

ABSTRACT
In lung cancer, the Epidermal Growth Factor Receptor (EGFR) is one of the main targets for clinical management of this disease. The effectiveness of therapies toward this receptor has already been linked to the expression of integrin receptor subunit β1 in NSCLC A549 cells. In this work we demonstrate that azurin, an anticancer therapeutic protein originated from bacterial cells, controls the levels of integrin β1 and its appropriate membrane localization, impairing the intracellular signaling cascades downstream these receptors and the invasiveness of cells. We show evidences that azurin when combined with gefitinib and erlotinib, tyrosine kinase inhibitors which targets specifically the EGFR, enhances the sensitivity of these lung cancer cells to these molecules. The broad effect of azurin at the cell surface level was examined by Atomic Force Microscopy. The Young 's module (E) shows that the stiffness of A549 lung cancer cells decreased with exposure to azurin and also gefitinib, suggesting that the alterations in the membrane properties may be the basis of the broad anticancer activity of this protein. Overall, these results show that azurin may be relevant as an adjuvant to improve the effects of other anticancer agents already in clinical use, to which patients often develop resistance hampering its full therapeutic response
KEYWORDS: atomic force microscoy, azurin, EGFR, gefitinib, NSCLC, β1, integrin
Introduction
In order to progress, malignant cancers need to invade surrounding tissues. To do so, cells need to establish effective connections with the surrounding extracellular matrix (ECM). Cell adhesion occurs mainly through cell-surface receptors, such as the integrin superfamily, which are composed of α- and β-subunits, forming 24 already known αβ heterodimers.1-3 These receptors contain sequences for physical attachment of cells to the ECM, and their signaling alters various processes, including cell shape and an appropriate response to directed cell migration.4 Increasing evidences suggest that cell adhesion controls not only proliferation and migration in cancer cells, but can also exert a broader modulation in the interaction with tumor microenvironment. For example, integrins can promote the intracellular signaling of other membrane proteins such as Growth Factor (GF) receptors. In the case of non-small cell lung cancer (NCSLC), increased expression of α5β1 integrin is a poor prognostic factor.5 NCSLC accounts for 80% of all lung cancer and is of an epithelial origin. It has been recently demonstrated that in lung cancer cells β1 integrin controls Epidermal Growth Factor receptor (EGFR) signaling and its tumorigenic properties,6 suggesting that this transmembrane protein may be a suitable target for therapies.
Azurin, a protein from the bacterium Pseudomonas aeruginosa, has demonstrated antitumor properties.7,8 This protein has been intensively studied in different in vitro and in vivo models, demonstrating its ability to interfere in different steps of tumor development.9-12 A peptide derived from this protein, composed of 28 amino acids, named p28, has recently completed a phase I clinical trial and is now undergoing a second phase I trial against pediatric brain tumors (https://clinicaltrials.gov/ct2/show/NCT01975116).12-16 In a previous work, we performed a genome-wide microarray analysis of azurin-treated breast cancer cells. Among the most represented classes of genes whose expression was down-regulated upon azurin exposure were the classes of genes associated to biological/cell adhesion and cell surface receptors linked to signal transduction. Based on that, we also observed a decrease in the protein levels of integrin subunit β1 in azurin-treated breast cancer cells and a decreased ability to adhere to different ECM components and to grow in anchorage-independent conditions.17
In this study, we demonstrate that these effects can be extended to a non-small cell lung carcinoma model and that azurin can affect the EGFR signaling in this model. Furthermore, we show that azurin potentiates the effects of EGFR-targeted therapy with gefitinib and erlotinib. We also demonstrate that azurin-treated lung cancer cells have altered morphological features analyzed by Atomic Force Microscopy (AFM) imaging and nanoindentation measurements, revealing significant alterations that can be the basis of the broad range anticancer effects of azurin.
Results
Azurin decreases adhesion of A549 to ECM components and β1 integrin subunit protein expression
A549 cell line is a model for NSCLC with expression of high levels of wt EGFR.6 We investigated if azurin had the ability to interfere with adhesion between A549 cells and protein constituents of the ECM, such as laminin-332, collagen type I, collagen type IV and fibronectin. Cells were exposed to azurin for 48h, after which they were left to adhere to ECM proteins. Adhesion was measured by the crystal-violet assay. In general, a decrease in the adhesion of azurin-treated cells to ECM proteins was observed. Adhesion was particularly reduced to collagen type I and fibronectin, where a 40–50% decrease was detected (Fig. 1A). BSA-coated wells were used as control, with no apparent alterations in the adhesive capacity of cells.
Figure 1.

A) Azurin alters adhesion of A549 lung cancer cells to some extracellular matrix (ECM) components. Azurin treatment (50 µM and 100 µM, 48h) caused a reduction in the percentage of adhesion of cells to laminin-332, fibronectin and collagen type-I and IV (adhesion time = 20 min; * p < 0.05). B) A single treatment with azurin at 100 µM for 48h (same conditions as for adhesion assays) reduces protein expression of integrin subunit β1 under normal plastic conditions (black bars), or a matrix formed by collagen type-I (white bars) in A549 lung cancer cells. In contrary, these cells exhibit higher levels of E-cadherin under the same conditions. In the right panel, results are presented as the ratio of band intensity of target protein between azurin treated samples and control samples, both normalized to their respective actin band intensity (* p < 0.05). C) Immunofluorescence staining of integrin β1 (green, upper panel) and E-cadherin (green, lower panel) under the same treatment conditions (nuclei – DAPI, blue). Treatment with azurin alters the normal membrane staining of this transmembrane protein causing a delocalozation to a diffuse pattern in the interior of cells; D) Co-immunoprecipitation of ubiquitin and integrin β1. An antibody to this integrin was incubated with total cell lysates and used to precipitate it from both control and azurin treated total cell lysates. Proteins were separated in SDS-Page gels transferred to membranes which were probed with anti-ubiquitin antibody. A band corresponding to ubiquitin was detected at the molecular weight correspondent to integrin β1.
The integrin subunit β1 is a critical player mediating adhesion to ECM protein components (integrins α2β1 and α5β1 bind to collagen and fibronectin, respectively). We have previously observed that azurin causes a decrease in the protein levels of this subunit in breast cancer cells.17 Here, we observed the same effect, with total protein levels consistently diminished when cells were grown in normal plastic conditions and, even more evident, when cells were grown on top of a solid matrix composed of collagen type I (1mg/mL) (Fig. 1B, white bars). We also tested the effects on E-cadherin protein levels. E-cadherin is a known tumor suppressor protein associated to the epithelial phenotype of non-cancerous cells.18 Interestingly, the total protein levels of E-cadherin were increased in the same treatment conditions.
A difference in the pattern of this integrin subunit was also observed after immunofluorescence staining. In untreated cells, β1 is located mainly at the cell membrane; however, upon treatment with azurin, the cells exhibit a much lower staining (Fig. 1C, upper panel). Based on this, we tested if azurin is targeting this protein for degradation, by studying its ubiquitination. Integrin β1 was immunoprecipitated from total protein extracts in the conditions previous described and the membranes were probed with anti-ubiquitin (FK2) to detect ubiquitin revealing a corresponding band (Fig. 1D). Furthermore, we also performed a immunostaining of E-cadherin in both untreated and treated cells, and it is possible to see that the cell membrane retains its integrity after azurin treatment (Fig. 1C, lower panel).
Azurin impairs invasion behavior and pro-tumorigenic signaling in A549 cells
The positive role of β1 integrin in the invasion of A549 cells has been previously demonstrated.6 We tested if azurin, by decreasing its protein levels, was also capable of decreasing the cells' ability to migrate directionally, through Matrigel in Transwell assays. Treatment with azurin lead to a decrease in the invasive capacity of the cells by around 30%, with no additional stimuli (Fig. 2), supporting the observed decrease in β1 integrin.
Figure 2.
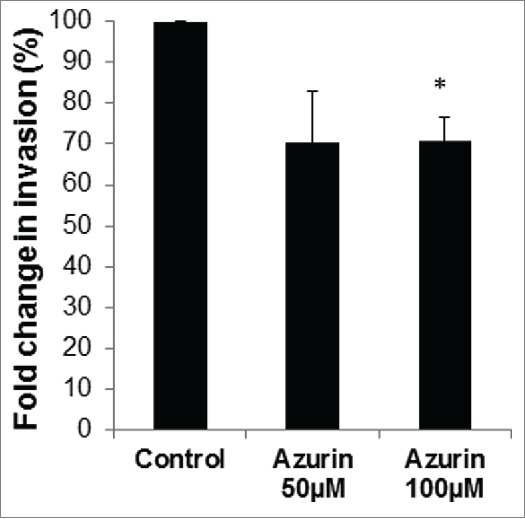
Azurin decreases invasion of A549 cells. Matrigel Invasion Assays showed that a single treatment with azurin 100 µM for 48 h significantly reduced the invasive behavior of breast cancer cells in Transwell assays (* p < 0.05).
In response to the physical attachment of integrins to the ECM, downstream intracellular signaling cascades are activated to improve cell proliferation and/or migratory and invasive capacities. Similar to other cell models,12,17 in A549 lung cancer cell lines, azurin decreases the phosphorylated levels of non-receptor tyrosine kinases, such as Src and Pi3K/Akt, downstream of EGFR (Fig. 3). A statistically significant decrease is observed in the phosphorylated forms of these 3 signaling intermediates after treatment with azurin, particularly when cells were treated with 100µM of azurin. For Akt, an addition of azurin at 100μM results in a decrease of about 10% in the total levels of this protein, but the phosphorylated form decreases to about 40% in the same conditions, indicating that the phosphorylated form is much more affected than the levels of both phosphorylated and non-phosphorylated protein.
Figure 3.
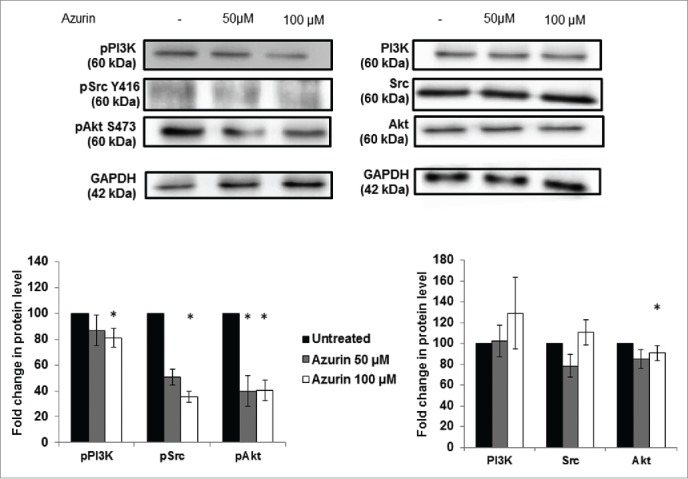
Effect of azurin in PI3K, Akt and Src signaling. Azurin at 50 µM and 100 µM decreased phosphorylation levels of pPI3K, pAkt and pSrc in both A549 cells. Total PI3K, Akt and Src levels were also analyzed. Results are presented as the ratio of band intensity of target protein between azurin treated samples and control samples, both normalized to their respective GAPDH band intensity (* p < 0.05).
Azurin leads to a defective response to EGF and enhanced sensitivity to EGFR-targeted therapy
In normal epithelial cells, the role of integrins in supporting the EGFR activation both in the presence and absence of EGF is already established.1 In A549, Morello and colleagues,6 have shown that EGFR phosphorylation is dependent on β1 integrin-adhesion events, suggesting a dependence for proper EGFR signaling on β1 integrin. When treated with azurin in the same conditions where β1 integrin protein levels are decreased (100 µM, 48h), an increase in the total levels of the EGFR are observed. The same has been observed when β1 integrin was silenced using siRNA; however, in that case β1-silenced cells had a defective response to soluble EGF. We tested then the ability of azurin-treated cells to respond to EGF. Two concentrations of EGF (20 and 50 ng/mL) were exposed to cells in the presence or absence of azurin for another 30 min, after 48h in the absence or presence of the protein, respectively. What we observed was that in the presence of azurin, the levels of phosphorylation in the EGFR on the signaling related residues Y1068 failed to increase such as those of the cells exposed to EGF in the absence of the pre-treatment with azurin (Fig. 4A). Also, upon binding of soluble EGF, EGFR internalization through its canonical pathways is essential for the downstream signaling cascade of this receptor. We observed that in the presence of azurin, despite the fact that the level of the receptor seems to be originally increased; the decrease of total EGFR is lower after EGF stimulation.
Figure 4.
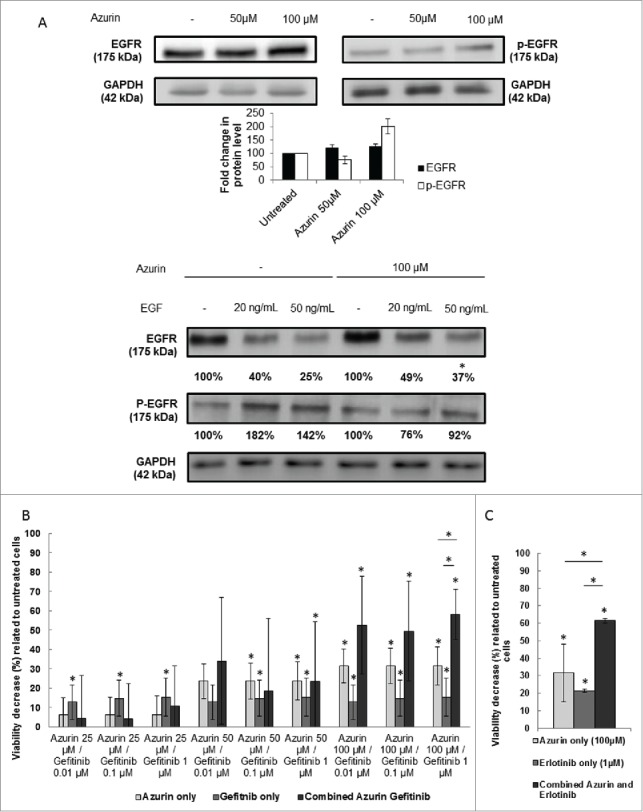
(A) Effect of azurin in EGFR signaling. Azurin at 50 µM and 100 µM decreased phosphorylation levels of pEGFR in both A549 cells. Total EGFR levels were also analyzed. Results are presented as the ratio of band intensity of target protein between azurin treated samples and control samples, both normalized to their respective actin band intensity (* p < 0.05) (upper panel); Azurin internalization impairs proper pEGFR response to soluble EGF. Cells were serum starved for 24h, pre-treated with azurin (100 µM, 48h) and treated with EGF (20 or 50 ng/ml) for 30 min. EGF-dependent signaling was evaluated in western blot with pEGFR Y1068 antibody. Results are presented as the ratio of band intensity of target protein between azurin-treated samples and control samples, both normalized to their respective GAPDH band intensity (* p < 0.05). B) Azurin potentiates the effects of gefitinib. 4 × 103 cells per well were plated in 96-well plates and left to adhere overnight. In the next day, cells were treated with either azurin (25, 50 or 100 µM), gefitinib (0.01, 0.1 or 1 µM) or a combination of both. After 72h, cell proliferation was determined by MTT assay. Results are expressed as percentage of cell death relative to the control (untreated cells). Values are presented as mean ± SD. White bars represent cell treated with azurin only, gray bars represent cells treated with gefitinib only, and dark gray bars represent cells treated with both agents. The asterisks over each bar represent statistical significance related to untreated cells; the asterisks over a line connecting 2 bars represent statistical significance between those 2 conditions (* p < 0.05). C) Azurin potentiates the effects of erlotinib. 4 × 103 cells per well were plated in 96-well plates and left to adhere overnight. In the next day, cells were treated with either azurin (100 µM), erlotinib (1 µM) or a combination of both. After 72h, cell proliferation was determined by MTT assay. Results are expressed as percentage of cell death relative to the control (untreated cells). Values are presented as mean ± SD. White bars represent cell treated with azurin only, gray bars represent cells treated with erlotinib only, and dark gray bars represent cells treated with both agents. The asterisks over each bar represent statistical significance related to untreated cells; the asterisks over a line connecting 2 bars represent statistical significance between those 2 conditions (* p < 0.05).
In this context, we exposed A549 lung cancer cells to a selective EGFR inhibitor, Gefitinib, at low concentrations, to assess the potential synergy of a co-treatment with azurin. The overexpression of β1 integrin in lung cancer has itself been associated to the resistance to the treatment with this drug, a process that is at least in part mediated by the signaling pathways that are attenuated by azurin.6,19,20 The combined treatment (Fig. 4B, dark gray bars) with different doses of azurin and gefitinib (at doses below the IC50) showed that the toxicity caused at the azurin concentrations where β1 integrin is decreased is much higher than upon exposure to either of the agents alone, indicating that the lowering of this integrin subunit and downstream signaling attenuation significantly enhances the effect of this pharmacological agent. In particular, when we combined the highest doses tested for each agent (azurin at 100μM and Gefitinib at 1μM, we observed an increase of about 15–20% in cell death when compared to the sum of each agent alone. To confirm this result, we tested another EGFR-targeted agent, namely erlotinib, demonstrating an identical behavior, i.e. a synergistic effect in comparison to the single treatments (Fig. 4C).
Azurin alters membrane properties in lung cancer cells
The observations presented in the previous sections regarding alterations in the adhesive properties of A549 cells to ECM, as well as defective signaling response to EGF by its receptor and associated intracellular signaling pathways, prompted us to study by Atomic Force Microscopy (AFM) possible alterations in the cellular biophysical and nanomechanic properties, which could explain the interference with cell attachment and response to growth factors. Cells are in constant need to adapt to changes in the surrounding environment, therefore adapting their physical and chemical properties, namely at the membrane level, the cell's physical barrier to its exterior. These changes impact important cellular processes such as adherence, signaling, invasion and proliferation. AFM is a technique with increased importance in detecting and quantifying these changes, namely on elasticity and deformability.21 By AFM imaging, we could detect differences both on cell height and area, induced by the treatments with azurin and the pharmacogical agent gefitinib (Fig. 5). These changes may be associated with differences in the cytoskeleton arrangement upon treatment, generated by the decreased intracellular signaling cascades that azurin and gefitinib produce. It is also noted that in control (untreated) condition, cells' nuclei appear thicker and better defined than upon treatment, particularly when both agents are added, in accordance with the highest toxicity observed.
Figure 5.
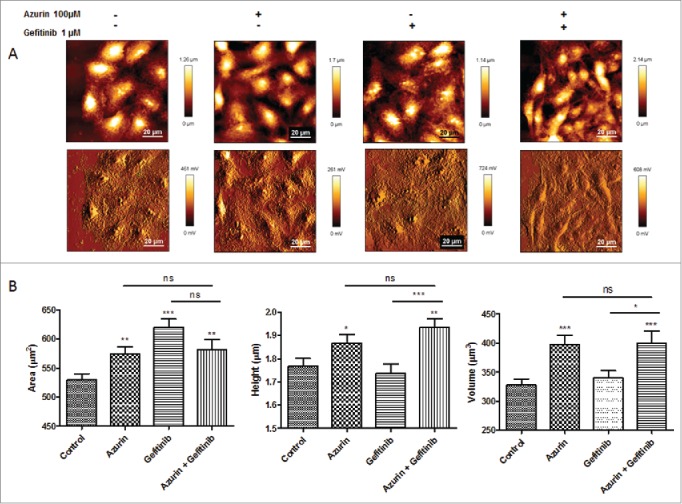
A) Representative height (upper panel) and error (lower panel) AFM images of A549 cells untreated (control), or treated with azurin (100 µM, 48h), gefitinib (1 µM, 72h) or both (same concentrations, 72h). B ) Morphological characteristics (cell area, height and volume) of A549 cells at the same conditions. Values are presented as mean ± SEM. * p < 0.05; ** p < 0.01; *** p < 0.001.
Variations in the elastic modulus can be used to assess the effect that external molecules can cause in the deformability of cells or on their stiffness. We measured alterations in the membrane elasticity through nanoindentation using the AFM tip of A549 cells untreated or treated with azurin (100 µM), gefitinib (1 µM) or both combined, for 72h. Individual force-displacement curves were obtained, converted to force-distance (F-d) curves and shifted to remove the offset. The Young's modulus (E) of treated cells was decreased upon treatment with either of the compounds (Fig. 6). For azurin alone, a decrease in E of approximately 30% was detected; gefitinib generated a decrease of the same magnitude, as well as the combined treatment. Interestingly, these results show that azurin induces the same type of decrease in membrane stiffness as gefitinib, suggesting that the same type of alterations may be occurring in those cells, like the decrease of intracellular signaling cascades that may lead to cell cycle arrest and/or inhibition of proliferation, reflected in the cell's phenotype and topography.
Figure 6.
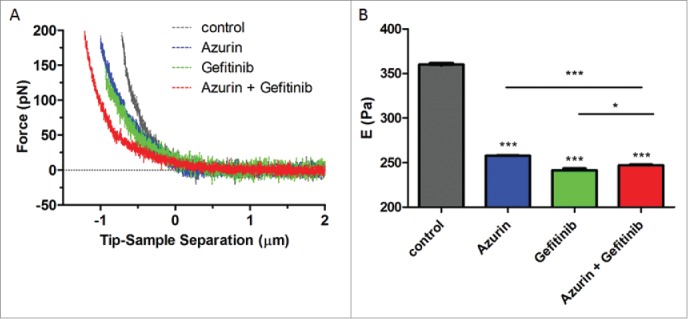
(A) Force indentation curves obtained from indentation of A549 cells untreated (control), or treated with azurin (100 µM, 48h), gefitinib (1 µM, 72h) or both (same concentrations, 72h); (B) Young's modulus for A549 cells untreated (control), or treated with azurin (100 µM, 48h), gefitinib (1 µM, 72h) or both (same concentrations, 72h). Results with standard deviations were obtained by using over 250 cells, in at least 3 different biological replicates, with 5 force curves per cell. Values are presented as the maximum Gaussian mean ± SEM. * p = 0.015; *** p < 0.001.
Discussion
The cooperation between integrins and growth factor receptors is well established as a coordinated mechanism used by cells to orchestrate an appropriate response to external stimuli and maintain tissue homeostasis.1,2 In the case of β1 integrin/EGFR, the importance of this coordinated response has been outlined for breast and lung cancers,6,22-24 therefore integrin subunit β1 is now recognized as a drug target in several cancer models. In fact, in lung cancer, its aberrant overexpression has been associated with reduced patient survival.5,25 In a recent study with breast cancer cells, we identified biological/cell adhesion as part of the response to azurin in a genome-wide transcription analysis. A decrease in the levels of β1 integrin was also observed in those models, along with decreased adhesion to ECM proteins and reduced mammosphere forming efficiency.17 In this work we demonstrate that azurin also causes a decrease in the levels of this protein in A549 non-small lung cancer cells and that it can also be associated to a decreased in the invasion through an artificial matrix such as Matrigel.
In an endothelial model of HUVEC cells, exposition to p28 for 30 min altered the migratory ability of those cells, as well as the kinase activity of VEGFR-2, in response to the soluble factor VEGFA.12 Also, the intracellular signaling response of non-receptor tyrosine kinases downstream of VEGFR-2, as FAK and PI3K, were also inhibited, suggesting an outside-in attenuated response to the soluble growth factor in the presence of p28. In breast cancer cells, addition of azurin for up to 24 or 48h, reduced phosphorylation of FAK and Src, also associated to decreased invasion through Matrigel, in models in which increased activity was activated by an overexpression of P-cadherin, also downregulated by azurin.26 In this work, the levels of p-Src Y416, pAkt S473 and pPI3K were severely attenuated, suggesting that the alterations of induced extend from the β1 integrin protein levels and appropriate membrane localization to the intracellular partners.
Our results show that, in addition to the decrease in β1 integrin, A549 lung cancer cells have a defective response to EGF and enhanced in vitro sensitivity to EGFR-targeted pharmacological agents. Exposition to EGF for 30 min in the presence of azurin prevented the phosphorylation of EGFR-Y1068 that occurred in the absence of the protein, implicating β1 integrin in the full response to EGF.
Recent reports have elucidated that the mechanisms by which lung cancer cells develop resistance to EFGR-targeted therapy often involve an elevated expression of β1 integrin, combined with increased activity of Src and PI3K/Akt pathways.19,20 Ju et al. have established a model of PC9-derived cells with increased resistance to gefitinib, PC9/AB2, in which elevated expression of this integrin subunit increased cells' adhesion and migration, which were reverted by the silencing of the receptor.20 In another study, Kanda and colleagues found that in lung cancer cells harboring EGFR activating mutations and increased resistance to erlotinib, there was an increased expression of Src and integrin β1. Likewise, silencing of integrin β1 restored sensitivity to the drug, further reducing the activation of Src and Akt. Other studies have shown that the interaction between cancer cells and the extracellular matrix proteins can also be a factor reducing the sensitivity to anticancer agents.27-29
Recently, we performed a global transcriptomic analysis in breast cancer cells, where an up-regulation in the expression of genes associated to membrane reorganization was observed. Another class of genes with increased expression related to untreated cells was endocytosis, suggesting that the mechanism by which azurin enters cells may cause alterations at the cell membrane level.17 One of the most interesting aspects of the anticancer activity displayed by azurin seems to be the broad range of molecular mechanisms it can affect in cancer cells.11,12,15,16 We hypothesized that such a broad range must be connected to an alteration in important features of cancer cells, such as its membrane properties, altering its ability to interact with the surrounding environment, its ability to respond to extracellular signaling and its response to therapeutic agents. Cell stiffness measured by Atomic Force Microscopy is gaining increased importance to study the characteristics of cancer cells and the way these properties may influence the response to therapeutic agents.21,30-32 On one hand, invasive cancer cells seem to have a lower membrane elastic modulus compared to non-invasive cells, which in turn, have even lower elastic modules than non-cancer cells in different models.33,34 On the other hand, there are other reports evidencing that cancer cells treated with different pharmacological agents exhibit a decrease in their membrane stiffness (decreased E), as observed in our study. Particularly, paclitaxel caused a decrease in the stiffness of Ishikawa and HeLa cells, reflecting the apoptotic changes occurred.31 Interestingly, 2 different anticancer peptides, human HNP-1 21 and a customized anticancer peptide CB1a,32 were recently reported to induce similar changes in different cancer models, in which the anticancer activity was accompanied with decrease membrane stiffness, reflecting the apoptotic changes induced. Also, the role of cytoskeleton rearrangement was recently associated with resistance to cisplatin and membrane stiffness in ovarian cancer. Cisplatin-resistant cell lines were found to have a higher Young's modulus than cisplatin-sensitive in a panel of 9 ovarian cancer cell lines.30 In our work, azurin alone led to a decrease of approximately 30% in the stiffness of A549 lung cancer cells, as observed for gefitinib alone, and for the combination of both agents. This result correlates with decreased invasion and the ability of cancer cells to adhere to the ECM protein components.
Tumor recurrence is a major problem that both clinicians and the scientific community face in combating cancer. New therapeutic strategies, more effective in killing cancer cells but also more selective, are needed, in order to increase the efficiency and decrease the toxic side effects associated with the current therapies in clinical use. Bacterial proteins and peptides are a class of new therapeutics with promising applications in this field. In recent years, azurin, a protein derived from the bacterium Pseudomonas aeruginosa, has been studied as a new therapeutic toward cancer. We provide evidences in this work that azurin alters the membrane biophysical properties of lung adenocarcinomas, targets the integrin subunit β1, as previously observed for breast cancer cells, and that these effects extend to signaling pathways that mediate resistance to a pharmacological agent, potentiating its effects at low doses. These effects may be of particular interest in drug resistant cancers, where the more rigid nature of the membrane was associated to increased resistance to the accumulation of anticancer drugs.35 In this context, a distinct membrane lipid composition in cancer cells relative to normal cells may be implicated, as well as the local organization in membrane microdomains, such as lipid rafts, that may be disrupted by azurin, potentiating the effects of other co-administrated drugs.
Materials and methods
Antibodies
The following antibodies were used: integrin subunit β1 (WB - 1:200, IF – 1:50, Santa Cruz Biotechnologies); E-cadherin (WB - 1:1000, IF – 1:100, clone HECD1, Takara Bio Inc.), β-actin (1:1000, Santa Cruz Biotechnologies) GAPDH (Glyceraldehyde 3-phosphate dehydrogenase 1:1000, Santa Cruz Biotechnology), Epidemal growth factor receptor (1:500, Cell Signaling), pEFGR -Y1068 (1:500, Cell signaling), total Src (1:500, Cell Signaling), pSrc Y416 (1:500, Cell Signaling), total Akt (1:1000, Santa Cruz Biotechnologies), pAkt S473 (1:500, Cell Signaling), pPI3K (1:500, Cell Signaling), and total PI3K (1:500, Cell Signaling).
Cell culture and growth conditions
Human lung adenocarcinoma cell line A549 was grown in DMEM (Gibco) supplemented with 10% heat-inactivated fetal bovine serum, penicillin and streptomycin (100U/ml and 100µg/ml, respectively), supplemented with 0.292 g/liter L-glutamine (Gibco) at 37°C in a 5% CO2 atmosphere.
Bacteria growth media and protein purification
Bacteria growth and protein expression and purification were performed as previously described.26
Adhesion assay to ECM substrates
Cell adhesion was performed in 96-well plates coated with laminin 332 (Sigma-Aldrich), fibronectin (Sigma-Aldrich), type-I or IV collagen (Sigma-Aldrich) (5µg/ml) overnight at 4°C. Afterwards, plates were washed 3 times with phosphate buffered saline (PBS) and non-specific binding sites were blocked with 0.5% BSA (w/v) in PBS containing PenStrep (Invitrogen) for 2h at 37°C. After washing again, 100 µL of untreated or azurin (50 or 100 µM; 48h) treated cells (106 cells/ml) were seeded in serum-free media for 30 min. Non-adherent cells were removed by washing plates 3 times with PBS, and the attached cells were fixed with acetone:methanol (1:1) for 10 minutes at 4°C. Adhesion was determined following the colorimetric method described by Busk et al.,36 measuring absorbance at 570 nm with a microplate reader. BSA and plastic uncoated wells were used as controls. Results are presented as the percentage of adhesion between azurin-treated and untreated cells (100%).
Protein extraction and western blot analysis
Cultured cells, treated with azurin (50 or 100 µM) or untreated, were lysed using catenin lysis buffer (1% Triton X-100 and 1% NP-40 in deionized PBS), supplemented with 1:7 protease inhibitor cocktail (Roche Diagnostics GmbH) and with 1:100 of phosphatase inhibitor cocktail 3 (Sigma). Cells were washed twice with PBS and lysed in 100 µL of catenin lysis buffer for 10 min, at 4°C. Lysed cells were collected and vortexed 3 times, for 10 s, prior to centrifugation at 14000 rpm for 10 min at 4°C. Total protein quantification was performed with BCATM Protein Assay kit (Pierce). Twenty µg (E -cadherin, total FAK, Src and Akt), 30 µg (β1 integrin, phosphorylated FAK, Src and Akt) or 40 µg (p-EGFR Y1068) of the total protein lysate was dissolved in sample buffer [Laemmli with 5% (v/v) 2-β-mercaptoethanol and 5% (v/v) bromophenol blue], boiled for 5 min at 95°C, and separated by SDS-PAGE. Proteins were transferred onto nitrocellulose membranes using the Trans-blot TurboTM Transfer System (BioRad). Membranes were blocked with 5% (w/v) non-fat dry milk in PBS containing 0.5% (v/v) Tween-20 (PBS-T) or 5% BSA (for phosphorylated protein detection) for 1 h, incubated with primary antibodies overnight at 4 °C and washed 3 times for 5 min with PBS-T. Membranes were then incubated for 1 h with secondary antibodies, conjugated with horseradish peroxidase. Proteins were detected through the addition of ECL reagent (Pierce) as a substrate and captured the chemiluminescense by Fusion Solo (Viber Lourmat) equipment. Three experiments were independently performed and representative results are shown. Signal quantifications were performed using ImageJ and results are presented as the ratio between the signal intensities in azurin-treated samples to untreated cells, both normalized to the respective actin band intensities.
Immunofluorescence
Cells were cultured on glass coverslips and treated with azurin (100 µM). After 48h, medium was collected and cells were washed twice with PBS. Fixation was performed with NH4Cl for 20 min at room temperature. Permeabilization was achieved with 1% Triton X-100 in PBS for 5 min at room temperature and coverslips were blocked with 5% BSA solution in PBS for 30 min. Primary antibody to β1 integrin was added for 1 h at room temperature (1:50). After this incubation, cells were washed 3 times for 5 min with PBS and incubated with secondary antibody for 1 hour, at room temperature (1:500 dilution, mouse polyclonal conjugated with Alexa-488). Each sample was washed with PBS after the incubation period and mounted with Vectashield (Vector Laboratories Inc.,) containing 4,6-diamidine-2-phenylindolendihydrochloride (DAPI). Samples were examined on a Leica TCS SP5 (Leica Microsystems CMS GmbH, Mannheim, Germany) inverted microscope (model DMI6000) with a 63× water (1.2-numerical-aperture) apochromatic objective.37
Matrigel invasion assays
Matrigel Invasion assay was performed according to the manufacturer´s instructions (BD Coat Matrigel Invasion Chambers, BD Biosciences). Briefly, Matrigel inserts containing an 8 µm pore size PET membrane with a thin layer of Matrigel Basement Membrane Matrix were pre-incubated with serum-free media for 2 h at 37°C. 5 × 104 A549 lung cancer cells were seeded in the upper compartment, with or without azurin (control). After 48h, invasive cells were colored with DAPI and counted under the microscope. In each condition, 10 independent fields were counted and the average of these fields considered as the mean number of invasive cells per condition. Results are presented as the fold change in invasion of cells in comparison with the untreated cells.
MTT cell proliferation assay
MTT [3-(4,5 dimethylthiazol-2-yl-2,5 tetrazolium bromide)] assays were used to determine the viability of breast cancer cells upon azurin exposure. Breast cancer cells were seeded in 96-well plates (3 replicates) (Orange Scientific) at a density of 4 × 103 cells. After 24h, medium was changed and fresh azurin, Gefitinib (Santa Cruz), a combination of both or an identical volume of media (100 µL) were added. After another 72h, 20 µL of MTT (5 mg/ml) were added to each well and incubated at 37°C for 3.5h. The reaction was stopped by the addition of 40mM HCl in isopropanol (150 µL). MTT formazan formed was spectrophotometrically read at 590 nm in a 96-well plate reader. Untreated cells were used as control, in order to determine the relative cell viability of treated cells.
Atomic force microscopy
An atomic force microscope NanoWizard II (JPK Instruments, Berlin, Germany) mounted on the top of an Axiovert 200 inverted microscope (Carl Zeiss, Jena, Germany) was used for cell imaging. The AFM head is equipped with a 15-μm z-range linearized piezoelectric scanner and an infrared laser. Cultured cells were washed with PBS, pH 7.4, and gently fixed with glutaraldehyde solution 2.0 % (v/v) for 10 min at room temperature. Cells were subsequently washed 3 times with PBS and MilliQ water and allowed to air dry at room conditions. The AFM imaging of the cells was performed in tapping mode, in air. Oxidized sharpened silicon tips (ACL tips from Applied Nanostructures, CA) with a tip radius of 6 nm, resonant frequency of about 190 kHz and spring constant of 45 N/m were used for the imaging. Imaging parameters were adjusted to minimize the force applied on the scanning of the topography of the sample. Scanning speed was optimized to 0.4 Hz, with 512 × 512 acquisition points. Imaging data were analyzed with the JPK image processing v. 5.1.8 (JPK Instruments, Germany). The height, area and volume of each imaged cell were quantified using the SPIP software (Image Metrology, Hørsholm, Denmark) v. 6.4.1. For each experimental condition approximately 7 high resolution AFM images were obtained in 4 different culture dishes.
Nanoindentation experiments, for cell elasticity assessment, were carried out on live cells, at 25°C, in serum free DMEM. For these measurements we used non-functionalized OMCL TR-400-type silicon nitride tips (Olympus, Japan). The softest triangular cantilevers, with a tip radius of 15 nm and a resonant frequency of 11 kHz, were used. The spring constants of the tips were calibrated by the thermal fluctuation method, having a nominal value of 0.02 N/m. For every contact between cell and cantilever, the distance between the cantilever and the cell was adjusted to maintain a maximum applied force of 200 pN before retraction. Cell elasticity was measured on one point of each cell adhered to the tissue culture dish (5 force-distance curves per cell), and on approximately 100 cells at 4 different cell dishes. Data collection for each force-distance cycle was performed at 1.5 Hz, with a Z-displacement range of 4 μm. Force curves were made at the center of the cell, on the top of its nucleus. Data acquired on the nanoindentation experiments (force curves) were analyzed to obtain the cells Young's modulus (E), using JPK Image Processing v. 5.1.8, by the application of the Hertzian model.38 The probe was modeled as a quadratic pyramid, with a tip angle of 35° (half-angle to face) and a Poisson ration of 0.50. Young's modulus histograms were constructed for each experimental condition studied. The ideal histogram bin size was chosen in order to achieve the best fitted Gaussian model peak length, yielding a selected binning size of 35 Pa. The maximum values of the Gaussian peaks represent different statistical measure of the Young's modulus of the cells.
Statistical analysis
Data are expressed as mean values of at least 3 independent experiments ± standard deviation (SD). Student's two-tailored t-tests were used to determine statistically significant differences (p < 0.05) between each condition analyzed and the respective untreated condition. In AFM measurements, statistical significance was determined with pair-wise comparisons with Student's t-test to compare the cells datasets, using a 5% confidence interval. Statistical analysis was carried out using the Graphpad Prism software, v. 5.0.
Disclosure of potential conflicts of interest
The authors declare no conflicts of interest.
Author contributions
This study was designed by AMF and NCS. NB, SA, FAC and FF performed the experiments. NB wrote the manuscript. All authors read and approved the final manuscript.
Funding
The work presented was supported by scientific projects (PTDC/EBBBIO/100326/2008, PTDC/QUI-BIQ/119509/2010 and RECI/CTM-POL/0342/2012) financed by the Portuguese Science and Technology Foundation (FCT). FCT also provides a post-doctoral research grants for Nuno Bernardes (SFRH/BPD/98162/2013) and Fábio Fernandes (SFRH/BPD/64320/2009). Funding received by iBB-Institute for Bioengineering and Biosciences from FCT (UID/BIO/04565/2013) from Programa Operacional Regional de Lisboa 2020 (Project N. 007317) is acknowledged.
References
- [1].Streuli CH, Akhtar N. Signal co-operation between integrins and other receptor systems. Biochem J 2009; 418:491-506; PMID:19228122; http://dx.doi.org/ 10.1042/BJ20081948 [DOI] [PubMed] [Google Scholar]
- [2].Desgrosellier JS, Cheresh DA. Integrins in cancer: biological implications and therapeutic opportunities. Nat Rev Cancer 2010; 10:9-22; PMID:20029421; http://dx.doi.org/ 10.1038/nrc2748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Rathinam R, Alahari SK. Important role of integrins in the cancer biology. Cancer 2010; 29:223-37; PMID:20492711 [DOI] [PubMed] [Google Scholar]
- [4].Pontier SM, Muller WJ. Integrins in mammary stem cell biology and breast cancer progression - a role in cancer stem cells? J Cell Sci 2009; 122:207-14; PMID:19118213; http://dx.doi.org/ 10.1242/jcs.040394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Dingemans A-MC, van den Boogaart V, Vosse BA, van Suylen R-J, Griffioen AW, Thijssen VL. Integrin expression profiling identifies integrin alpha5 and beta1 as prognostic factors in early stage non-small cell lung cancer. Mol Cancer 2010; 9:152; PMID:20565758; http://dx.doi.org/ 10.1186/1476-4598-9-152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Morello V, Cabodi S, Sigismund S, Camacho-Leal MP, Repetto D, Volante M, Papotti M, Turco E, Defilippi P. β1 integrin controls EGFR signaling and tumorigenic properties of lung cancer cells. Oncogene 2011; 30:4087-96; PMID:21478906; http://dx.doi.org/ 10.1038/onc.2011.107 [DOI] [PubMed] [Google Scholar]
- [7].Yamada T, Hiraoka Y, Ikehata M, Kimbara K, Avner BS, Gupta TK Das, Chakrabarty AM. Apoptosis or growth arrest: modulation of tumor suppressor p53 specificity by bacterial redox protein azurin. PNAS 2004; 101:4770-5; PMID:15044691; http://dx.doi.org/ 10.1073/pnas.0400899101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Punj V, Bhattacharyya S, Saint-dic D, Vasu C, Cunningham EA, Graves J. Bacterial cupredoxin azurin as an inducer of apoptosis and regression in human breast cancer. Oncogene 2004; 23:2367-78; PMID:14981543; http://dx.doi.org/ 10.1038/sj.onc.1207376 [DOI] [PubMed] [Google Scholar]
- [9].Yamada T, Fialho AM, Punj V, Bratescu L, Gupta TK Das, Chakrabarty AM. Internalization of bacterial redox protein azurin in mammalian cells: entry domain and specificity. Cell Microbiol 2005; 7:1418-31; PMID:16153242; http://dx.doi.org/ 10.1111/j.1462-5822.2005.00567.x [DOI] [PubMed] [Google Scholar]
- [10].Chaudhari A, Mahfouz M, Fialho AM, Yamada T, Granja AT, Zhu Y, Hashimoto W, Schlarb-Ridley B, Cho W, Das Gupta TK, et al.. Cupredoxin-cancer interrelationship: azurin binding with EphB2, interference in EphB2 tyrosine phosphorylation, and inhibition of cancer growth. Biochemistry 2007; 46:1799-810; PMID:17249693; http://dx.doi.org/ 10.1021/bi061661x [DOI] [PubMed] [Google Scholar]
- [11].Taylor BN, Mehta RR, Yamada T, Lekmine F, Christov K, Chakrabarty AM, Green A, Bratescu L, Shilkaitis A, Beattie CW, et al.. Noncationic peptides obtained from azurin preferentially enter cancer cells. Cancer Res 2009; 69:537-46; PMID:19147567; http://dx.doi.org/ 10.1158/0008-5472.CAN-08-2932 [DOI] [PubMed] [Google Scholar]
- [12].Mehta RR, Yamada T, Taylor BN, Christov K, King ML, Majumdar D, Lekmine F, Tiruppathi C, Shilkaitis A, Bratescu L, et al.. A cell penetrating peptide derived from azurin inhibits angiogenesis and tumor growth by inhibiting phosphorylation of VEGFR-2, FAK and Akt. Angiogenesis 2011; 14:355-69; PMID:21667138; http://dx.doi.org/ 10.1007/s10456-011-9220-6 [DOI] [PubMed] [Google Scholar]
- [13].Yamada T, Christov K, Das Gupta TK BC. Mechanism of action of p28, a first-in-class, non-HDM2 mediated peptide inhibitor of p53 ubiquitination. In: J Clin Oncol 2011; 29. page suppl; abstr e13513. [Google Scholar]
- [14].Warso M a, Richards JM, Mehta D, Christov K, Schaeffer C, Rae Bressler L, Yamada T, Majumdar D, Kennedy S a, Beattie CW, et al.. A first-in-class, first-in-human, phase I trial of p28, a non-HDM2-mediated peptide inhibitor of p53 ubiquitination in patients with advanced solid tumours. Br J Cancer 2013; 108:1061-70; PMID:23449360; http://dx.doi.org/ 10.1038/bjc.2013.74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Yamada T, Christov K, Shilkaitis A, Bratescu L, Green A, Santini S, Bizzarri AR, Cannistraro S, Gupta TKD, Beattie CW. p28, A first in class peptide inhibitor of cop1 binding to p53. Br J Cancer 2013; 108:2495-504; PMID:23736031; http://dx.doi.org/ 10.1038/bjc.2013.266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Yamada T, Mehta RR, Lekmine F, Christov K, King ML, Majumdar D, Shilkaitis A, Green A, Bratescu L, Beattie CW, et al.. A peptide fragment of azurin induces a p53-mediated cell cycle arrest in human breast cancer cells. Mol Cancer Ther 2009; 8:2947-58; PMID:19808975; http://dx.doi.org/ 10.1158/1535-7163.MCT-09-0444 [DOI] [PubMed] [Google Scholar]
- [17].Bernardes N, Ribeiro AS, Abreu S, Vieira AF, Carreto L, Santos M, Seruca R, Paredes J, Fialho AM. High-throughput molecular profiling of a P-cadherin overexpressing breast cancer model reveals new targets for the anti-cancer bacterial protein azurin. Int J Biochem Cell Biol 2014; 50:1-9; PMID:24509127; http://dx.doi.org/ 10.1016/j.biocel.2014.01.023 [DOI] [PubMed] [Google Scholar]
- [18].Paredes J, Figueiredo J, Albergaria A, Oliveira P, Carvalho J, Ribeiro AS, Caldeira J, Costa AM, Simões-Correia J, Oliveira MJ, et al.. Epithelial E- and P-cadherins: Role and clinical significance in cancer. Biochim Biophys Acta 2012; 1826:297-311; PMID:22613680 [DOI] [PubMed] [Google Scholar]
- [19].Kanda R, Kawahara A, Watari K, Murakami Y, Sonoda K, Maeda M, Fujita H, Kage M, Uramoto H, Costa C, et al.. Erlotinib Resistance in Lung Cancer Cells Mediated by Integrin β1/Src/Akt-Driven Bypass Signaling. Cancer Res 2013; 73:6243-53; PMID:23872583; http://dx.doi.org/ 10.1158/0008-5472.CAN-12-4502 [DOI] [PubMed] [Google Scholar]
- [20].Ju L, Zhou C, Li W, Yan L. Integrin beta1 over-expression associates with resistance to tyrosine kinase inhibitor gefitinib in non-small cell lung cancer. J Cell Biochem 2010; 111:1565-74; PMID:21053345; http://dx.doi.org/ 10.1002/jcb.22888 [DOI] [PubMed] [Google Scholar]
- [21].Gaspar D, Freire JM, Pacheco TR, Barata JT, Castanho MARB. Apoptotic human neutrophil peptide-1 anti-tumor activity revealed by cellular biomechanics. Biochim Biophys Acta - Mol Cell Res 2015; 1853:308-16; PMID:25447543; http://dx.doi.org/ 10.1016/j.bbamcr.2014.11.006 [DOI] [PubMed] [Google Scholar]
- [22].Ju L, Zhou C. Integrin beta 1 enhances the epithelial- mesenchymal transition in association with gefitinib resistance of non-small cell lung cancer. Cancer Biomark 2013; 13:329-36; PMID:24440972 [DOI] [PubMed] [Google Scholar]
- [23].Zhang X, Fournier M V, Ware JL, Bissell MJ, Yacoub A, Zehner ZE. Inhibition of vimentin or beta1 integrin reverts morphology of prostate tumor cells grown in laminin-rich extracellular matrix gels and reduces tumor growth in vivo. Mol Cancer Ther 2009; 8:499-508; PMID:19276168; http://dx.doi.org/ 10.1158/1535-7163.MCT-08-0544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Carpenter PM, Dao AV, Arain ZS, Chang MK, Nguyen HP, Arain S, Wang-Rodriguez J, Kwon S-Y, Wilczynski SP. Motility induction in breast carcinoma by mammary epithelial laminin 332 (laminin 5). Mol Cancer Res 2009; 7:462-75; PMID:19351903; http://dx.doi.org/ 10.1158/1541-7786.MCR-08-0148 [DOI] [PubMed] [Google Scholar]
- [25].Okamura M, Yamaji S, Nagashima Y, Nishikawa M, Yoshimoto N, Kido Y, Iemoto Y, Aoki I, Ishigatsubo Y. Prognostic value of integrin beta 1-ILK-pAkt signaling pathway in non-small cell lung cancer. Hum Pathol 2007; 38:1081-91; PMID:17442374; http://dx.doi.org/ 10.1016/j.humpath.2007.01.003 [DOI] [PubMed] [Google Scholar]
- [26].Bernardes N, Ribeiro AS, Abreu S, Mota B, Matos RG, Arraiano CM, Seruca R, Paredes J, Fialho AM. The bacterial protein azurin impairs invasion and FAK/Src signaling in P-cadherin-overexpressing breast cancer models. PLoS One 2013; 19:e69023; http://dx.doi.org/ 10.1371/journal.pone.0069023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Giannelli G, Azzariti a, Fransvea E, Porcelli L, Antonaci S, Paradiso a. Laminin-5 offsets the efficacy of gefitinib (‘Iressa’) in hepatocellular carcinoma cells. Br J Cancer 2004; 91:1964-9; PMID:15545972; http://dx.doi.org/ 10.1038/sj.bjc.6602231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Rintoul RC, Sethi T. Extracellular matrix regulation of drug resistance in small-cell lung cancer. Clin Sci (Lond) 2002; 102:417-24; PMID:11914104; http://dx.doi.org/ 10.1042/cs1020417 [DOI] [PubMed] [Google Scholar]
- [29].Helleman J, Jansen MPHM, Burger C, van der Burg MEL, Berns EMJJ. Integrated genomics of chemotherapy resistant ovarian cancer: A role for extracellular matrix, TGFbeta and regulating microRNAs. Int J Biochem Cell Biol 2010; 42:25-30; PMID:19854294; http://dx.doi.org/ 10.1016/j.biocel.2009.10.016 [DOI] [PubMed] [Google Scholar]
- [30].Sharma S, Santiskulvong C, Rao J, Gimzewski JK, Dorigo O. The role of Rho GTPase in cell stiffness and cisplatin resistance in ovarian cancer cells. Integr Biol (Camb) 2014; 6:611-7; PMID:24718685; http://dx.doi.org/ 10.1039/c3ib40246k [DOI] [PubMed] [Google Scholar]
- [31].Kim KS, Cho CH, Park EK, Jung MH, Yoon KS, Park HK. AFM-Detected apoptotic changes in morphology and biophysical property caused by paclitaxel in Ishikawa and HeLa cells. PLoS One 2012; 7:e30066; PMID:22272274; http://dx.doi.org/ 10.1371/journal.pone.0030066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Kao FS, Pan YR, Hsu RQ, Chen HM. Efficacy verification and microscopic observations of an anticancer peptide, CB1a, on single lung cancer cell. Biochim Biophys Acta - Biomembr 2012; 1818:2927-35; PMID:22846508; http://dx.doi.org/ 10.1016/j.bbamem.2012.07.019 [DOI] [PubMed] [Google Scholar]
- [33].Corbin EA, Kong F, Lim CT, King WP, Bashir R. Biophysical properties of human breast cancer cells measured using silicon MEMS resonators and atomic force microscopy. Lab Chip 2015; 15:839-47; PMID:25473785; http://dx.doi.org/ 10.1039/C4LC01179A [DOI] [PubMed] [Google Scholar]
- [34].Prabhune M, Belge G, Dotzauer A, Bullerdiek J, Radmacher M. Comparison of mechanical properties of normal and malignant thyroid cells. Micron 2012; 43:1267-72; PMID:22522060; http://dx.doi.org/ 10.1016/j.micron.2012.03.023 [DOI] [PubMed] [Google Scholar]
- [35].Peetla C, Vijayaraghavalu S, Labhasetwar V. Biophysics of cell membrane lipids in cancer drug resistance: implications for drug transport and drug delivery with nanoparticles. Adv Drug Deliv Rev 2013; 65:1686-98; PMID:24055719; http://dx.doi.org/ 10.1016/j.addr.2013.09.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Busk M, Pytelasv R, Sheppardsil D. Characterization of the Integrin avP6 as a Fibronectin-binding Protein. J Biol Chem 1992; 267(9):5790-6; PMID:1532572 [PubMed] [Google Scholar]
- [37].Pinto SN, Silva LC, de Almeida RFM, Prieto M. Membrane domain formation, interdigitation, and morphological alterations induced by the very long chain asymmetric C24:1 ceramide. Biophys J 2008; 95:2867-79; PMID:18586849; http://dx.doi.org/ 10.1529/biophysj.108.129858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Johnson KL, Kendall K, Roberts aD. Surface Energy and the Contact of Elastic Solids. Proc R Soc A Math Phys Eng Sci 1971; 324:301-13; http://dx.doi.org/ 10.1098/rspa.1971.0141 [DOI] [Google Scholar]