

Abstract
Toll-like receptor (TLR) agonists represent potentially useful cancer vaccine adjuvants in their ability to stimulate antigen-presenting cells (APCs) and subsequently amplify the cytotoxic T-cell response. The purpose of this study was to characterize APC responses to TLR activation and to determine the subsequent effect on lymphocyte activation. We exposed murine primary bone marrow-derived macrophages to increasing concentrations of agonists to TLRs 2, 3, 4, and 9. This resulted in a dose-dependent increase in production of not only tumor necrosis factor–alpha (TNF-α), a surrogate marker of the proinflammatory response, but also interleukin 10 (IL-10), a well-described inhibitory cytokine. Importantly, IL-10 secretion was not induced by low concentrations of TLR agonists that readily produced TNF-α. We subsequently stimulated lymphocytes with anti-CD3 antibody in the presence of media from macrophages activated with higher doses of TLR agonists and observed suppression of interferon gamma release. Use of both IL-10 knockout macrophages and IL-10 small-interfering RNA (siRNA) ablated this suppressive effect. Finally, IL-10 siRNA was successfully used to suppress CpG-induced IL-10 production in vivo. We conclude that TLR-mediated APC stimulation can induce a paradoxical inhibitory effect on T-cell activation mediated by IL-10.
Keywords: toll-like receptors, innate immunity, IL-10
Introduction
Immunotherapy remains an exciting, yet largely unfulfilled, modality in the treatment of malignant disease. Myriad treatment strategies have been explored, including vaccination against viral precursors, cytokine manipulation, adoptive T-cell transfer, and dendritic cell administration.1 Obvious theoretical potential and astonishing anecdotal successes continue to fuel investigation.
The development of tumor-specific vaccines for either adjuvant or neoadjuvant therapy represents an extremely attractive approach; however, the challenges associated with their development have been starkly illustrated as the intricacies of the so-called “escape” phase of cancer immunoediting have been elucidated. These include loss of antigen-presenting machinery, decreased expression of tumor antigens, reduced sensitivity to immune effector molecules, expression of inhibitory molecules, and induction of regulatory cells including regulatory T-cells (Tregs).1
Stimulation of Toll-like receptors (TLRs), components of the innate immune system, are extensively studied as a means for overcoming tumor escape mechanisms. TLRs are the best-characterized class of pattern recognition receptors in mammalian species,2 detecting unique nonself-molecular patterns conserved throughout entire classes of pathogens.3 The reasoning behind their use as anticancer agents is twofold. First, activation of the innate immune system and subsequent production of an appropriate cytokine and chemokine milieu will promote a direct cytotoxic antitumor response.4 Second, their activation also augments the development of adaptive responses that can be exploited to accelerate vaccines for tumor-specific immunity.5
There are likely between 10–15 unique TLRs in mammalian species.2 At least 11 TLRs are identified in humans, and these are well conserved in mouse species.6 TLRs recognize multiple pathogen-associated molecular patterns, including bacterial lipoproteins (TLR2); double-stranded RNA (TLR3); lipopolysaccharide (LPS) (TLR4); flagellin (TLR5); single-stranded viral RNA (TLR7); and unmethylated CpG DNA (TLR9), that are unique to bacteria and viruses.
In order to augment the development of a cancer vaccine, our laboratory has explored the use of TLR agonists as vaccine adjuvants. In so doing, we sought to examine the murine APC cytokine response to TLR activation and the subsequent effect of this activation on lymphocytes. Here, we identify a dose-response curve of cytokine production in bone marrow-derived macrophages (BMM) after the activation of TLRs 2, 3, 4, and 9. This cytokine production is characterized by increasing levels of not only the proinflammatory cytokine tumor necrosis factor–alpha (TNF-α), but also the inhibitory cytokine interleukin 10 (IL-10) that inhibited lymphocyte activation in vitro. Finally, we show that this inhibitory effect can be overcome by targeting IL-10 with small-interfering RNA (siRNA) both in vitro and in vivo.
Materials and methods
Mice
Female A/J, C57BL/6, and IL-10 deficient (C57BL/6 background) mice were purchased at 4 weeks of age from The Jackson Laboratory (Bar Harbor, ME). After arrival, mice were acclimated for at least 4 days prior to use. Animals were housed in specific pathogen-free conditions in the Animal Laboratory at Children’s National Medical Center. All mice were handled in accordance with the guidelines of the Institutional Animal Care and Use Committee.
Reagents
OK-432 was obtained from Chugai Pharmaceutical Co (Tokyo, Japan). CpG-ODN 1826 was purchased from Coley Pharmaceutical Group (Wellesley, MA; Lot# C44-0604-AE1A). Polyinosinic:polycytidylic acid sodium salt (Poly I:C) was purchased from Sigma-Aldrich (St Louis, MO; Product #PO913).
Macrophage harvest and growth
Primary bone marrow macrophages were directly harvested from sacrificed A/J and C57BL/6 mice. Femurs and tibias were dissected and the intramedullary canals were flushed with PD solution (phosphate-buffered saline containing 1% penicillin–streptomycin) via a 26 g needle until the bones were clear. Cells were resuspended in PD solution and centrifuged. The cells were then cultured in canted neck flasks (CellStar 658190 T75 Flask, Greiner Bio-One, Monroe, NC; suspension culture, sterile, plug seal cap, 250 mL) containing 30% L929 cell-conditioned media in a humidified incubator at 37°C with 5% CO2. Cells were refed on either day 2 or 3, then plated between days 6 and 9 into 24-well plates. 2 × 105 cells were plated in each well with 350 µL of D10 solution (Dulbecco’s modified Eagle’s medium [DMEM], supplemented with 10% fetal bovine serum [FBS], penicillin [100 IU/mL], streptomycin [100 µg/mL] [all Sigma-Aldrich], and 1% L-glutamine [Mediatech Inc, Manassas, VA]). The 24-well plates were then returned to the incubator and macrophages were given at least 16 hours to acclimate prior to stimulation. The obtained cell population was 99.6% positive for CD11b, 88.7% for F4/80, 83.9% for CD14, 30.2% for CD11c, and <1% for Gr1 (data not shown).
Flow cytometry
Cultured bone marrow cells were collected and stained with the following fluorochrome-conjugated murine antibodies (mAbs) obtained from eBioscience (San Diego, CA): phycoerythrin:cyanine-5 (PE-Cy5)-conjugated CD11b (M1/70), allophycocyanin (APC)-conjugated CD11c (N418), phycoerythrin (PE)-conjugated F4/80 (BM8), fluorescein isothiocyanate (FITC)-conjugated CD14 (Sa2-8), and allophycocyanin (APC)-conjugated Gr1 (RB6-8C5). Flow cytometry was performed using FACS Calibur (BDBioscience, San Jose, CA); results were analyzed using FlowJo software (Treestar, Ashland, OR).
Macrophage stimulation
At the conclusion of the acclimation period, varying amounts of CpG, Poly I:C, and OK-432 were administered to the cells. The macrophages were then reincubated for 24 hours before either cytokine assay or splenocyte stimulation was performed.
Splenocyte harvest
Spleens of sacrificed A/J and C57BL/6 mice were removed and crushed through cell strainers (BD Biosciences, San Jose, CA). Strained cells were then centrifuged and re-suspended in R10 Solution (RPMI media containing 10% FBS, penicillin [100 IU/mL], streptomycin [100 µg/mL], 1% L-glutamine, 25 nM HEPES buffer [Mediatech Inc], and 0.005 mM B-mercaptoethanol [Sigma-Aldrich]). A cell-count was performed using trypan blue to ensure cell viability. 2 × 105 cells per well were plated into 96-well round-bottom plates; each well contained 150 µL of RPMI solution.
Splenocyte stimulation
Immediately after the splenocytes were plated, 150 µL of stimulated macrophage supernatant was added to each well in combination with 1.0 µg/mL of purified anti-CD3 (BD Biosciences; Cat # 553058, αCD3e) for lymphocyte activation. Splenocytes were then incubated at 37°C for 48 hours prior to cytokine assay.
siRNA oligonucleotide design and validation
All RNA oligonucleotides used in this study were synthesized and high-pressure liquid chromatography (HPLC)-purified by Integrated DNA Technologies (Coralville, IA). Compound identity was verified by electrospray ionization mass spectrometry (ESI-MS) and compound purity was measured to be better than 90% by analytical HPLC. Final duplexes were prepared in sodium salt form. Ten Dicer-substrate siRNAs (DsiRNAs)7,8 targeting murine IL-10 (NM_010548) were tested for functional potency as described below. The most potent DsiRNA (mIL10-1) was an asymmetric duplex with a 25-base passenger strand and a 27-base guide strand with sequence 5′-pGGAUCUUAGCUACGGAAACAACtc-3′ (S-strand) and 5′-pGAGUUGUUUCCGUUAGCUAAGAU CCCU-3′, where RNA bases are uppercase, DNA bases are lowercase, and p is phosphate. For all experiments, the negative control DsiRNA NC1 was employed with sequence 5′-p CGUUAAUCGCGUAUAAUACGCGUat-3′ (S-strand) and 5′-pAUACGCGUAUUAUACGCGAUUAACGAC-3′ (AS-strand) and for some experiments the positive control anti-HPRT1 (NM_000194) DsiRNA was employed with sequence 5′-pGCCAGACUUUGUUGGAUUUGAAAtt-3′ (S-strand) and pAAUUUCAAAUCCAACAAAGUCUGGCUU (AS-strand). The anti-HPRT1 DsiRNA works as a positive control for human, mouse, and rat cells lines equally well.
The murine macrophage cell line RAW 264.7 was employed for all DsiRNA validation experiments (ATCC, Manassas, VA; Cat #TIB-71). Cells were split into 48-well plates at 60% confluency (250,000 cells per well) at the time of transfection in 250 µL DMEM containing 10% FBS (ATCC). RNA duplexes were transfected at 10 nM, 1 nM, and 0.1 nM final concentrations with 2.5 µL TransIT-TKO® (Mirus, Madison, WI) per 50 µL OptiMEM™ I (Invitrogen, Carlsbad, CA). The HPRT1 DsiRNA was used as an internal positive control to assess transfection efficiency and the NC1 DsiRNA was a nontargeting negative control. All transfections were performed in triplicate. Twenty-four hours post-transfection, all transfected wells were stimulated with 50 ng/mL Escherichia coli K12 LPS (InvivoGen, San Diego, California) for 8 hours to induce IL-10 production. RNA was harvested 32 hours post-transfection using the SV96 Total RNA Isolation Kit (Promega, Madison, WI) and cDNA was synthesized using 150 ng total RNA with SuperScript™-II Reverse Transcriptase (Invitrogen) per the manufacturer’s instructions using both random hexamer and oligo-dT priming.
Quantitative real-time polymerase chain reaction (PCR) was performed using 10 ng cDNA per 10 µL reaction, Immolase™ DNA Polymerase (Bioline, Randolph, MA), 200 nM primers, and 200 nM probe. IL-10 specific primers were IL-10-For 5′-GTACAGCCGGGAAGACAATAAC, IL-10-Rev 5′-TTGGCAACCCAAGTAACCC, and probe IL-10-P 5′ FAM-TGCCTTCAGCCAGGTGAAGACTTT-IBFQ (Iowa Black FQ, dark quencher). HPRT1-specific primers were HPRT-For 5′-GACTTTGCTTTCCTTGGTCAGGCA, HPRT-Rev 5′-GGCTTATATCCAACACTTCGTGGG and probe HPRT-P 5′-MAXN-ATGGTCAAGGT CGCAAGCTTGCTGGT-IBFQ. Cycling conditions employed were: 95°C for 10 minutes followed by 40 cycles of two-step PCR with 95°C for 15 seconds and 60°C for 1 minute. PCR and fluorescence measurements were done using an ABI Prism™ 7900 Sequence Detector (Applied Biosystems Inc, Foster City, CA). All reactions were performed in triplicate. Expression data were normalized to levels of an internal control gene, RPL23 (NM_022891). RPL23 specific primers were RPL23-For 5′ CTGTGAAGGGAATCAAGGGA, RPL23-Rev 5′ TGTCGAATTACCACTGCTGG, and probe RPL23-P 5′ FAM-CTGAACAGACTTCCTGCTGC TGGTG-IBFQ. Copy number standards were run in parallel using linearized cloned amplicons for qualitative PCR assays. Unknowns were extrapolated against standards to establish absolute quantitative measurements.
siRNA transfection
Primary bone marrow macrophages were harvested as described above. Cells were plated into 24-well plates and allowed to acclimate for 16–24 hours. Lipofectamine 2000 (Invitrogen) was combined with siRNA as directed by the manufacturer’s guidelines. The final concentration of siRNA added to the cells was 33 nM. Cells were incubated in serum-free transfection medium for 8 hours, at which time the supernatants were removed and replaced with growth media. The cells were stimulated after 16–24 hours of reacclimation.
To test targeting of cytokine response in vivo, we targeted IL-10 by delivering IL-10 siRNA loaded into poly(lactic-co-glycolic acid) (PLGA) microparticles (MPs) (Division of Pharmaceutics, University of Iowa, Iowa City IA). Nonspecific siRNA (DsiRNA NC1) and empty PLGA MPs were used as controls. A/J mice (four per group) received intraperitoneal administration of siRNA in PLGA MPs (2.5 µg per mouse suspended in 100 µL of phosphate-buffered solution). No signs of distress were observed in any of the groups.
Peritoneal exudates were collected at 6, 12, and 18 hours after PLGA injection. Peritoneal exudates were centrifuged and cell pellets suspended and cultured overnight at 37°C in DMEM with 10% serum (D10). After incubation, the supernatant was discarded and adherent cells were placed in triplicate in 24-well plates with 250 µL of D10 media and allowed to attach for 6 hours. Cells were then stimulated with 1 µg/mL CpG. After 12 hours of stimulation, supernatants were recovered and analyzed by enzyme-linked immunosorbent assay (ELISA) for their IL-10 and TNF-α concentrations.
Enzyme-linked immunosorbent assay
Cell culture supernatants were collected from triplicate wells after stimulation. TNF-α and IL-10 secreted by BMM and peritoneal macrophages, and interferon-gamma (IFN-γ) secreted by lymphocytes were measured by ELISA using purified capture and biotinylated detection antibody pairs (BD Biosciences). The ELISA plates were read using the Multiskan FC microplate photometer (Thermo Scientific, Hudson, NH) at 450 nm. The data was analyzed using SkanIT software (Thermo Fisher Scientific, Vantaa, Finland).
Statistical analysis
Experiments were performed in triplicates. Error bars indicate standard error of the mean (SEM). Results are representative of three independent experiments. For reverse transcription-PCR experiments, error bars represent SEM for the triplicate quantitative PCR reactions performed on three biological replicates. The statistical significance was evaluated using Student’s t-test.
Results
TLR agonists stimulate bone marrow macrophages in a dose-dependent fashion
To characterize the responses to TLR stimulation, primary BMM were exposed to varying concentrations of specific TLR compounds and IL-12, TNF-α and IL-10 responses were measured. We anticipated identifying an optimal TLR agonist and dosage; however, irrespective of the TLR agonist used, the BMM response was characterized by increasing production and eventual plateau of the cytokine measured. In all TLRs tested, production of IL-10 characteristically lagged behind that of TNF-α (Figure 1) or IL-12 (data not shown) and only emerged at higher concentrations of TLR-agonist. This indicates that an optimal proinflammatory dose of each TLR-agonist can be identified to maximize the production of TNF-α prior to induction of IL-10 secretion. Importantly, secretion of IL-10 reaches maximal values at the same or slightly higher TLR agonist concentrations relative to those that induce maximal TNF-α production. Furthermore, no combination of two TLR agonists was capable of selectively stimulating proinflammatory responses, as evident from the ratios of TNF-α and IL-10 production (Figure 1D). These observations suggest that in vivo TLR agonist administration may be impractical if the goal is to obtain a predominantly proinflammatory response.
Figure 1.

Macrophage TNF-α and IL-10 production after exposure to varying concentrations of TLR agonists. Primary bone marrow macrophages were harvested from A/J mice, plated, and stimulated with Poly I:C, a TLR3 agonist (A), CpG, a TLR9 agonist (B), OK 432, a TLR2/4 agonist (C), or various combinations of two agonists, as indicated. Cytokine levels were measured by ELISA 16–24 hours after stimulation and expressed as pg/mL (A–C) or TNF-α/IL-10 ratios (D, mean and standard deviation of three separate experiments).
Notes: The differences between untreated and treated cultures are statistically significant (**P < 0.01; ***P < 0.001).
Abbreviations: TLR, Toll like receptor; TNF, Tumor Necrosis Factor; IL-10, Interleukin 10; ELISA, Enzyme linked Immunosorbent Assay; Poly I:C, Polyinosinic:polycytidylic acid; CpG, Cytosine-phosphate-Guanin.
Macrophage IL-10 production inhibits in vitro lymphocyte activation
Given significant IL-10 production at higher doses of TLR agonists, we hypothesized that a paradoxical anti-inflammatory response to TLR agonist could occur. Therefore, we developed an in vitro model to investigate the effect on lymphocyte activation. Primary murine BMM were first exposed to TLR agonists. After exposure, the activated macrophage supernatant was collected and transferred to a culture of autologous murine splenocytes stimulated with anti-CD3 antibody. Lymphocyte activation was determined by IFN-γ production. Remarkably, exposure of autologous splenocytes to the cytokine milieu of TLR-activated macrophages consistently led to lymphocyte suppression in comparison with controls exposed to anti-CD3 antibody alone (Figure 2A). Profound suppression of T cell activation was achieved even with supernatants with cultures containing relatively lower levels of IL-10 (eg, supernatants from OK-432-stimulated macrophages). Importantly, IFN-γ production was induced only by anti-CD3, and not by any agent transferred from the primary culture (Figure 2B).
Figure 2.
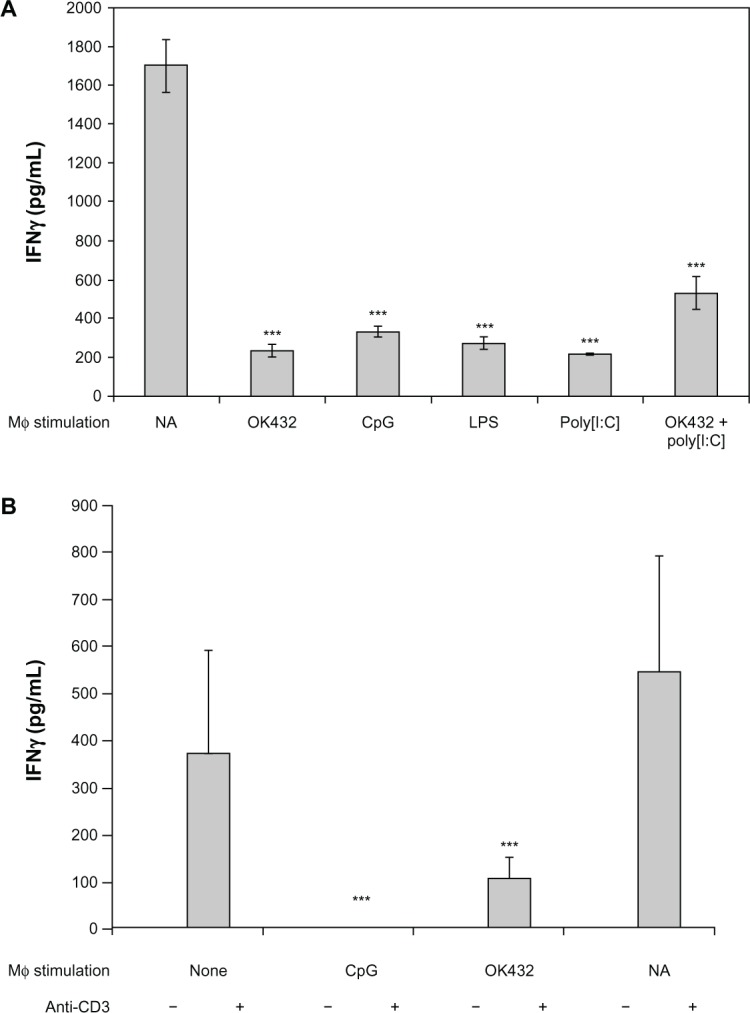
Modulation of anti-CD3-induced IFN-γ production by TLR-stimulated macrophage supernatants. Whole A/J (A) or C57BL/6 (B) spleen cells were plated in the presence of 1.0 µg/mL of anti-CD3 antibody.
Notes: The activation was performed in the presence of 100% supernatants of macrophages stimulated with OK-432 (10 µg/mL), CpG (3 µg/mL), LPS (10 µg/mL), or Poly I:C (50 µg/mL), as indicated. Concentrations of combination of OK-432 and Poly I:C were also 10 µg/mL and 50 µg/mL, respectively. Not applicable (NA) represents splenocytes plated with anti-CD3 antibody alone in the absence of any macrophage supernatant. Cytokine levels were measured by ELISA 48 hours after stimulation. The differences between cultures containing supernatants from untreated and treated macrophages are statistically significant (***P < 0.001).
Abbreviations: TLR, Toll like receptor; TNF, Tumor Necrosis Factor; IL-10, Interleukin 10; ELISA, Enzyme linked Immunosorbent Assay; Poly I:C, Polyinosinic:polycytidylic acid; CpG, Cytosine-phosphate-Guanin; LPS, Lipopolysaccharide; IFN, Interferon.
To evaluate the role of IL-10 in this suppressive lymphocyte response, the experiment was repeated using BMM from IL-10-knockout mice. Supernatants from TLR stimulated IL-10 KO BMM did not inhibit IFN-γ production from autologous splenocytes stimulated with anti-CD3 antibody (Figure 3). These observations suggest that IL-10 is a critical cytokine inhibiting lymphocyte activation in this model.
Figure 3.

Modulation of anti-CD3-induced IFN-γ production by supernatants of TLR-stimulated IL-10-deficient macrophages.
Notes: Methods and TLR agonist concentrations are identical to those listed in Figure 2. Macrophage control represents supernatant from unstimulated macrophages in combination with splenocytes and anti-CD3. Splenocyte control represents splenocytes incubated in the presence of anti-CD3 without macrophage supernatant. The differences between cultures containing supernatants from untreated and treated macrophages are statistically significant (**P < 0.01; ***P < 0.001).
Abbreviations: TLR, Toll like receptor; IL-10, Interleukin 10; CpG, Cytosine-phosphate-Guanin; LPS, Lipopolysaccharide; IFN, Interferon.
IL-10 siRNA targeting in macrophages reversed the suppression of lymphocyte activation induced by TLR-stimulated macrophage supernatants
To directly and selectively target IL-10 production, we investigated the use of IL-10 siRNA to suppress macrophage IL-10 production. siRNA constructs were designed and subsequently tested for their ability to suppress IL-10 secretion from a RAW macrophage cell line by quantitative reverse transcription-PCR. IL-10 mRNA knockdown by the most effective construct is shown in Figure 4. Lipofectamine 2000 was used to transfect BMM with this IL-10 siRNA construct. Subsequent CpG-mediated activation of transfected BMM led to a notable decrease in secreted levels of IL-10 (Figure 5A) with minimal effect on TNF-α production (Figure 5B).
Figure 4.
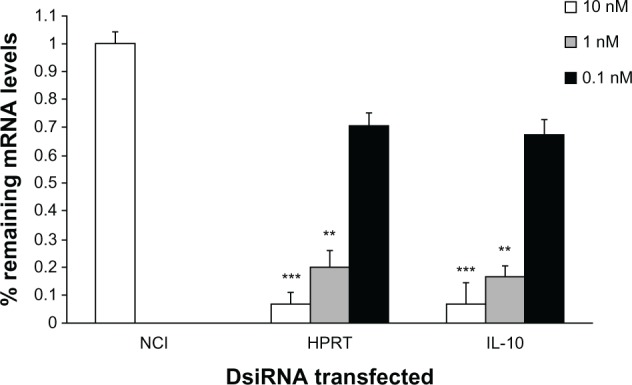
Validation of anti-IL-10 DsiRNA. NC1, HPRT, and mIL10-1 DsiRNAs were transfected into RAW264.7 cells at 10 nM, 1 nM, and 0.1 nM concentrations.
Notes: At 24 hours, cells were stimulated with 50 ng/mL LPS and RNA was harvested 8 hours later. RT-qPCR was performed and relative expression of the HPRT mRNA (in the HPRT DsiRNA transfected cultures) and IL-10 mRNA (in the IL-10-1 DsiRNA transfected cultures) normalized to the RPL23 are shown, setting the control NC1 transfection to 100%. The differences between NC1- and HPRT- or IL-10-specific siRNA are statistically significant (**P < 0.01; ***P < 0.001).
Abbreviations: IL-10, Interleukin 10; HPRT, Hypoxantine-guanine phosphorybosyl-transferase; RT-qPCR, Reverse transcriptase quantitative polymerase chain reaction.
Figure 5.

IL-10 siRNA decreases IL-10 production and abolishes suppression in vitro. A/J macrophages were transfected with IL-10 siRNA using Lipofectamine 2000 reagent and stimulated with 1.0 µg/mL CpG. Shown are levels of IL-10 (A), or TNF-α and IL-10 in primary overnight cultures (B), or levels of anti-CD3-induced IFN-γ production by spleen cells in the presence of primary culture supernatants (C).
Notes: The differences between Lipofectamine 2000-treated and IL-10 siRNA-treated cultures are statistically significant for IL-10, but not TNF-α production (*P < 0.05; ***P < 0.001).
Abbreviations: TNF, Tumor Necrosis Factor; IL-10, Interleukin 10; CpG, Cytosine-phosphate-Guanin; IFN, Interferon; HPRT, Hypoxantine-guanine phosphorybosyltransferase.
After demonstrating that BMM IL-10 production could be constrained through the administration of IL-10 siRNA, we applied this finding to the in vitro model of macrophage-mediated lymphocyte activation described above. Supernatant from the siRNA lipofected and stimulated macrophages was exposed to autologous splenocytes in the presence of R10 media and anti-CD3 antibody. Subsequent lymphocyte activation was measured via IFN-γ ELISA. As seen in Figure 5C, lymphocytes exposed to IL-10 siRNA lipofected macrophage supernatant demonstrated an approximately fourfold increase in IFN-γ activity. These findings show that suppression of IL-10 following macrophage TLR stimulation induces a cytokine milieu conducive to T-cell activation.
IL-10 production is suppressed in vivo following treatment with IL-10 siRNA-loaded PLGA microparticles
PLGA MPs loaded with IL-10 or control siRNA were injected intraperitoneally at a dose of 2.5 µg per mouse and peritoneal exudate was harvested 6 hours later. This time point was chosen because after 6 hours of in vitro exposure, more than 90% of peritoneal exudate cells engulfed PLGA MPs (Figure 6) and longer incubations were associated with concomitant nonspecific effects (data not shown). CpG stimulation (1 µg/mL) of peritoneal exudate cells resulted in significantly reduced IL-10 levels following treatment with IL-10 siRNA-loaded PLGA-MPs, while TNF-α levels were not affected (Figure 7A). Furthermore, IFN-γ response following T lymphocyte activation was higher when stimulated in the presence of supernatants of in vivo IL-10 siRNA-loaded macrophages (Figure 7B).
Figure 6.

Uptake of PLGA MPs in vivo. A/J mice were injected with PLGA MPs and peritoneal exudate cells were examined at indicated time intervals (1, 2, 6 and 12 hours post injection) by light microscopy. Shown are representative images for each time point (A), percentage of cells loaded with PLGA MPs (B) and average number of PLGA MPs per cell (C).
Abbreviation: PLGA MPs, Poly lactic-co-glycolic acid microparticles.
Figure 7.

Inhibition of IL-10 production in vivo by siRNA. A/J mice were injected with empty, IL-10 siRNA-loaded, or control NC siRNA-loaded PLGA MPs. Peritoneal exudate cells were isolated, stimulated with CpG and examined for production of TNF-α and IL-10 (A). Supernatants of these cultures were added to anti-CD3-stimulated spleen cells and production of IFN-γ was assessed (B).
Note: The differences between empty PLGA-treated and IL-10 siRNA-treated cells are statistically significant (***P < 0.001).
Abbreviations: IL-10, Interleukin 10; CpG, Cytosine-phosphate-Guanin; PLGA, MPs-Poly lactic-co-glycolic acid microparticles; IFN, Interferon.
Discussion
Our findings demonstrate the paradoxical anti-inflammatory response of murine BMM to TLR stimulation. This effect is mediated by IL-10 production, a well-described suppressor of immune responses. IL-10’s suppressive effects are mediated through a variety of mechanisms; including downregulation of macrophage and dendritic cell antigen-presenting capacity, blocking secretion of cytokines and chemokines, preventing expression of costimulatory molecules and chemokine receptors, increasing generation of free-radicals and upregulating expression of β2-integrin ligand (intracellular adhesion molecule 1).9 In addition, the current work shows that IL-10 produced by macrophage stimulation is responsible for suppressing T-cell activation. In direct contrast, the production of proinflammatory cytokines, such as TNF-α drive the polarization of CD4+ T-cells towards a Th-1 phenotype. Thus, the pro- vs anti-inflammatory cytokine balance is crucial in the control of immune homeostasis,10 and their relative expression in response to TLR stimulation has implications for the clinical application of TLR agonists to cancer treatment, including their use as cancer vaccine adjuvants.
Recent evidence supports the concept of a paradoxical anti-inflammatory response to TLR stimulation. Murine bone marrow dendritic cells stimulated with TLR 2 (Pam3CSK4) and TLR 4 (LPS) agonists demonstrate a dose-dependent increase in IL-10 secretion.10 Moreover, high-dose CpG induces CD19+ dendritic cell-mediated activation of suppressor T-cells via an indoleamine 2,3-dioxygenase (IDO) pathway, a response that is particularly prominent in areas of chronic inflammation and resultant constitutive IDO expression, such as the tumor microenviornment.11,12 Taken together, these findings highlight the variability of the immunologic response to TLR agonists, ostensibly as mechanisms to allow the avoidance of unregulated local inflammation. Unfortunately, they add additional layers of intricacy to the use of TLR agonists as vaccine adjuvants. Our finding that the transition from pro- to anti-inflammatory cytokine secretion occurs following relatively small increases of TLR agonist concentration additionally complicates their use. Beyond optimizing the target microenvironment for TLR agonist administration, simple variation in individual pharmacokinetics could make accurate adjuvant dosing challenging.
Dendritic cells are the most important antigen presenting cell type in promoting immune responses.13,14 Just like macrophages, dendritic cells produce IL-10 in response to TLR stimulation.10 In fact, inhibition of IL-10 production by dendritic cells is one of the many strategies being attempted with the hope of successful immunotherapy of tumors.15 However, IL-10 is produced by and affects the function of many cell types,16 and the ultimate fate of immune response induction does not rest solely on dendritic cells. In fact, the immunosuppressive potential of macrophages and myeloid-derived suppressor cells is well known.17 Hence, targeting of IL-10 production as part of raising anti-tumor immunity should be inclusive of as many cell types as possible. Our siRNA approach is designed to efficiently target cells with antigen-processing abilities, which includes both dendritic cells and macrophages.
By constraining macrophage TLR-mediated IL-10 production through administration of siRNA and, consequently, augmenting activated splenocyte IFN-γ production, we have demonstrated an ability to augment the efficacy of TLR-agonists in vitro. In order for this finding to have clinical utility, in vivo IL-10 modulation will be necessary. Several potential strategies exist in which investigators have explored the use of anti-IL-10 antibodies; one in particular has demonstrated its efficacy in humans in the treatment of systemic lupus erythematosus.18 The “paradoxically beneficial” effect of anti-IL-10 is due to an effect of IL-10 on B cells helping produce (auto)antibodies in lupus. The use of IL-10 signaling pathway inhibitors are also shown to have in vitro efficacy, as demonstrated in the above-mentioned study by Hirata et al, where inhibitors of the p38 mitogen-activated protein kinase (MAPK), and c-jun N-terminal kinase (JNK)1/2 pathways effectively constrained IL-10 production.10 Finally, a recent study utilized JC virus-like particles for the targeted delivery of IL-10 siRNA in mice.19 While the first of these three options has been the most extensively studied, the last two may prove to be more effective, as TNF-α and IL-10 secretion can exhibit a synergistic relationship in which the presence of the former amplifies secretion of the latter.20 As demonstrated by our data, the converse does not seem to be true; moreover, previous studies on human monocytes have demonstrated that IL-10 can in fact inhibit TNF-α secretion.21 De novo prevention of IL-10 production is therefore less likely to prevent TNF-α secretion than post hoc ablation of its effects.
Previous studies have both detected secretion of anti-inflammatory cytokines and demonstrated techniques to constrain their production. However, our study takes comparable findings a step further by developing an in-vitro model to demonstrate their effects on T-cell activation. The fact that TLR 9 in humans is expressed in B-cells and plasmacytoid dendritic cells,22 but not monocytes or macrophages like in the mouse, appears to limit the impact of our findings to the mouse. However, our findings were not specific to TLR9 stimulation, but pertained to all TLR receptors tested.
Additionally, we demonstrated that siRNA constructs can be effectively delivered by PLGA MPs both in vitro and in vivo. The clinical use of PLGA MPs as a drug-delivery system is expanding as it is a biocompatible polymer without systemic toxicity.23 We demonstrated that the uptake of PLGA MPs by peritoneal macrophages reached at least 90% at 6 hours and that the uptake continued beyond that time point as indicated by the rising number of MPs per cell (Figure 6). This load was not deleterious as no suppression of cytokine production was observed after a 12- and 24-hour exposure to empty MPs (data not shown). Slow biodegradable materials such as PLGA could improve the stability of siRNA and promote a sustained and localized effect on the target organ or tissue.24 Macrophages preferentially phagocytose the MPs, thus making these PLGA polymers a useful vehicle for targeting macrophage function. Our in vivo experiments indicated that macrophage exposure to siRNA-loaded PLGA MPs was effective in suppressing IL-10 production, an effect that could potentially be used in vaccine formulations to augment host immunity.
In conclusion, our data demonstrates that the secretion of proinflammatory cytokines in response to TLR activation is balanced by the anti-inflammatory effects of IL-10 secretion in a dose-dependent fashion. The production of IL-10 prevents the development of the cytotoxic T-cell response in an in vitro model of TLR activation. Targeting macrophage IL-10 production through the administration of siRNA abolishes the suppressive T-cell effect. As this applies to the development of cancer vaccines, strategies to either constrain or ablate IL-10 and enhance the proinflammatory response may improve the efficacy of TLR agonists as vaccine adjuvants. Finally, in vivo suppression of macrophage IL-10 was successfully accomplished by IL-10 siRNA-loaded PLGA-MP. MPs loaded with siRNA appear to present a potential method for targeting of IL-10 production. These findings should have important implications for modulating immunity with siRNA strategies.
Acknowledgments
This work was supported in part by the Sheikh Zayed Institute of Pediatric Surgical Innovation and the Michael Sandler Cancer Research Fund at Children’s National Medical Center.
Footnotes
Disclosure
The authors report no conflict of interest in this work.
References
- 1.Bhardwaj N. Harnessing the immune system to treat cancer. J Clin Invest. 2007;117(5):1130–1136. doi: 10.1172/JCI32136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Iwasaki A, Medzhitov R. Toll-like receptor control of the adaptive immune responses. Nat Immunol. 2004;5(10):987–995. doi: 10.1038/ni1112. [DOI] [PubMed] [Google Scholar]
- 3.Janeway CA, Jr, Medzhitov R. Introduction: the role of innate immunity in the adaptive immune response. Semin Immunol. 1998;10(5):349–350. doi: 10.1006/smim.1998.0142. [DOI] [PubMed] [Google Scholar]
- 4.Krieg AM. Therapeutic potential of Toll-like receptor 9 activation. Nat Rev Drug Discov. 2006;5(6):471–484. doi: 10.1038/nrd2059. [DOI] [PubMed] [Google Scholar]
- 5.Pasare C, Medzhitov R. Toll-like receptors and acquired immunity. Semin Immunol. 2004;16(1):23–26. doi: 10.1016/j.smim.2003.10.006. [DOI] [PubMed] [Google Scholar]
- 6.So EY, Ouchi T. The application of Toll like receptors for cancer therapy. Int J Biol Sci. 2010;6(7):675–681. doi: 10.7150/ijbs.6.675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kim DH, Behlke MA, Rose SD, Chang MS, Choi S, Rossi JJ. Synthetic dsRNA Dicer substrates enhance RNAi potency and efficacy. Nat Biotechnol. 2005;23(2):222–226. doi: 10.1038/nbt1051. [DOI] [PubMed] [Google Scholar]
- 8.Rose SD, Kim DH, Amarzguioui M, et al. Functional polarity is introduced by Dicer processing of short substrate RNAs. Nucleic Acids Res. 2005;33(13):4140–4156. doi: 10.1093/nar/gki732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pestka S, Krause CD, Sarkar D, Walter MR, Shi Y, Fisher PB. Interleukin-10 and related cytokines and receptors. Annu Rev Immunol. 2004;22:929–979. doi: 10.1146/annurev.immunol.22.012703.104622. [DOI] [PubMed] [Google Scholar]
- 10.Hirata N, Yanagawa Y, Ebihara T, et al. Selective synergy in anti-inflammatory cytokine production upon cooperated signaling via TLR4 and TLR2 in murine conventional dendritic cells. Mol Immunol. 2008;45(10):2734–2742. doi: 10.1016/j.molimm.2008.02.010. [DOI] [PubMed] [Google Scholar]
- 11.Baban B, Chandler PR, Sharma MD, et al. IDO activates regulatory T cells and blocks their conversion into Th17-like T cells. J Immunol. 2009;183(4):2475–2483. doi: 10.4049/jimmunol.0900986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mellor AL, Baban B, Chandler PR, Manlapat A, Kahler DJ, Munn DH. Cutting edge: CpG oligonucleotides induce splenic CD19+ dendritic cells to acquire potent indoleamine 2,3-dioxygenase-dependent T cell regulatory functions via IFN Type 1 signaling. J Immunol. 2005;175(9):5601–5605. doi: 10.4049/jimmunol.175.9.5601. [DOI] [PubMed] [Google Scholar]
- 13.Steinman RM. The dendritic cell system and its role in immunogenicity. Ann Rev Immunol. 1991;9:271–296. doi: 10.1146/annurev.iy.09.040191.001415. [DOI] [PubMed] [Google Scholar]
- 14.Steinman RM. Decisions about dendritic cells: past, present, and future. Annu Rev Immunol. 2012;30:1–22. doi: 10.1146/annurev-immunol-100311-102839. [DOI] [PubMed] [Google Scholar]
- 15.Huang FP, Chen YX, To CK. Guiding the “misguided” – functional conditioning of dendritic cells for the DC-based immunotherapy against tumours. Eur J Immunol. 2011;41(1):18–25. doi: 10.1002/eji.201040543. [DOI] [PubMed] [Google Scholar]
- 16.Moore KW, de Waal Malefyt R, Coffman RL, O’Garra A. Interleukin-10 and the interleukin-10 receptor. Annu Rev Immunol. 2001;19:683–765. doi: 10.1146/annurev.immunol.19.1.683. [DOI] [PubMed] [Google Scholar]
- 17.Allavena P, Mantovani A. Immunology in the clinic review series; focus on cancer: tumour-associated macrophages: undisputed stars of the inflammatory tumour microenvironment. Clin Exp Immunol. 2012;167(2):195–205. doi: 10.1111/j.1365-2249.2011.04515.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Llorente L, Richaud-Patin Y, Garcia-Padilla C, et al. Clinical and biologic effects of anti-interleukin-10 monoclonal antibody administration in systemic lupus erythematosus. Arthritis Rheum. 2000;43(8):1790–1800. doi: 10.1002/1529-0131(200008)43:8<1790::AID-ANR15>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 19.Chou MI, Hsieh YF, Wang M, et al. In vitro and in vivo targeted delivery of IL-10 interfering RNA by JC virus-like particles. J Biomed Sci. 2010;17:51. doi: 10.1186/1423-0127-17-51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hirata N, Yanagawa Y, Ogura H, et al. The role of tumor necrosis factor-alpha for interleukin-10 production by murine dendritic cells. Cell Immunol. 2011;266(2):165–171. doi: 10.1016/j.cellimm.2010.09.012. [DOI] [PubMed] [Google Scholar]
- 21.Donnelly RP, Freeman SL, Hayes MP. Inhibition of IL-10 expression by IFN-gamma up-regulates transcription of TNF-alpha in human monocytes. J Immunol. 1995;155(3):1420–1427. [PubMed] [Google Scholar]
- 22.Krieg AM. Toll-like receptor 9 (TLR9) agonists in the treatment of cancer. Oncogene. 2008;27(2):161–167. doi: 10.1038/sj.onc.1210911. [DOI] [PubMed] [Google Scholar]
- 23.Zhang Z, Huang G. Intra-articular lornoxicam loaded PLGA microspheres: enhanced therapeutic efficiency and decreased systemic toxicity in the treatment of osteoarthritis. Drug Deliv. 2012;19(5):255–263. doi: 10.3109/10717544.2012.700962. [DOI] [PubMed] [Google Scholar]
- 24.Khan A, Benboubetra M, Sayyed PZ, et al. Sustained polymeric delivery of gene silencing antisense ODNs, siRNA, DNAzymes and ribozymes: in vitro and in vivo studies. J Drug Target. 2004;12(6):393–404. doi: 10.1080/10611860400003858. [DOI] [PubMed] [Google Scholar]