92; PMID:25512341; http://dx.doi.org/ 10.1038/cr.2014.165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Carpenter B, McKay M, Dundas SR, Lawrie LC, Telfer C, Murray GI. Heterogeneous nuclear ribonucleoprotein K is over expressed, aberrantly localised and is associated with poor prognosis in colorectal cancer. Br J Cancer 2006; 95:921-7; PMID:16953238; http://dx.doi.org/ 10.1038/sj.bjc.6603349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Chen LC, Chung IC, Hsueh C, Tsang NM, Chi LM, Liang Y, Chen CC, Wang LJ, Chang YS. The antiapoptotic protein, FLIP, is regulated by heterogeneous nuclear ribonucleoprotein K and correlates with poor overall survival of nasopharyngeal carcinoma patients. Cell Death Differ 2010; 17:1463-73; PMID:20224598; http://dx.doi.org/ 10.1038/cdd.2010.24 [DOI] [PubMed] [Google Scholar]
- [16].Ciarlo M, Benelli R, Barbieri O, Minghelli S, Barboro P, Balbi C, Ferrari N. Regulation of neuroendocrine differentiation by AKT/hnRNPK/AR/β-catenin signaling in prostate cancer cells. Int J Cancer 2012; 131:582-90; PMID:22015967; http://dx.doi.org/ 10.1002/ijc.26402 [DOI] [PubMed] [Google Scholar]
- [17].Wen F, Shen A, Shanas R, Bhattacharyya A, Lian F, Hostetter G, Shi J. Higher Expression of the Heterogeneous Nuclear Ribonucleoprotein K in Melanoma. Annals Surg Oncol 2010; 17:2619-27; http://dx.doi.org/ 10.1245/s10434-010-1121-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Wu CS, Chang KP, Chen LC, Chen CC, Liang Y, Hseuh C, Chang YS. Heterogeneous ribonucleoprotein K and thymidine phosphorylase are independent prognostic and therapeutic markers for oral squamous cell carcinoma. Oral Oncology 2012; 48:516-22; PMID:22321252; http://dx.doi.org/ 10.1016/j.oraloncology.2012.01.005 [DOI] [PubMed] [Google Scholar]
- [19].Dayyani F, Wang J, Yeh JRJ, Ahn EY, Tobey E, Zhang DE, Bernstein ID, Peterson RT, Sweetser DA. Loss of TLE1 and TLE4 from the del(9q) commonly deleted region in AML cooperates with AML1-ETO to affect myeloid cell proliferation and survival. Blood 2008; 111:4338-47; PMID:18258796; http://dx.doi.org/ 10.1182/blood-2007-07-103291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Krönke J, Bullinger L, Teleanu V, Tschürtz F, Gaidzik VI, Kühn MWM, Rücker FG, Holzmann K, Paschka P, Kapp-Schwörer S, et al.. Clonal evolution in relapsed NPM1-mutated acute myeloid leukemia. Blood 2013; 122:100-8; PMID:23704090; http://dx.doi.org/ 10.1182/blood-2013-01-479188 [DOI] [PubMed] [Google Scholar]
- [21].Sweetser DA, Peniket AJ, Haaland C, Blomberg AA, Zhang Y, Zaidi ST, Dayyani F, Zhao Z, Heerema NA, Boultwood J, et al.. Delineation of the minimal commonly deleted segment and identification of candidate tumor-suppressor genes in del(9q) acute myeloid leukemia. Genes Chromosomes Cancer 2005; 44:279-91; PMID:16015647; http://dx.doi.org/ 10.1002/gcc.20236 [DOI] [PubMed] [Google Scholar]
- [22].Cancer Genome Atlas Research Network . Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N Engl J Med 2013; 368:2059-74; PMID:23634996; http://dx.doi.org/ 10.1056/NEJMoa1301689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Bullinger L, Kronke J, Schon C, Radtke I, Urlbauer K, Botzenhardt U, Gaidzik V, Cario A, Senger C, Schlenk RF, et al.. Identification of acquired copy number alterations and uniparental disomies in cytogenetically normal acute myeloid leukemia using high-resolution single-nucleotide polymorphism analysis. Leukemia 2009; 24:438-49; PMID:20016533; http://dx.doi.org/ 10.1038/leu.2009.263 [DOI] [PubMed] [Google Scholar]
- [24].Bomsztyk K, Denisenko O, Ostrowski J. hnRNP K: One protein multiple processes. BioEssays 2004; 26:629-38; PMID:15170860; http://dx.doi.org/ 10.1002/bies.20048 [DOI] [PubMed] [Google Scholar]
- [25].Ostareck-Lederer A, Ostareck DH. Precision mechanics with multifunctional tools: How HnRNP K and HnRNPs E1/E2 contribute to post-transcriptional control of gene expression in hematopoiesis. Curr Protein Peptide Sci 2012; 13:391-400; http://dx.doi.org/ 10.2174/138920312801619484 [DOI] [PubMed] [Google Scholar]
- [26].Barboro P, Ferrari N, Balbi C. Emerging roles of heterogeneous nuclear ribonucleoprotein K (hnRNP K) in cancer progression. Cancer Letters 2014; 352:152-9; PMID:25016060; http://dx.doi.org/ 10.1016/j.canlet.2014.06.019 [DOI] [PubMed] [Google Scholar]
- [27].Ritchie SA, Pasha MK, Batten DJP, Sharma RK, Olson DJH, Ross ARS, Bonham K. Identification of the SRC pyrimidine‐binding protein (SPy) as hnRNP K: implications in the regulation of SRC1A transcription. Nucleic Acids Res 2003; 31:1502-13; PMID:12595559; http://dx.doi.org/ 10.1093/nar/gkg246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Michelotti EF, Michelotti GA, Aronsohn AI, Levens D. Heterogeneous nuclear ribonucleoprotein K is a transcription factor. Mol Cell Biol 1996; 16:2350-60; PMID:8628302; http://dx.doi.org/ 10.1128/MCB.16.5.2350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Shnyreva M, Schullery DS, Suzuki H, Higaki Y, Bomsztyk K. Interaction of two multifunctional proteins: Heterogeneous nuclear ribonucleoprotein K and Y-box-binding protein. J Biol Chem 2000; 275:15498-503; PMID:10809782; http://dx.doi.org/ 10.1074/jbc.275.20.15498 [DOI] [PubMed] [Google Scholar]
- [30].Dimitrova N, Zamudio Jesse R, Jong Robyn M, Soukup D, Resnick R, Sarma K, Ward Amanda J, Raj A, Lee Jeannie T, Sharp Phillip A, et al.. LincRNA-p21 Activates p21 In cis to Promote Polycomb Target Gene Expression and to Enforce the G1/S Checkpoint. Mol Cell 2014; 54:777-90; PMID:24857549; http://dx.doi.org/ 10.1016/j.molcel.2014.04.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Huarte M, Guttman M, Feldser D, Garber M, Koziol MJ, Kenzelmann-Broz D, Khalil AM, Zuk O, Amit I, Rabani M, et al.. A Large Intergenic Noncoding RNA Induced by p53 Mediates Global Gene Repression in the p53 Response. Cell 2010; 142:409-19; PMID:20673990; http://dx.doi.org/ 10.1016/j.cell.2010.06.040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Samuel SK, Spencer VA, Bajno L, Sun J-M, Holth LT, Oesterreich S, Davie JR. In situ cross-linking by cisplatin of nuclear matrix-bound transcription factors to nuclear DNA of human breast cancer cells. Cancer Res 1998; 58:3004-8; PMID:9679963 [PubMed] [Google Scholar]
- [33].Denisenko ON, Bomsztyk K. The product of the murine homolog of the Drosophila extra sex combs gene displays transcriptional repressor activity. Mol Cell Biol 1997; 17:4707-17; PMID:9234727; http://dx.doi.org/ 10.1128/MCB.17.8.4707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Evans JR, Mitchell SA, Spriggs KA, Ostrowski J, Bomsztyk K, Ostarek D, Willis AE. Members of the poly (rC) binding protein family stimulate the activity of the c-myc internal ribosome entry segment in vitro and in vivo. Oncogene 2003; 22:8012-20; PMID:12970749; http://dx.doi.org/ 10.1038/sj.onc.1206645 [DOI] [PubMed] [Google Scholar]
- [35].Lee M-H, Mori S, Raychaudhuri P. trans-Activation by the hnRNP K Protein Involves an Increase in RNA Synthesis from the Reporter Genes. J Biol Chem 1996; 271:3420-7; PMID:8631943; http://dx.doi.org/ 10.1074/jbc.271.7.3420 [DOI] [PubMed] [Google Scholar]
- [36].Naarmann-de Vries IS, Urlaub H, Ostareck DH, Ostareck-Lederer A. Caspase-3 cleaves hnRNP K in erythroid differentiation. Cell Death Dis 2013; 4:e548; PMID:23519117; http://dx.doi.org/ 10.1038/cddis.2013.75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Ostareck DH, Ostareck-Lederer A, Wilm M, Thiele BJ, Mann M, Hentze MW. mRNA silencing in erythroid differentiation: hnRNP K and hnRNP E1 Regulate 15-lipoxygenase translation from the 3′ End. Cell 1997; 89:597-606; PMID:9160751; http://dx.doi.org/ 10.1016/S0092-8674(00)80241-X [DOI] [PubMed] [Google Scholar]
- [38].Lee SW, Lee MH, Park JH, Kang SH, Yoo HM, Ka SH, Oh YM, Jeon YJ, Chung CH. SUMOylation of hnRNP‐K is required for p53‐mediated cell‐cycle arrest in response to DNA damage. EMBO J 2012; 31:4441-52; PMID:23092970; http://dx.doi.org/ 10.1038/emboj.2012.293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Pelisch F, Pozzi B, Risso G, Muñoz MJ, Srebrow A. DNA damage-induced heterogeneous nuclear ribonucleoprotein K sumoylation regulates p53 transcriptional activation. J Biol Chem 2012; 287:30789-99; PMID:22825850; http://dx.doi.org/ 10.1074/jbc.M112.390120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Yano M, Okano HJ, Okano H. Involvement of Hu and heterogeneous nuclear ribonucleoprotein K in neuronal differentiation through p21 mRNA post-transcriptional regulation. J Biol Chem 2005; 280:12690-9; PMID:15671036; http://dx.doi.org/; http://dx.doi.org/ 10.1074/jbc.M411119200 [DOI] [PubMed] [Google Scholar]
- [41].Gallardo M, Lee Hun J, Zhang X, Bueso-Ramos C, Pageon Laura R, McArthur M, Multani A, Nazha A, Manshouri T, Parker-Thornburg J, et al.. hnRNP K Is a Haploinsufficient Tumor Suppressor that Regulates Proliferation and Differentiation Programs in Hematologic Malignancies. Cancer Cell 2015; 28:486-99; PMID:26412324; http://dx.doi.org/ 10.1016/j.ccell.2015.09.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Au PYB, You J, Caluseriu O, Schwartzentruber J, Majewski J, Bernier FP, Ferguson M, Valle D, Parboosingh JS, Sobreira N, et al.. Gene Matcher aids in the identification of a new malformation syndrome with intellectual disability, unique facial dysmorphisms, and skeletal and connective tissue abnormalities caused by de novo variants in hnrnpk. Human Mutation 2015; 36:1009-14; PMID:26173930; http://dx.doi.org/ 10.1002/humu.22837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Liu Y, Gervasi C, Szaro BG. A crucial role for hnRNP K in axon development in Xenopus laevis. Development 2008; 135:3125-35; PMID:18725517; http://dx.doi.org/ 10.1242/dev.022236 [DOI] [PubMed] [Google Scholar]
- [44].de Oca Luna RM, Wagner DS, Lozano G. Rescue of early embryonic lethality in mdm2-deficient mice by deletion of p53. Nature 1995; 378:203-6; PMID:7477326; http://dx.doi.org/ 10.1038/378203a0 [DOI] [PubMed] [Google Scholar]
- [45].Forbes SA, Beare D, Gunasekaran P, Leung K, Bindal N, Boutselakis H, Ding M, Bamford S, Cole C, Ward S, et al.. COSMIC: exploring the world's knowledge of somatic mutations in human cancer. Nucleic Acids Res 2015; 43:D805-D11; PMID:25355519; http://dx.doi.org/ 10.1093/nar/gku1075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Piñol-Roma S, Choi YD, Matunis MJ, Dreyfuss G. Immunopurification of heterogeneous nuclear ribonucleoprotein particles reveals an assortment of RNA-binding proteins. Genes Dev 1988; 2:215-27; http://dx.doi.org/ 10.1101/gad.2.2.215 [DOI] [PubMed] [Google Scholar]