

Cancer stem cells are commonly defined as those cells that constitute a very low fraction of cells in a tumor, and which are uniquely capable of generating a new tumor following transfer or migration into a new environment. For certain cancers such as colorectal, breast and certain leukemias, the existence of cancer stem cells is well-documented.1 Such cells are believed to be largely responsible for metastases, and for relapse following chemotherapy. The recent findings by some groups that some commonly-used chemotherapeutic agents can actually enrich for cancer stem cells2 has prompted the emerging concept that in order to successfully combat a tumor, both the bulk tumor and the cancer stem cells need to be targeted. The difficulty lies in finding the target: a protein expressed in both the bulk tumor and the stem cells, on which both the tumor and the cancer stem cell rely for survival. Two recent publications have identified an isoform of the p53 family member p73 as such a protein (Fig. 1).
Figure 1.
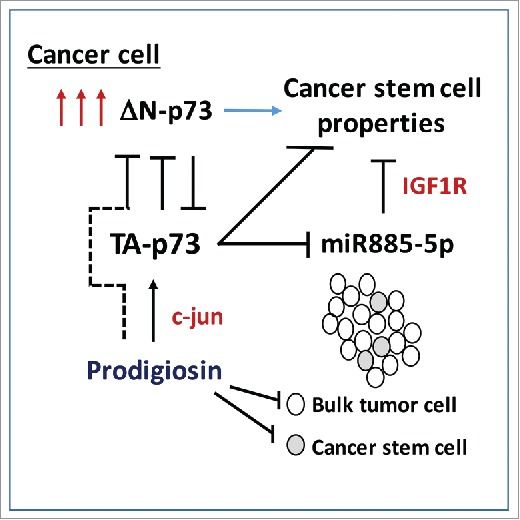
Combined mechanism(s) whereby the ΔN-p73 variant increases cancer stem cell function. The dashed line connecting c-jun to inhibition of deltaN-p73 reflects the speculative nature of this particular regulation.
p53 has long been established as a negative regulator of stem cells.3 Additionally, p53 has 2 ‘siblings’, p63 and p73, which are homologous in structure and function. However, p63 and p73 have “Jekyll and Hyde” isoforms when it comes to tumor growth: namely, the full length isoforms, so-called TA p63 and TA p73, are predominantly tumor suppressive, while the N-terminal truncation variants, so-called ΔN-p63 and ΔN-p73, are typically tumor-promoting, and not surprisingly are frequently overexpressed in tumors. Two groups recently discovered that silencing ΔN-p73 causes decreased expression of stem cell markers, decreased anchorage-independent growth (a hallmark of cancer stem cell function) and decreased ability to engraft as tumors. In the publication by Pützer and colleagues, these researchers found that overexpression of ΔN-p73 in the melanoma Sk-Mel-29 led to higher CD133, Nanog and Oct4, along with increased anchorage independent growth and ability to form tumors in xenografts.4 Mechanistically this group found that the ability of ΔN-p73 to confer cancer stem cell function relied on its ability to negatively regulate the microRNA miR885-5p. They went on to show that the target of this microRNA is the IGF1 receptor. In sum, overexpression of ΔN-p73 variant led to greatly enhanced IGF1 signaling, which then functioned in a feedback loop to increase cancer stem cell function.
El-Deiry and colleagues found a similar impact of ΔN-p73 on cancer stem cells, but this time focused on colorectal cancer stem cells. This group started with an innovative screen to search for compounds that could restore p53 function.5 These researchers discovered a compound called prodigiosin, which turned out to restore p53 function by inhibiting its negative regulator, ΔN-p73. Mechanistically, this group found that the ability of prodigiosin to inhibit the function of ΔN-p73 occurred by virtue of the ability of this compound to increase the level of c-jun; the latter transcription factor upregulates TA-p73, which in turn inhibits ΔN-p73. El-Deiry and colleagues found that treatment of colorectal tumors with prodigiosin led to decreased colonosphere formation, decreased levels of cells positive for the stem cell markers ALDH, CD44, ID3 and E-cadherin, and decreased ability of colorectal cancer stem cells to initiate tumors as xenografts.6 Notably, when treatment with prodigiosin was stopped, tumor growth did not renew, supporting the premise that prodigiosin led to a cure in treated mice. These findings suggest that prodigiosin possesses unique anti-cancer functions.
Altogether these exciting findings need to be followed up by addressing several questions: Do all cancer stem cells rely on ΔN-p73, or might ΔN-p63 play a role in other tumor types? Can the drug pramlintide, which targets both ΔN-p73 and ΔN-p637 be useful to target cancer stem cells? And finally, are there other mechanisms by which ΔN-variants of p53 family members confer stem cell properties? These are likely to be exciting times for therapeutic strategies targeting p53 family members.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
References
- [1].Beck B, Blanpain C. Nat Rev Cancer 2013; 13:727-38; PMID:24060864; http://dx.doi.org/ 10.1038/nrc3597 [DOI] [PubMed] [Google Scholar]
- [2].Gupta PB, et al.. Cell 2009; 138:645-59; PMID:19682730; http://dx.doi.org/ 10.1016/j.cell.2009.06.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Mizuno H, et al.. Proc Natl Acad Sci U S A 2010; 107:22745-50; PMID:21149740; http://dx.doi.org/ 10.1073/pnas.1017001108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Meier C, et al.. Cancer Res 2016; 76:197-205; PMID:26554827; http://dx.doi.org/ 10.1158/0008-5472.CAN-15-1228 [DOI] [PubMed] [Google Scholar]
- [5].Hong B, et al.. Cancer Res 2014; 74:1153-65; PMID:24247721; http://dx.doi.org/ 10.1158/0008-5472.CAN-13-0955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Prabhu VV, et al.. Cancer Res 2016; PMID:2675923926759239 [Google Scholar]
- [7].Venkatanarayan A, et al.. Cell Cycle 2016; 15:164-71; PMID:26652033; http://dx.doi.org/ 10.1080/15384101.2015.1121333 [DOI] [PMC free article] [PubMed] [Google Scholar]