

Abstract
Natural recombination combines pieces of pre-existing proteins to create new tertiary structures and functions. We describe a computational protocol, called SEWING, which is inspired by this process and builds new proteins from connected or disconnected pieces of existing structures. Helical proteins designed with SEWING contain structural features absent from other de novo designed proteins and in some cases remain folded to over 100 °C. High resolution structures of the designed proteins CA01 and DA05R1 were solved by X-ray crystallography (2.2 Å resolution) and NMR respectively, and there was excellent agreement with the design models. This method provides a new strategy to rapidly create large numbers of diverse and designable protein scaffolds.
Most efforts in de novo protein design have been focused on creating idealized proteins composed of canonical structural elements. Examples include the design of coiled-coils, repeat proteins, TIM barrels and Rossman folds (1–6). These studies elucidate the minimal determinants of protein structure, but they do not aggressively explore new regions of structure space. Additionally, idealized structures may not always be the most effective starting points for engineering novel protein functions. Functional sites in proteins are often created from non-ideal structural elements, such as kinks, pockets and bulges.
The lack of non-ideal structural elements from de novo designed proteins highlights a key difference between natural protein evolution and current design methods. Specifically, that protein design methods universally begin with a target structure in mind. Therefore, the space of designable structures that can accommodate these non-ideal protein elements is limited by the imagination of the designer. In contrast, natural evolution is based not on design, but on cellular fitness provided by the evolved protein function. This lack of a predetermined target fold is a powerful feature of protein evolution that holds significant potential for the design of novel structures and functions. In an effort to tap this potential, we sought to develop a method of computational protein design inspired by mechanisms of natural protein evolution.
Gene duplication and homologous recombination mix and match elements of protein structure to give rise to new structures and functions (7–9). This phenomenon is most evident at the level of independently folding protein domains (10–12), but recent studies have shown that these same principles function at a smaller scale during the evolution of distinct, globular protein folds (13). Insertions, deletions and replacement of secondary and supersecondary structural elements sample alternative tertiary structures (14–16). Our design strategy, called SEWING (Structure Extension With Native-substructure Graphs), is motivated by this process and builds new protein structures from pieces of naturally occurring protein domains. The process is not dictated by the need to adopt a specific target fold, but rather aimed at creating large sets of alternative structures that satisfy predefined design requirements. One of the strengths of this approach is that it ensures that all of the structural elements of the protein are inherently designable, while at the same time allowing for the incorporation of structural oddities unlikely to be found in idealized proteins. Here, we apply SEWING to the design of helical proteins. We show that designed structures are diverse, and contain structural features absent from alternative design strategies.
SEWING begins with the extraction of small structural motifs, or substructures, from existing protein structures. These serve as the basic building blocks for all generated models. We aimed to identify substructures that were both large enough to carry information regarding structural preference, yet small enough to allow combinations that can generate novel globular structures. Ultimately, we chose to extract two distinct types of substructures. The first is composed of continuous stretches of protein structure that encompass two secondary structural elements separated by a loop (Fig. 1). These substructures capture the relative orientation between adjacent secondary structure elements and maintain local packing interactions. Additionally, there is evidence that substructures of this size adopt a relatively limited number of conformations that have already been sampled exhaustively in known protein structures (14). The second are composed of groups of 3–5 secondary structural elements, where each element makes van der Waals contacts with every other element but are not necessarily continuous in primary sequence (Fig. 1, supplementary methods). Non-adjacent, or discontinuous substructures maintain longer-range tertiary interactions that provide valuable stability, and are often conserved during protein evolution (17).
Fig. 1.

Overview of the SEWING method. (A) Continuous SEWING workflow for CA01. (B) Discontinuous SEWING workflow for DA05. Each panel, from left to right: parental PDBs with extracted substructures; Graph schematic – colored nodes indicate substructures contained in final design model, superimposed structures show structural similarity indicated by adjacent edges; Design model before sequence optimization and loop design; Final design models.
The goal of SEWING is to combine and modify these extracted components in order to develop new tertiary structures. Naturally occurring homologous recombination, in which sequence similarity between DNA leads to the combination of the genetic material, guides the formation of new protein chimeras. This process enriches for proteins that are more likely to be well-folded and functional, as sequence similarity filters for segments that are structurally compatible. In the case of SEWING, we know the three dimensional structures of the building blocks, and therefore, we can directly use structural information to probe which substructures are well suited for combination. During SEWING, continuous substructures are eligible for combination if the C-terminal region of one substructure shares high structure similarity with the N-terminal region of another substructure, and superposition of the two regions does not create any steric clashes between other regions in the two substructures. This type of superposition ensures that the three-dimensional spacing between all pairs of secondary structural elements adjacent in primary sequence is similar to that observed in the PDB. During discontinuous SEWING, combinations are created by superimposing two elements (helices in this study) from one substructure with two elements from another substructure. For both continuous and discontinuous SEWING, structure similarity is identified using a fast geometric hashing approach that ensures that the regions of interest can align with low-RMSD (18).
Once pairwise structural similarity is calculated between all substructures, these data are used to generate a large graph (Fig. 1). The nodes in this graph represent the substructures, and the edges indicate a level of structural similarity that allows recombination. Novel structures are generated from this graph by traversing a path, where each followed edge adds new structural elements to the design model. The number of edges included in the sampled paths can control the approximate size of the generated structures. Unlike previously described methods of de novo backbone generation, no target structure is required, and output structures span a diverse set of globular folds.
Previous studies have demonstrated that protein fragments can adopt alternative structures when placed in new environments (19–21), and thus similar to natural evolution, the next step in the design process was to further stabilize the protein through mutagenesis. This optimization step was achieved using iterative steps of sidechain packing and backbone minimization available in the Rosetta molecular modeling suite (22). Preference for the amino acid sequence present in the parental substructure was used to better preserve the structural interactions inherent to the parent substructures.
To test SEWING, we designed a diverse set of helical proteins using graphs composed of continuous substructures or discontinuous substructures. Continuous and discontinuous substructures were extracted from non-redundant subsets of the protein data bank (PDB) (23, 24). In total, 33,928 continuous substructures, and 4,584 discontinuous substructures were extracted. Design models from the continuous graph were generated using 3-edge paths, and were therefore composed of substructures extracted from 4 existing structures from the PDB (Fig. 1). The continuous graph contained 345 million edges, allowing an estimated 7*1016 backbones that can be filtered and optimized in later designs steps (Supplemental methods). Initially, 50,000 alternative tertiary structures were created and used as templates for rotamer-based sequence optimization and energy minimization. These models were filtered and sorted using metrics that evaluate predicted energy (normalized by sequence length), sidechain packing, buried polar groups, and sequence/structure agreement (Supplementary methods) (25). When examining the models, we noticed that the naïve SEWING procedure was biased towards creating low contact order models, i.e. structures with few contacts between residues distant in primary sequence. To overcome this bias, we filtered for models with contact orders more representative of naturally occurring helical proteins (Fig. S1). We have subsequently demonstrated that Monte Carlo sampling of the SEWING graph with a score function that favors long-range contacts can be used to build high-contact models with high frequency (Fig. S1). This illustrates one way that directed sampling of the SEWING graph can be used to enforce design requirements.
In total, 11 designs based on continuous SEWING were selected for experimental characterization (Table S1). Each region of the final designs shared between 45% and 65% sequence identity with the substructure they were built from (Fig. S2, S3), but when performing a BLAST search with the full-length sequences, no matches were identified that align over the full length of the proteins. 8 designs expressed well in E. coli and were readily purified from the soluble fraction of 1 L cultures. 4 of the 8 proteins were monomeric via Size Exclusion Chromotagraphy/Multi-angle light scattering, had a circular dichroism (CD) spectra characteristic of a helical protein and unfolded cooperatively upon thermal denaturation (Fig. 2, S4, S5). Two of the designed proteins are hyperthermophiles and require high concentrations of chemical denaturant in order to observe thermal unfolding (Fig. 2B). For one design, CA01, several thermodynamic parameters were determined by fitting a modified Gibbs-Helmholtz equation to the thermal and chemical denaturation surface (Fig. 3B, Table S2) (26). The extrapolated melting temperature of 126 °C places it among the top 0.01% of values in the ProTherm database of protein stabilities (27). The crystal structure of CA01 was solved to 2.2 Å and shows excellent agreement with the design model, with a Cα RMSD of 0.8 Å. Similarly, the side-chain packing of the protein core is nearly identical between the design model and experimental structure (Fig. 3, S6, Table S3).
Fig. 2.
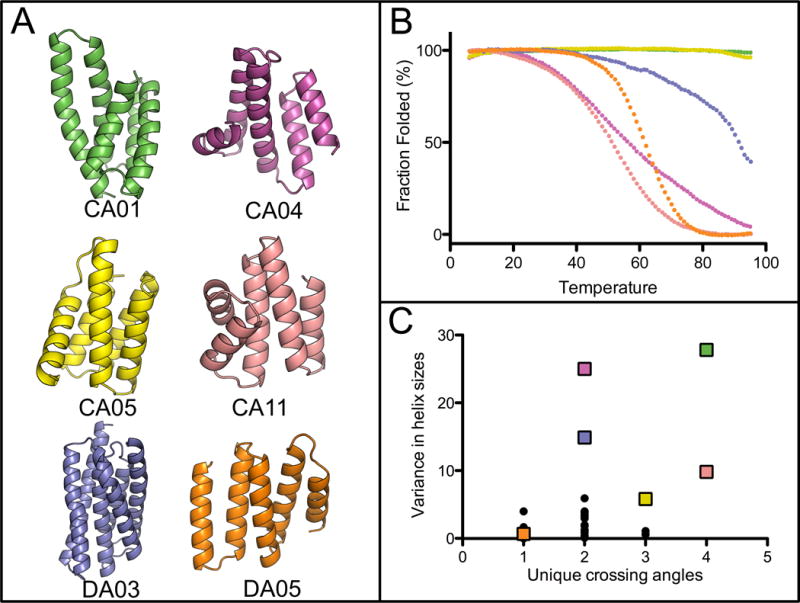
Well-folded SEWING designs. (A) Design models obtained with continuous (CA) and discontinuous (DA) SEWING. (B) Temperature denaturation curves for well-folded SEWING designs, colored to match design models. (C) A comparison of previously designed helical structures (black dots), to SEWING models (colored squares) demonstrates the structural complexity of SEWING designs. Crossing angles between all pairs of helices in each structure were calculated. Crossing angles were considered unique if they differed by >20 degrees from all other calculated angles in the same structure. Variance in helix size describes the calculated variance in the number of residues/helix for all helices in single structure. A complete list of helix and crossing-angle definitions for de novo designs can be found in Table S5 and S6.
Fig. 3.
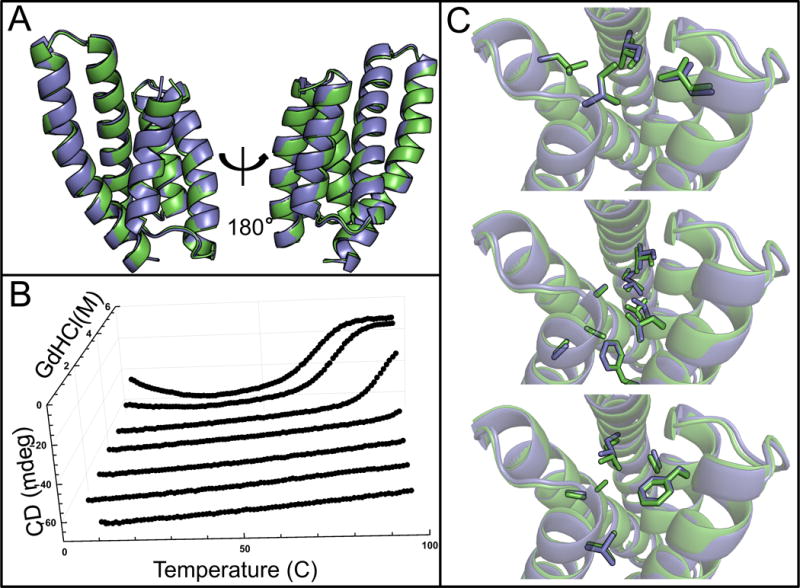
Structural and biophysical characterization of CA01. (A) Backbone superimposition of the design model (green) and crystal structure (blue). (B) In the chemical and temperature denaturation experiment a sharp unfolding transition is observed at 5M GdHCl and 75 °C (C) Comparison of sidechain packing between design model (green) and crystal structure (blue) at three different layers of the structure.
The structural variety in the design models for the well-folded proteins is of particular note (Fig. 2). The SEWING generated models include kinked and curved helices (Fig. S7, S8), cavities and clefts (Fig. S9, S10), and a large range of helix-crossing angles (Fig. 2). The topologies of the SEWING models are varied when compared to previously designed alpha-helical proteins, which are restricted to coiled-coils, repeat proteins and up-down four helix bundles (Fig. 2C). To compare SEWING models with naturally occurring protein structures, we searched for structurally similar domains using the DALI server (28). In general, large portions of the models aligned to regions of existing protein structures. However, the sequence identities across the alignments were typically below 20% and the positions of the unaligned residues frequently diverged (Fig. S11). For instance, the fifth helix of CA01 is shifted by ~9 Å relative to fifth helix in the top DALI match. These sequences and structural differences provide unique surfaces which may serve as templates for future design goals.
To test discontinuous SEWING, models were generated from 2-edge paths, and thus were composed of structural elements from 3 parent structures. The variable number of helical elements in the discontinuous substructures therefore allowed design models to be composed of 5 to 11 helices. Unlike models from the continuous-substructure graph, discontinuous models require the addition of loops between consecutive helices. Loops were designed using a database of fragments from the PDB (29). Each loop fragment was superimposed onto the design model and optimized using gradient-based minimization in Cartesian space. Any path that created structures for which no loop-fragment could be found was eliminated from the set of designs. Design models were filtered and optimized in the same way as models from the continuous graph. In total, 10 were selected for experimental characterization (Table S1).
Of these 10 designs, 2 expressed at levels sufficient for purification. Both purified proteins were helical and folded as evidenced by CD (Fig. 2, S4). Similar to the results from the continuous designs, one discontinuous design, DA03, demonstrated high thermostability, requiring high levels of denaturant to completely unfold. For this design, a 181 residue 6-helix bundle, unfolding appears to follow a three-state model (Fig. S12).
The structure of the other well-folded discontinuous design, DA05, was solved using nuclear magnetic resonance (NMR) spectroscopy as the protein did not readily crystallize (Fig. S13, S14, Table S4). The first 4 helices of the design model match the lowest energy member of the NMR ensemble very closely, with a Cα RMSD of 0.8 Å (Fig. 4, S6). However, the NMR data indicate the final helix of the protein is disordered in solution. In an effort to identify the errors in the design model that led to the unstructured region, structural preference for the designed sequence was evaluated with fragment analysis as described previously (1). The fragments extracted for the unstructured region showed especially poor preference for the designed helical structure (Fig. S15). We attempted to design a new final helix for the DA05 design using the continuous SEWING method. The final helix of the initial design model was removed and the remainder of the model was added as a node to the continuous graph. New helices were evaluated by following a single edge from this new node (Fig. 4A). Three models designed in this way were selected for experimental testing. Two of the tested designs, DA05R1 and DA05R2, show a significant increase in melting temperature relative to the initial DA05 design (Fig. S16). The NMR structure of DA05R1 shows the newly designed helix adopts the designed conformation, highlighting the utility of combining the continuous and discontinuous graphs (Fig. 4B, S17, S18, Table S4).
Fig. 4.

Result for discontinuous assembly DA05 and DA05R1. (A) From left to right: Backbone superimposition of the DA05 design model (orange) with a member of the NMR ensemble (dark blue). Example continuous substructure graph for the design of a new final helix onto DA05. Superimposition of three design models containing new helices. (B) From left to right: Backbone superimposition of the DA05R1 design model (light blue) with a member of the DA05R1 NMR ensemble (dark blue). Comparison of sidechain packing between the DA05R1 design model and the NMR structure for DA05R1.
The additional step of loop-building is a critical difference between discontinuous and continuous SEWING. The accurate design of loops is a long-standing challenge for protein design, and this additional step may have contributed to the relatively lower success rate observed for discontinuous SEWING. In contrast, continuous SEWING maintains the relative orientation between adjacent helices allowing many of the designed loop sequences to be taken directly from the native substructure. The power of this strategy is seen in the high structural accuracy achieved for the loops in the CA01 design (Fig 3, S2).
Our results show that computational adaptations of basic evolutionary principles, such as recombination and mutation, can be used to accurately and rapidly design a diverse set of helical protein structures. The diversity of SEWING designs will further increase when alternative types of substructures are included, such as β-α motifs and β-hairpins. Furthermore, discontinuous and continuous SEWING can be merged, as in the DA05R1 design, creating additional diversity. We anticipate that this structural diversity will be advantageous for functional design, as every backbone generated with SEWING has new surface and pocket features that provide potential binding sites for ligands or macromolecules. Additionally, SEWING offers an approach for stitching together functional motifs from naturally occurring proteins, an evolutionary mechanism to generate multi-functional proteins and allosteric systems.
Supplementary Material
Acknowledgments
This work was supported by National Institutes of Health Grants RO1GM073960 and R01GM117968 (to B.K.), and GM094597 (to T.S.). Use of the Advanced Photon Source was supported by the U. S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under Contract No. W-31-109-Eng-38. Coordinates and structure factors have been deposited in the Protein Data Bank with the accession codes 5E6G (CA01), 2N8I (DA05), and 2N8W (DA05R1). T.M.J. and B.K. designed the research. Chemical shifts have been deposited in the Biological Magnetic Resonance Bank (BMRB) with the accession codes 25850 (DA05) and 25868 (DA05R1). T.M.J. wrote the backbone assembly code. T.M.J. and B.W. carried out the backbone assembly and design simulations. T.M.J. conducted the biophysical analysis. T.W. and T.M.J. solved the structure of CA01. X.X., A.E., and J.F., under the advisement of T.S., solved the NMR structure of DA05 and DA05R1.
References and Notes
- 1.Koga N, et al. Principles for designing ideal protein structures. Nature. 2012;491:222–7. doi: 10.1038/nature11600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Joh NH, et al. De novo design of a transmembrane Zn2+-transporting four-helix bundle. Science. 2014;346:1520–4. doi: 10.1126/science.1261172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Huang P-S, et al. High thermodynamic stability of parametrically designed helical bundles. Science (80-) 2014;346:481–485. doi: 10.1126/science.1257481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Doyle L, et al. Rational design of α-helical tandem repeat proteins with closed architectures. Nature. 2015;528:585–588. doi: 10.1038/nature16191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brunette T, et al. Exploring the repeat protein universe through computational protein design. Nature. 2015;528:580–584. doi: 10.1038/nature16162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kuhlman B, et al. Design of a novel globular protein fold with atomic-level accuracy. Science. 2003;302:1364–8. doi: 10.1126/science.1089427. [DOI] [PubMed] [Google Scholar]
- 7.Hughes AL. Gene duplication and the origin of novel proteins. Proc Natl Acad Sci U S A. 2005;102:8791–8792. doi: 10.1073/pnas.0503922102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Blake CCF. Do genes-in-pieces imply proteins-in-pieces? Nature. 1978;273:267–267. [Google Scholar]
- 9.Bashton M, Chothia C. The Generation of New Protein Functions by the Combination of Domains. Structure. 2007;15:85–99. doi: 10.1016/j.str.2006.11.009. [DOI] [PubMed] [Google Scholar]
- 10.Koide S. Generation of new protein functions by nonhomologous combinations and rearrangements of domains and modules. Curr Opin Biotechnol. 2009;20:398–404. doi: 10.1016/j.copbio.2009.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Eisenbeis S, et al. Potential of fragment recombination for rational design of proteins. J Am Chem Soc. 2012;134:4019–22. doi: 10.1021/ja211657k. [DOI] [PubMed] [Google Scholar]
- 12.Vogel C, Bashton M, Kerrison ND, Chothia C, Teichmann SA. Structure, function and evolution of multidomain proteins. Curr Opin Struct Biol. 2004;14:208–216. doi: 10.1016/j.sbi.2004.03.011. [DOI] [PubMed] [Google Scholar]
- 13.Grishin NV. Fold change in evolution of protein structures. J Struct Biol. 2001;134:167–185. doi: 10.1006/jsbi.2001.4335. [DOI] [PubMed] [Google Scholar]
- 14.Fernandez-Fuentes N, Dybas JM, Fiser A. Structural characteristics of novel protein folds. PLoS Comput Biol. 2010;6:e1000750. doi: 10.1371/journal.pcbi.1000750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Söding J, Lupas AN. More than the sum of their parts: On the evolution of proteins from peptides. BioEssays. 2003;25:837–846. doi: 10.1002/bies.10321. [DOI] [PubMed] [Google Scholar]
- 16.Reeves GA, Dallman TJ, Redfern OC, Akpor A, Orengo CA. Structural Diversity of Domain Superfamilies in the CATH Database. J Mol Biol. 2006;360:725–741. doi: 10.1016/j.jmb.2006.05.035. [DOI] [PubMed] [Google Scholar]
- 17.Aronson HE, Royer WE, Hendrickson WA. Quantification of tertiary structural conservation despite primary sequence drift in the globin fold. Protein Sci. 1994;3:1706–1711. doi: 10.1002/pro.5560031009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nussinov R, Wolfson HJ. Efficient detection of three-dimensional structural motifs in biological macromolecules by computer vision techniques. Proc Natl Acad Sci U S A. 1991;88:10495–9. doi: 10.1073/pnas.88.23.10495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schellenberg MJ, et al. Context-Dependent Remodeling of Structure in Two Large Protein Fragments. J Mol Biol. 2010;402:720–730. doi: 10.1016/j.jmb.2010.08.020. [DOI] [PubMed] [Google Scholar]
- 20.Bharat TAM, Eisenbeis S, Zeth K, Höcker B. A beta alpha-barrel built by the combination of fragments from different folds. Proc Natl Acad Sci U S A. 2008;105:9942–7. doi: 10.1073/pnas.0802202105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.de Bono S, Riechmann L, Girard E, Williams RL, Winter G. A segment of cold shock protein directs the folding of a combinatorial protein. Proc Natl Acad Sci U S A. 2005;102:1396–1401. doi: 10.1073/pnas.0407298102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Leaver-Fay A, et al. Rosetta3: An Object-Oriented Software Suite for the Simulation and Design of Macromolecules. Methods Enzymol. 2011;487:545–574. doi: 10.1016/B978-0-12-381270-4.00019-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang G, Dunbrack RL. PISCES: A protein sequence culling server. Bioinformatics. 2003;19:1589–1591. doi: 10.1093/bioinformatics/btg224. [DOI] [PubMed] [Google Scholar]
- 24.Richardson JS, Richardson DC. Invited review: Studying and polishing the PDB’s macromolecules. Biopolymers. 2013;99:170–182. doi: 10.1002/bip.22108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sheffler W, Baker D. RosettaHoles2: a volumetric packing measure for protein structure refinement and validation. Protein Sci. 2010;19:1991–5. doi: 10.1002/pro.458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kuhlman B, Raleigh DP. Global analysis of the thermal and chemical denaturation of the N-terminal domain of the ribosomal protein L9 in H2O and D2O. Determination of the thermodynamic parameters, deltaH(o), deltaS(o), and deltaC(o)p and evaluation of solvent isotope effects. Protein Sci. 1998;7:2405–12. doi: 10.1002/pro.5560071118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sarai A, et al. Thermodynamic databases for proteins and protein-nucleic acid interactions. Biopolymers. 2001;61:121–126. doi: 10.1002/1097-0282(2002)61:2<121::AID-BIP10077>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- 28.Holm L, Rosenström P. Dali server: conservation mapping in 3D. Nucleic Acids Res. 2010;38:W545–9. doi: 10.1093/nar/gkq366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tyka MD, Jung K, Baker D. Efficient sampling of protein conformational space using fast loop building and batch minimization on highly parallel computers. J Comput Chem. 2012;33:2483–91. doi: 10.1002/jcc.23069. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.