

Abstract
Hepatitis B virus (HBV) infection-related hepatocellular carcinoma (HCC) represents a major health problem worldwide. HBV X (HBx) protein is the most common open reading frame that may undergo mutations, resulting in the development of HCC. This study aimed to determine specific HBx mutations that differentiate the central- and para-tumor tissues, and identify their association with HCC development. HBx gene from HCC tumor and para-tumor tissues of 47 HCC patients was amplified, sequenced and statistically analyzed. A novel combination of 2 mutations at residues 10 and 144 was identified which might play a significant role in HCC development. Expression vectors carrying HBx with the specific mutations were constructed and transfected into HepG2 and p53-null HepG2 cells. Compared to wild type (WT) and single mutation of HBx at residue 10 or 144, the 10/144 double mutations strongly up-regulated p21 expression and prolonged G1/S transition in WT- and p53-null HepG2 cells. Apoptosis was also inhibited by HBx harboring 10/44 double-mutation. Binding of 10/144 double-mutant HBx to p53 was lower than WT HBx. Conclusively, the 10/144 double mutation of HBx might play a crucial role in HCC formation.
Keywords: Hepatitis B virus, Mutation, Hepatocellular carcinoma
Introduction
Hepatocellular carcinoma (HCC), the fifth common cancer worldwide, is mainly caused by hepatitis B virus (HBV) [1]. In China, 7.8% of the population has HBV infection with positive serum hepatitis B surface antigen (HBsAg) [2]. Moreover, most Chinese individuals are infected by HBV genotype C, another risk factor for HCC development [3–7].
Three factors, including integration of viral DNA into host genome, chronic inflammation of host immune response, and HBV-encoded proteins, have been implicated in hepatocarcinogenesis [8–12]. HBx is one of the four structural proteins of HBV. The coding gene of this protein may integrate into host DNA and play a key role in HCC development [13,14]. HBx modulates transcription, responses to genotoxic stress, protein degradation, and other signaling pathways [15], affecting viral replication and proliferation, cell cycle checkpoints [16], apoptosis [17], and carcinogenesis [18]. HBx mutations, including double-substitution K130M-V131I, found preferentially in HCC patients [19], may be a risk factor for HCC development [20]. Furthermore, HBx gene disruptions were more frequent in tumor than in non-tumor liver [21,22]. Various HBx mutants showed different effects on cell proliferation and cell cycle progression [23], indicating their close relation to HCC.
Materials and methods
Samples
Central- and para-tumor tissues from 47 HCC patients (40 males, 7 females, aged 22–78 years with average of 51 years), who underwent surgical tumor resection at Beijing Youan Hospital during 2009–2010, were included. This study was approved by the Ethical Committee of Beijing Youan Hospital and in compliance with the Declaration of Helsinki. All participants provided with written consent. All participants were diagnosed with HCC and HBsAg-seropositiveness. Their clinical characteristics and demographics were listed in Table 1.
Table 1.
Demographics and clinical characteristics of patients included in the study.
| Item | n | Median | Value range |
|---|---|---|---|
| Age (yrs) | 47/47 | 51 | 22–78 |
| Gender (male/n) | 40/47 | N/A | N/A |
| Cirrhosis (n) | 28/47 | N/A | N/A |
| Anti-HBV therapy | 35/47 | N/A | N/A |
| Anti-HBV therapy time (mon) | 35/47 | 24 | 6–96 |
| Alanine aminotransferase (IU/L) | 47/47 | 42.3 | 13.6–1,094.9 |
| Aspartate aminotransferase (IU/L) | 47/47 | 48.3 | 17.8–691.7 |
| Total bilirubin (μmol/L) | 47/47 | 35.8 | 11.6–125.6 |
| Direct bilirubin (μmol/L) | 47/47 | 12.7 | 3.5–45.3 |
| Total protein (g/L) | 47/47 | 52.5 | 48.9–72.8 |
| Albumin (g/L) | 47/47 | 30.2 | 26.9–46.5 |
| Positive e antigen (n) | 27/47 | N/A | N/A |
| e antigen titer (coi) | 27/47 | 124.5 | 10.6–268.4 |
| Surface antigen titer (coi) | 47/47 | 668.7 | 125.8–6,834.5 |
| HBV DNA (IU/mL) | 47/47 | 2560 | <100–12,300,000 |
| HBV genotype | 47/47 | C1 | C1 |
| Alpha-fetoprotein | 47/47 | 15.63 | 1.3–18,930 |
Tissue histology, polymerase chain reaction (PCR), and population-based sequencing
Frozen central- and para-tumor tissues collected after tumor-resection were histologically distinguished by a certified pathologist. Genomic DNA was extracted as described [24]. HBx gene from nt 1373 to 1838 was amplified by nested PCR with external primers, 5′-atggctgctagggtgtgct-3′ (sense) and 5′-aacatgagatgattaggcagaggt-3′ (anti-sense), and internal primers, 5′-catggctgctaggctgtgc-3′ (sense) and 5′-agaggcagaggtgaaaaagtt-3′ (anti-sense). After denature at 94 °C for 3 min, amplification was performed with 30 cycles at 94 °C, 30 sec, annealing at 56 °C, 30 sec, and extension at 72 °C, 1 min, followed by extension at 72 °C, 5 min. PCR-amplified products were sequenced using ABI Prism Big Dye kit version 3.0 with ABI 3730XL DNA automated sequencer (Applied Biosystems (ABI), Foster City, CA). All sequences were deposited in GenBank with accession numbers KC814852–KC814928.
Data analysis
Initial sequences were aligned using Clustal W, with default gap parameters and ‘IUB’ DNA weight matrix. Alignment was edited in Bioedit v7.0 to preserve frame insertions and deletions. Sequences were analyzed with bioinformatic tools as described [25] below:
Sequences were analyzed for possible mutation patterns using Viral Epidemiology Signature Pattern Analysis) (http://www.hiv.lanl.gov/cgi-bin/P-vespa/vespa.cgi) [26].
Selective pressure on individual codons was assessed via Single Likelihood Ancestor Counting analysis [27], as implemented in HyPhy software package (http://www.datamonkey.org/). Likelihood-based selection analyses were performed on sequence alignments for each codon with a conservative significance threshold of P = 0.1.
Shannon entropy was calculated to identify HBx signatures from central tumor, using Entropy (http://www.hiv.lanl.gov/content/sequence/ENTROPY/entropy.html). Residue-specific entropy was computed from frequency f(Ai) of amino acid A at position I according to –ΣAf(Ai)ln(f[Ai]) [28].
Classification experiments were conducted using Waikato Environment for Knowledge Analysis [29]. J48 decision tree inducer, based on C4.5 algorithm [30], was implemented with the parameter ‘MinNumObj’ set at a value of 11 to limit theoretical complexity and minimized over-fitting risk. Classifiers were evaluated using 100 iterations of stratified ten-fold cross-validation, which reflects performance of classification models on novel datasets.
Plasmid construction, cell culture and DNA transfection
WT HBx gene, A10R, S144R, and A10/S144 double-mutated variants were amplified from corresponding liver tissues by nested PCR with external and internal primers, and amplified products were cloned respectively into the EcoRI site of pcDNA3.1 (His) vector (Invitrogen). HepG2 and HepG2 p53-null cells (gifts from Dr. James Ou, USC, LA, CA) were grown in high glucose–DMEM supplemented with 10% fetal bovine serum, which were transfected using Fugene HD Transfection Reagent (Promega).
Cell cycle analysis
Cell-cycle was analyzed with flow cytometry as described [31]. Cells were trypsinized, washed twice with PBS, fixed in 80% ethanol, washed with PBS and resuspended in 50 μg/mL propidium iodide containing 125 U/mL RNase A. DNA contents were assayed on FACSCanto II and analyzed using Mod-Fit software version 3.2 (BD).
Luciferase assay
Cells were transfected with p21 (pGL3-p21) or Bax luciferase reporter plasmid (pGL3-Bax), together with indicated amounts of HBx-expressing vector, pGL4.70-hRluc expression vector (0.02 μg), and appropriate amount of empty plasmid vector, for a total of 0.2 μg of plasmid DNA per transfection. Luciferase assays were performed 36 hr post-transfection followed with manuals of Dual-Luciferase Reporter Assay System (Promega). Each experiment was repeated at least 3 times.
Real-time PCR
Total RNA was isolated from tissues 24 hr post-transfection using RNeasy Mini Kit (Qiagen, Hilden, Germany). Reverse-transcription was performed using SuperScriptII First-Strand Synthesis System (Invitrogen). RT-qPCR was performed on ViiA 7DX RT-PCR system using Fast SYBR Green Master Mix (ABI). Relative transcript levels of target genes were normalized with GAPDH mRNA levels. Specific primers used for RT-qPCR were: p21, 5′-GACAGCAGAGGAAGACCATGTGGA-3′ (forward) and 5′-CGTTTTCGACCCTGAGAGTCTCCA-3′ (reverse); Bax, 5′-CCGAGTCACTGAAGCGACTGATGT-3′ (forward) and 5′-ACAAAGATGGTCACGGTCTGCCAC-3′ (reverse); and GAPDH, 5′-ACAGTCCATGCCATCACTGCCA-3′ (forward) and 5′-AGGCAGGGATGATGTTCTGGAGAG-3′ (reverse).
Cell apoptosis assay
48 hr post transfection, cells were washed twice with PBS, resuspended in Annexin-V binding buffer (Southern Biotech) and analyzed by flow cytometry (BD).
Western-blot analysis and co-immunoprecipitation
48 hr post transfection, cells were lysed in RIPA buffer, and protein concentrations were measured using BCA Protein Assay Kit (Biomed Biology, Beijing, China). Cell lysates (10 μg) were separated by 10–15% SDS–PAGE gels, transferred onto a PVDF membrane (Millipore) and blotted with monoclonal antibodies for p21 (clone-DCS60) (Cell signal Technology), p53, His, CDK-2 (clone D-12) (all three, Santa Cruz), cyclin-E (clone HE-12) (Calbiochem, Cambridge, MA, USA), and anti-β-actin (clone-C2) (Cell Signal Technology). Specific proteins were detected with enhanced chemiluminescence (Pierce SuperSignal, Thermo Fisher Scientific Inc., Rockford, IL, USA).
Cell lysates (500 μg of protein) were incubated with 2 μg of His antibody for 3 hr at 4 °C and then with protein A/G agarose (Santa Cruz) for 2 hr with gentle shaking. Beads were collected by centrifugation, washed 3 times with RIPA buffer, and eluted by incubation with SDS loading buffer at 95 °C for 5 min. Immunoblot assay was performed with anti-p53 monoclonal antibody.
Results
Amino acid residues 10 and 144 of HBx protein anchored signature sequence and might play a role in HCC development
For site specific analyses, there was no difference between viral variants sampled inside (central) and surrounding (para) liver tumors by VESPA and no consistent pattern for site specific selection (SLAC). We identified high frequency of K130M mutation and A1762T/G1764A double-mutation in 47 para-tumor and central-tumor samples, similar to previous reports [20,32,33]. But no statistically significant differences in these mutation sites were observed between central- and para-tumor tissues, although there were differences in entropy between both sequence datasets. Specifically, positions 10, 11, 12, 14 and 144 showed higher entropy in central-tumor tissue dataset (red bars in Fig. 1). Machine learning analysis revealed that positions 10 and 144 were associated with the location of sampled tissue (Fig. 2). These positions anchored the signature sequence which is able to classify variants with 75.5% accuracy, suggesting that A10R and S144R mutants are involved in HBV-related hepatocarcinogenesis. HBV mutation is also reported to be a risk factor of liver cirrhosis, another HBV related liver disease [34]. So we compared the frequency of A10/S44 double-mutation between HCC with liver cirrhosis and HCC without liver cirrhosis (only chronic hepatitis B, CHB) in this study. The results showed that A10/S44 double-mutation only occurred in tumor tissues but not para-tumor tissues and Fisher’s exact test confirmed that no significant difference was found between these 2 groups (p = 0.102, data did not reveal).
Fig. 1.
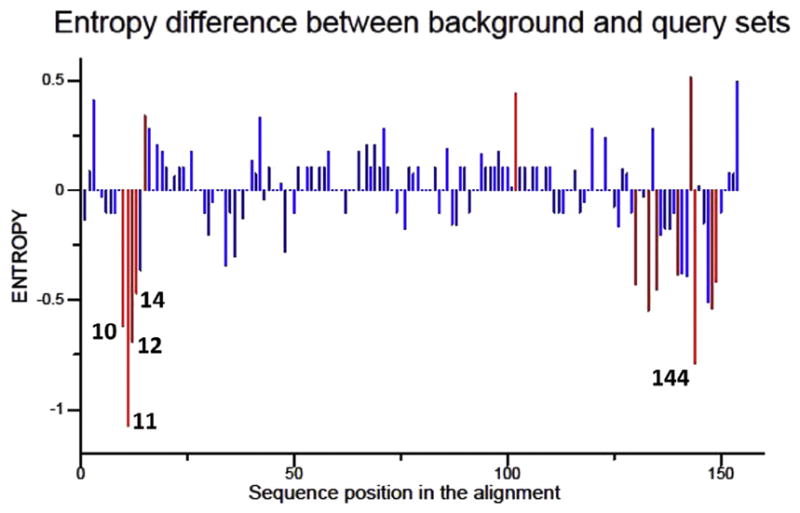
Difference in Shannon entropy between central-tumor and para-tumor at sites across the hepatitis B virus X gene. Residue specific entropy was computed and significant sites with p ≤ 0.05 are shown in red in the difference plot. Sites with significant differences were distinguished by red bars, and the five sites with the highest net differences were labeled with position numbers (numbered according to hepatitis B virus X). (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
Fig. 2.

Genetic signature associated with central-tumor and para-tumor derived sequences from HCC patients. Decision tree classifying X sequences were based on tissue of origin with 75.5% accuracy. p, para-tumor; c, central-tumor. The values in parentheses are the number of instances/number of incorrect classifications. Residues are numbered starting from methionine at the beginning of the hepatitis B virus X protein.
HBx A10R/S144R double-mutation prolonged G1/S transition
According to our analysis, HBxA10/S144 double-mutation was unique in tumor tissues other than para-tumor tissues. To elucidate the function of HBx A10/S144 double-mutant, we first focused on HBx’s function in cell cycle regulation. HBx expression vectors were constructed and transfected into HepG2 (p53+/+) and HepG2 p53-null cells for 24 hours. HBx expression vector harboring K130M-V131I mutation was also constructed and used as control (named Xcon blow). HBx and p53 expression levels were detected with Western blot (Fig. 3A); the cell cycle stages were analyzed by flow cytometry to identify their roles in cell cycle arrest (Fig. 3B and C). The results showed that S144R, Xcon and both A10R and S144R (XD) overexpression dramatically increased the proportion of cells at G1 phase compared with wild-type HBx or HBx A10R overexpression alone in HepG2 p53-null cells. The proportion of G1 phase at HBx A10R or S144R (XD) overexpression was significantly higher than that of A10R or S144R alone and that of Xcon overexpression in HepG2 p53-null cells. The same significant effect of A10R/S144R overexpression on G1 phase was also found in HepG2 cells with WT p53. These results suggested that HBx A10R mutation found in clinical specimens might play a critical role in HCC process.
Fig. 3.

Hepatitis B virus X A10/S144 double mutation prolonged G1-S transition and up-regulated the expression of p21 in a p53-independent manner. (A) HepG2 p53 null/HepG2 cells were transfected with indicated hepatitis B virus X expression vector together or control plasmid. Hepatitis B virus X, p53 and actin expression were analyzed by Western blotting. (B) Effect of hepatitis B virus X mutation on modifications of cell cycle parameters. 36 hours after transfection with indicated plasmids, cell cycle parameters were stained with propidium iodide (PI) and analyzed by flow cytometry. (C) Effect of hepatitis B virus X on cell-cycle progression. The percentage difference in the size of the G1 cell population was determined by the comparison of hepatitis B virus X expression vector transfected Hep3B/HepG2 cells with control plasmid transfected cells.
Association of p21 up-regulation by HBX A10R/S144R overexpression with p53 status
It was found that HBx restrained p21 expression in a p53-independent manner by interfering with Sp1 activity [35]. Hence, we detected whether HBX A10R/S144R mutation interfered p21 expression. First, we ran p21-luciferase report gene assay to identify the role of HBX A10R/S144R in p21 transactivation. The results showed that only HBX A10R/S144R double-mutation transactivated p21 promoter (Fig. 4A). p21-luciferase report gene activity was significantly higher in HepG2 cells than in HepG2-p53 null cells, suggesting that HBX A10R/S144R mutant transactivated p21 in p53-dependent and p53-independent ways. We further investigated how HBX A10R/S144R mutant regulated endogenous p21 transcription by detecting p21 mRNA expression. As shown in Fig. 4B, p21 and mRNA levels were up-regulated among wild type, Xcon, A10R, S144R and A10R/S144R overexpressed HepG2 and HepG2 p53 null cells. More importantly, the highest level of p21 expression was detected in both p53 null and p53 WT cells overexpressing A10R/S144R double mutations. Cyclin E and cyclin-dependent kinase (CDK2) are known to form a complex and contribute to cell cycle arrest. So we examined how overexpression of wild type, Xcon, A10R, S144R, and A10R/S144R HBx affected cyclin E and CDK2 expression using Western blot. The protein expression of p21 was also detected in this condition. As shown in Fig. 4C, p21 and CDK2 were increased whereas cyclin E was decreased in XD-transfected cells in both p53 wild type and p53 null HepG2 cells, indicating that A10R-S144R double mutation prolonged G1/S transition via its regulation of cell cycle arrest complex p21, cyclin E and CDK2.
Fig. 4.

Hepatitis B virus X A10R/S144R double mutation increased p21 expression in p53 independent manner. (A) p21 relative luciferase activity. Cells were co-transfected with pGL3-p21 promoter vector, pGL4.70-hRluc expression vector and hepatitis B virus X vectors. Luciferase assays were performed 36 hours post-transfection by using dual-Luciferase reporter assay system. (B) qRT-PCR analysis of p21 mRNA in hepatitis B virus X vectors treated H1299 cells, with GAPDH mRNA as internal control. (C) Immunoblot analysis of p21, cyclin E and cyclin-dependent kinase 2 expression in hepatitis B virus X vectors transfected cells.
HBx A10R/S144R double-mutation decreased Bax expression and inhibited apoptosis
We also explored the apoptotic function of HBx mutants. As shown in Fig. 5A, the percentage of apoptotic cells increased slightly in cells transfected with WT-HBx, Xcon, XA10R and XS144R, respectively, but decreased significantly in XD-transfected p53 null HepG2 cells (Fig. 5A, gray bars). However, the percentage of apoptotic cells decreased in all the wild-type HepG2 cells transfected with HBx-expressing plasmids, especially in XD-transfected cells (Fig. 5A, black bars). These results showed that in HepG2 and p53-null HepG2 cells, wild-type-, A10R- and S144R-mutated HBx had different effects on apoptosis, but XD-HBx expression definitively inhibited apoptosis independent of the p53 status. Further, we observed that Bax promoter transcription and mRNA expression were higher in cells transfected with wild-type and A10R-mutated HBx than with control plasmid, but decreased in cells transfected with S144R and XD HBx in both HepG2 and p53-null HepG2 cells (Fig. 5B and C). The same trend existed in Bax protein expression with Western blot analysis (Fig. 5D). HBx interacts with p53 [36,37], inhibiting p53 transcription activity [38] and p53-induced apoptosis [39]. HBx’s ability to sequester p53 can influence hepatocyte transformation and HCC development. This study showed that although HBx variants had slightly different effects on cell cycle and apoptosis in HepG2 and p53-null HepG2 cells, the outcomes of HBx variant-transfected cells had no substantial difference relevant to the p53 status. We performed immunoprecipitation to investigate interaction between HBx variants and p53 and found that the binding of A10R/S144R double-mutation to p53 decreased compared with WT-HBx (Fig. 5E). This reduced binding with p53 indicates that HBx A10R/S144R double-mutant-mediated apoptosis inhibition does not require p53 to participate. HBxA10R/S144R double-mutant decreased cell apoptosis of infected cells and facilitated tumor formation in p53 independent manner.
Fig. 5.

Hepatitis B virus X A10R/S144R double mutation decreased Bcl-2 associated X protein (Bax) expression and inhibited apoptosis. (A) The percentage of Annexin V+/PI+ cells in hepatitis B virus X vector transfected cells analyzed by flow cytometry; (B) Bcl-2 associated X protein relative luciferase activity. Cells were co-transfected with pGL3-Bax promoter vector, pGL4.70-hRluc expression vector and hepatitis B virus X vectors. Luciferase assays were performed 36 hours post-transfection by using dual-Luciferase reporter assay system; (C) qRT-PCR analysis of Bax mRNA in HBx vectors treated H1299 cells, with GAPDH mRNA as internal control. (D) Immunoblot analysis of Bcl-2 associated X protein expression in hepatitis B virus X vectors transfected cells. (E) HepG2 cells were co-transfected with hepatitis B virus X vectors and p53 vector. 48 hours later, the cell lysates were immunoprecipitated (IP) with anti-His antibody, separated on polyacrylamide gel electrophoresis (SDS–PAGE), transferred to polyvinylidene fluoride (PVDF) membrane and incubated with anti-p53 antibody followed by enhanced chemiluminescence detection.
Discussion
Mutations in the X region of HBV have been reported to be associated with progression of liver disease into cirrhosis and HCC [21,40]. In a meta-analysis, site mutations, including C1653T, T1753V, and A1762T/G1764A, were key predictors of HCC formation [34,41–43]. While functional changes caused by these mutations have been investigated [44–46], the previous investigations focused only on viral populations in blood [40,47–50]. Virus in local tissues may have more direct effects on tumor development. Integrated HBV DNA is more frequently associated with rearrangement of both viral and cellular genes, further affecting tumor development and progression [51,52].
This study detected a high frequency of K130M mutation and A1762T/G1764A double-mutation (approximately 2/3), consistent with previous reports [20,32,33]. But by using conventional data analysis method, no difference in the calculated amino acid frequency or selection between central- and para-tumor isolates was found, which is also consistent with previous study [53]. Analyses with statistical and machine learning tools revealed a difference in a novel HBx A10-S144 double mutation between central- and para-tumor isolates. These 2 sites also showed the highest entropy net differences. To our knowledge, this is the first report of A10-S144 double mutation. Only one study mentioned that site 144 was a novel HBx mutation associated with HCC development [54].
HBX’s functions in transcriptional regulation, DNA repair, cell cycle regulation, and apoptosis can be a potential mechanism of HCC formation [55–57]. This study revealed that A10R/S144R double-mutation arrested cell cycle and decreased apoptosis, which coordinate the timing of repair processes, accumulate genomic mutations, and facilitate HCC formation. An important mechanism of HBV-induced hepatocarcinogenesis is that HBx binds p53 and decreases p53-mediated transcription activation and p53-dependent apoptosis [38,39,58]. This study showed that A10R/S144R double-mutant binding with p53 was lower than WT-HBx, partly explaining why this double-mutant decreased p53-related apoptosis and indirectly demonstrating its relationship with HCC formation. While individual mutation of residue 10 or 144 had different effects on cell cycle regulation and apoptosis compared to WT-HBx, this double-mutation exerted a prominent effect, suggesting potential synergy between two mutations, which can be elucidated by structural study on HBx mutant-protein.
In summary, HBx10-144 double mutation was a novel signature sequence, which distinguished central- from para-tumor tissues, increased p21 expression, prolonged G1/S transition, and inhibited cell apoptosis. Further studies are needed to verify its functions on host HCC-related gene expression and define its roles in HCC formation, which likely shed light on the mechanisms of HCC development.
Acknowledgments
This research was supported by grants from the National Natural Science Foundation of China (81371399, 81571178, 81272266, 81071843, 81470098 and 81100288), CFAR Translational Virology Core (AI36214), HNRC Neurovirology Core (MH62512 and MH83552), Twelfth Key Science and Technology Five Year Plan of China (2012BAI15B08), and the Project of Construction of Innovative Teams and Teacher Career Development for Universities and Colleges under Beijing Municipality (IDHT20150502).
References
- 1.Nguyen VT, Law MG, Dore GJ. Hepatitis B-related hepatocellular carcinoma: epidemiological characteristics and disease burden. J Viral Hepat. 2009;16:453–463. doi: 10.1111/j.1365-2893.2009.01117.x. [DOI] [PubMed] [Google Scholar]
- 2.Chinese Society of Liver Cancer, Chinese Anti-Cancer Association; Chinese Society of Clinical Oncology, Chinese Anti-Cancer Association; Liver Cancer Study Group, Chinese Society of Hepatology, Chinese Medical Association; Chinese Pathological Group of Hepatobiliary Tumor and Liver Transplantation. Expert consensus on the scheme of pathological diagnosis of primary liver cancer. Zhonghua Gan Zang Bing Za Zhi. 2011;19:254–256. [PubMed] [Google Scholar]
- 3.Chan HL, Hui AY, Wong ML, Tse AM, Hung LC, Wong VW, et al. Genotype C hepatitis B virus infection is associated with an increased risk of hepatocellular carcinoma. Gut. 2004;53:1494–1498. doi: 10.1136/gut.2003.033324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kao JH, Chen PJ, Chen DS. Recent advances in the research of hepatitis B virus-related hepatocellular carcinoma: epidemiologic and molecular biological aspects. Adv Cancer Res. 2010;108:21–72. doi: 10.1016/B978-0-12-380888-2.00002-9. [DOI] [PubMed] [Google Scholar]
- 5.Lin CL, Kao JH. Hepatitis B virus genotypes and variants. Cold Spring Harb Perspect Med. 2015;5:a021436. doi: 10.1101/cshperspect.a021436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tsubota A, Arase Y, Ren F, Tanaka H, Ikeda K, Kumada H. Genotype may correlate with liver carcinogenesis and tumor characteristics in cirrhotic patients infected with hepatitis B virus subtype adw. J Med Virol. 2001;65:257–265. doi: 10.1002/jmv.2028. [DOI] [PubMed] [Google Scholar]
- 7.Yang HI, Yeh SH, Chen PJ, Iloeje UH, Jen CL, Su J, et al. Associations between hepatitis B virus genotype and mutants and the risk of hepatocellular carcinoma. J Natl Cancer Inst. 2008;100:1134–1143. doi: 10.1093/jnci/djn243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Berasain C, Castillo J, Perugorria MJ, Latasa MU, Prieto J, Avila MA. Inflammation and liver cancer: new molecular links. Ann N Y Acad Sci. 2009;1155:206–221. doi: 10.1111/j.1749-6632.2009.03704.x. [DOI] [PubMed] [Google Scholar]
- 9.Bertoletti A, Gehring A. Immune response and tolerance during chronic hepatitis B virus infection. Hepatol Res. 2007;37(Suppl 3):S331–S338. doi: 10.1111/j.1872-034X.2007.00221.x. [DOI] [PubMed] [Google Scholar]
- 10.Bonilla Guerrero R, Roberts LR. The role of hepatitis B virus integrations in the pathogenesis of human hepatocellular carcinoma. J Hepatol. 2005;42:760–777. doi: 10.1016/j.jhep.2005.02.005. [DOI] [PubMed] [Google Scholar]
- 11.Bouchard MJ, Navas-Martin S. Hepatitis B and C virus hepatocarcinogenesis: lessons learned and future challenges. Cancer Lett. 2011;305:123–143. doi: 10.1016/j.canlet.2010.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Feitelson MA, Lee J. Hepatitis B virus integration, fragile sites, and hepatocarcinogenesis. Cancer Lett. 2007;252:157–170. doi: 10.1016/j.canlet.2006.11.010. [DOI] [PubMed] [Google Scholar]
- 13.Paterlini P, Poussin K, Kew M, Franco D, Brechot C. Selective accumulation of the X transcript of hepatitis B virus in patients negative for hepatitis B surface antigen with hepatocellular carcinoma. Hepatology. 1995;21:313–321. [PubMed] [Google Scholar]
- 14.Unsal H, Yakicier C, Marcais C, Kew M, Volkmann M, Zentgraf H, et al. Genetic heterogeneity of hepatocellular carcinoma. Proc Natl Acad Sci USA. 1994;91:822–826. doi: 10.1073/pnas.91.2.822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kekule AS, Lauer U, Weiss L, Luber B, Hofschneider PH. Hepatitis B virus transactivator HBx uses a tumour promoter signalling pathway. Nature. 1993;361:742–745. doi: 10.1038/361742a0. [DOI] [PubMed] [Google Scholar]
- 16.Park US, Park SK, Lee YI, Park JG. Hepatitis B virus-X protein upregulates the expression of p21waf1/cip1 and prolongs G1→S transition via a p53-independent pathway in human hepatoma cells. Oncogene. 2000;19:3384–3394. doi: 10.1038/sj.onc.1203674. [DOI] [PubMed] [Google Scholar]
- 17.Diao J, Khine AA, Sarangi F, Hsu E, Iorio C, Tibbles LA, et al. X protein of hepatitis B virus inhibits Fas-mediated apoptosis and is associated with up-regulation of the SAPK/JNK pathway. J Biol Chem. 2001;276:8328–8340. doi: 10.1074/jbc.M006026200. [DOI] [PubMed] [Google Scholar]
- 18.Murakami S. Hepatitis B virus X protein: a multifunctional viral regulator. J Gastroenterol. 2001;36:651–660. doi: 10.1007/s005350170027. [DOI] [PubMed] [Google Scholar]
- 19.Fan W, Shi B, Wei H, Du G, Song S. Comparison of hepatitis B X gene mutation between patients with hepatocellular carcinoma and patients with chronic hepatitis B. Virus Genes. 2011;42:162–170. doi: 10.1007/s11262-010-0557-5. [DOI] [PubMed] [Google Scholar]
- 20.Lee JH, Han KH, Lee JM, Park JH, Kim HS. Impact of hepatitis B virus (HBV) x gene mutations on hepatocellular carcinoma development in chronic HBV infection. Clin Vaccine Immunol. 2011;18:914–921. doi: 10.1128/CVI.00474-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chen WN, Oon CJ, Leong AL, Koh S, Teng SW. Expression of integrated hepatitis B virus X variants in human hepatocellular carcinomas and its significance. Biochem Biophys Res Commun. 2000;276:885–892. doi: 10.1006/bbrc.2000.3562. [DOI] [PubMed] [Google Scholar]
- 22.Takahashi K, Akahane Y, Hino K, Ohta Y, Mishiro S. Hepatitis B virus genomic sequence in the circulation of hepatocellular carcinoma patients: comparative analysis of 40 full-length isolates. Arch Virol. 1998;143:2313–2326. doi: 10.1007/s007050050463. [DOI] [PubMed] [Google Scholar]
- 23.Liu X, Zhang S, Lin J, Feitelson MA, Gao H, Zhu M. Hepatitis B virus X protein mutants exhibit distinct biological activities in hepatoma Huh7 cells. Biochem Biophys Res Commun. 2008;373:643–647. doi: 10.1016/j.bbrc.2008.06.087. [DOI] [PubMed] [Google Scholar]
- 24.Cho SW, Shin YJ, Hahm KB, Jin JH, Kim YS, Kim JH, et al. Analysis of the precore and core promoter DNA sequence in liver tissues from patients with hepatocellular carcinoma. J Korean Med Sci. 1999;14:424–430. doi: 10.3346/jkms.1999.14.4.424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Choi JY, Hightower GK, Wong JK, Heaton R, Woods S, Grant I, et al. Genetic features of cerebrospinal fluid-derived subtype B HIV-1 tat. J Neurovirol. 2012;18:81–90. doi: 10.1007/s13365-011-0059-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Korber B, Myers G. Signature pattern analysis: a method for assessing viral sequence relatedness. AIDS Res Hum Retroviruses. 1992;8:1549–1560. doi: 10.1089/aid.1992.8.1549. [DOI] [PubMed] [Google Scholar]
- 27.Poon AF, Frost SD, Pond SL. Detecting signatures of selection from DNA sequences using Datamonkey. Methods Mol Biol. 2009;537:163–183. doi: 10.1007/978-1-59745-251-9_8. [DOI] [PubMed] [Google Scholar]
- 28.Reza FM. An Introduction to Information Theory. Dover; New York: 1994. [Google Scholar]
- 29.Frank E, Witten IH. Data Mining: Practical Machine Learning Tools and Techniques with Java Implementations. Morgan Kaufmann; San Francisco: 2000. [Google Scholar]
- 30.Quinlan JR. C4.5: Programs for Machine Learning. Morgan Kaufmann Publishers; San Mateo, CA: 1993. [Google Scholar]
- 31.Ahn JY, Jung EY, Kwun HJ, Lee CW, Sung YC, Jang KL. Dual effects of hepatitis B virus X protein on the regulation of cell-cycle control depending on the status of cellular p53. J Gen Virol. 2002;83:2765–2772. doi: 10.1099/0022-1317-83-11-2765. [DOI] [PubMed] [Google Scholar]
- 32.Iyer S, Groopman JD. Interaction of mutant hepatitis B X protein with p53 tumor suppressor protein affects both transcription and cell survival. Mol Carcinog. 2011;50:972–980. doi: 10.1002/mc.20767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Leon B, Taylor L, Vargas M, Luftig RB, Albertazzi F, Herrero L, et al. HBx M130K and V131I (T-A) mutations in HBV genotype F during a follow-up study in chronic carriers. Virol J. 2005;2:60. doi: 10.1186/1743-422X-2-60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tseng TC, Liu CJ, Yang HC, Chen CL, Yang WT, Tsai CS, et al. Higher proportion of viral basal core promoter mutant increases the risk of liver cirrhosis in hepatitis B carriers. Gut. 2015;64:292–302. doi: 10.1136/gutjnl-2014-306977. [DOI] [PubMed] [Google Scholar]
- 35.Ahn JY, Chung EY, Kwun HJ, Jang KL. Transcriptional repression of p21(waf1) promoter by hepatitis B virus X protein via a p53-independent pathway. Gene. 2001;275:163–168. doi: 10.1016/s0378-1119(01)00604-7. [DOI] [PubMed] [Google Scholar]
- 36.Feitelson MA, Zhu M, Duan LX, London WT. Hepatitis B x antigen and p53 are associated in vitro and in liver tissues from patients with primary hepatocellular carcinoma. Oncogene. 1993;8:1109–1117. [PubMed] [Google Scholar]
- 37.Wang XW, Forrester K, Yeh H, Feitelson MA, Gu JR, Harris CC. Hepatitis B virus X protein inhibits p53 sequence-specific DNA binding, transcriptional activity, and association with transcription factor ERCC3. Proc Natl Acad Sci USA. 1994;91:2230–2234. doi: 10.1073/pnas.91.6.2230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lee SG, Rho HM. Transcriptional repression of the human p53 gene by hepatitis B viral X protein. Oncogene. 2000;19:468–471. doi: 10.1038/sj.onc.1203312. [DOI] [PubMed] [Google Scholar]
- 39.Wang XW, Gibson MK, Vermeulen W, Yeh H, Forrester K, Sturzbecher HW, et al. Abrogation of p53-induced apoptosis by the hepatitis B virus X gene. Cancer Res. 1995;55:6012–6016. [PubMed] [Google Scholar]
- 40.Chen GG, Li MY, Ho RL, Chak EC, Lau WY, Lai PB. Identification of hepatitis B virus X gene mutation in Hong Kong patients with hepatocellular carcinoma. J Clin Virol. 2005;34:7–12. doi: 10.1016/j.jcv.2005.01.006. [DOI] [PubMed] [Google Scholar]
- 41.Kao JH, Chen PJ, Lai MY, Chen DS. Basal core promoter mutations of hepatitis B virus increase the risk of hepatocellular carcinoma in hepatitis B carriers. Gastroenterology. 2003;124:327–334. doi: 10.1053/gast.2003.50053. [DOI] [PubMed] [Google Scholar]
- 42.Liu S, Zhang H, Gu C, Yin J, He Y, Xie J, et al. Associations between hepatitis B virus mutations and the risk of hepatocellular carcinoma: a meta-analysis. J Natl Cancer Inst. 2009;101:1066–1082. doi: 10.1093/jnci/djp180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tatsukawa M, Takaki A, Shiraha H, Koike K, Iwasaki Y, Kobashi H, et al. Hepatitis B virus core promoter mutations G1613A and C1653T are significantly associated with hepatocellular carcinoma in genotype C HBV-infected patients. BMC Cancer. 2011;11:458. doi: 10.1186/1471-2407-11-458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fang ZL, Sabin CA, Dong BQ, Wei SC, Chen QY, Fang KX, et al. The association of HBV core promoter double mutations (A1762T and G1764A) with viral load differs between HBeAg positive and anti-HBe positive individuals: a longitudinal analysis. J Hepatol. 2009;50:273–280. doi: 10.1016/j.jhep.2008.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Li MS, Lau TC, Chan SK, Wong CH, Ng PK, Sung JJ, et al. The G1613A mutation in the HBV genome affects HBeAg expression and viral replication through altered core promoter activity. PLoS ONE. 2011;6:e21856. doi: 10.1371/journal.pone.0021856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Parekh S, Zoulim F, Ahn SH, Tsai A, Li J, Kawai S, et al. Genome replication, virion secretion, and e antigen expression of naturally occurring hepatitis B virus core promoter mutants. J Virol. 2003;77:6601–6612. doi: 10.1128/JVI.77.12.6601-6612.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jang JW, Chun JY, Park YM, Shin SK, Yoo W, Kim SO, et al. Mutational complex genotype of the hepatitis B virus X/precore regions as a novel predictive marker for hepatocellular carcinoma. Cancer Sci. 2012;103:296–304. doi: 10.1111/j.1349-7006.2011.02170.x. [DOI] [PubMed] [Google Scholar]
- 48.Kim JK, Chang HY, Lee JM, Baatarkhuu O, Yoon YJ, Park JY, et al. Specific mutations in the enhancer II/core promoter/precore regions of hepatitis B virus subgenotype C2 in Korean patients with hepatocellular carcinoma. J Med Virol. 2009;81:1002–1008. doi: 10.1002/jmv.21501. [DOI] [PubMed] [Google Scholar]
- 49.Liao Y, Hu X, Chen J, Cai B, Tang J, Ying B, et al. Precore mutation of hepatitis B virus may contribute to hepatocellular carcinoma risk: evidence from an updated meta-analysis. PLoS ONE. 2012;7:e38394. doi: 10.1371/journal.pone.0038394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lin CL, Kao JH. Perspectives and control of hepatitis B virus infection in Taiwan. J Formos Med Assoc. 2015;114:901–909. doi: 10.1016/j.jfma.2015.06.003. [DOI] [PubMed] [Google Scholar]
- 51.Koch S, von Loringhoven AF, Hofschneider PH, Koshy R. Amplification and rearrangement in hepatoma cell DNA associated with integrated hepatitis B virus DNA. EMBO J. 1984;3:2185–2189. doi: 10.1002/j.1460-2075.1984.tb02111.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Matsubara K, Tokino T. Integration of hepatitis B virus DNA and its implications for hepatocarcinogenesis. Mol Biol Med. 1990;7:243–260. [PubMed] [Google Scholar]
- 53.Iavarone M, Trabut JB, Delpuech O, Carnot F, Colombo M, Kremsdorf D, et al. Characterisation of hepatitis B virus X protein mutants in tumour and non-tumour liver cells using laser capture microdissection. J Hepatol. 2003;39:253–261. doi: 10.1016/s0168-8278(03)00217-4. [DOI] [PubMed] [Google Scholar]
- 54.Wang Q, Zhang T, Ye L, Wang W, Zhang X. Analysis of hepatitis B virus X gene (HBx) mutants in tissues of patients suffered from hepatocellular carcinoma in China. Cancer Epidemiol. 2012;36:369–374. doi: 10.1016/j.canep.2011.11.006. [DOI] [PubMed] [Google Scholar]
- 55.Clippinger AJ, Gearhart TL, Bouchard MJ. Hepatitis B virus X protein modulates apoptosis in primary rat hepatocytes by regulating both NF-kappaB and the mitochondrial permeability transition pore. J Virol. 2009;83:4718–4731. doi: 10.1128/JVI.02590-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wang HD, Trivedi A, Johnson DL. Regulation of RNA polymerase I-dependent promoters by the hepatitis B virus X protein via activated Ras and TATA-binding protein. Mol Cell Biol. 1998;18:7086–7094. doi: 10.1128/mcb.18.12.7086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yoo YG, Oh SH, Park ES, Cho H, Lee N, Park H, et al. Hepatitis B virus X protein enhances transcriptional activity of hypoxia-inducible factor-1alpha through activation of mitogen-activated protein kinase pathway. J Biol Chem. 2003;278:39076–39084. doi: 10.1074/jbc.M305101200. [DOI] [PubMed] [Google Scholar]
- 58.Cheng AS, Yu J, Lai PB, Chan HL, Sung JJ. COX-2 mediates hepatitis B virus X protein abrogation of p53-induced apoptosis. Biochem Biophys Res Commun. 2008;374:175–180. doi: 10.1016/j.bbrc.2008.06.098. [DOI] [PubMed] [Google Scholar]