

Abstract
Background and purpose
Data from both humans and animal models suggest that most recovery from motor impairment occurs in a sensitive period that lasts only weeks after stroke and is mediated in part by an increased responsiveness to training. Here we used a mouse model of focal cortical stroke to test two hypotheses. First we investigated if responsiveness to training decreases over time after stroke. Second, we tested whether fluoxetine, which can influence synaptic plasticity and stroke recovery, can prolong the period over which large training-related gains can be elicited after stroke.
Methods
Mice were trained to perform a skilled prehension task to an asymptotic level of performance after which they underwent stroke induction in the caudal forelimb area (CFA). The mice were then retrained after a 1-day or 7-day delay with and without fluoxetine.
Results
Recovery of prehension after a CFA stroke was complete if training was initiated one day after stroke but incomplete if it was delayed by 7 days. In contrast, if fluoxetine was administered at 24 hours after stroke, then complete recovery of prehension was observed even with the 7-day training delay. Fluoxetine appeared to mediate its beneficial effect by reducing inhibitory interneuron expression in intact premotor cortex rather than through effects on infarct volume or cell death.
Conclusions
There is a gradient of diminishing responsiveness to motor training over the first week after stroke. Fluoxetine can overcome this gradient and maintain maximal levels of responsiveness to training even 7 days after stroke.
Keywords: motor recovery, ischemic stroke, fluoxetine, post-stroke plasticity
Introduction
The largest amount of motor recovery at the both the impairment and functional levels occurs in the first 4 weeks after ischemic stroke both in humans1–4 and in rat models.5–8 We have previously referred to this period of spontaneous recovery and increased responsiveness to motor training as the post-stroke “sensitive period.”9 The sensitive period is a unique, time-limited environment of heightened plasticity characterized by molecular,8, 10 physiological,11, 12 and structural changes13, 14 that are qualitatively and quantitatively distinct to plasticity mechanisms in the absence of stroke or in the presence of a chronic stroke.6, 9 That there is a causal link between the unique short-lived plasticity milieu after stroke and the amount of recovery from hemiparesis in this same period is supported by rodent experiments that have manipulated plasticity in the sensitive period, for example by increasing15, 16 or decreasing BDNF,17 which augments or prevents recovery, respectively. Here we asked whether there might be a to augment or prolong the sensitive period. Specifically, is it possible to initiate training later after stroke but still maintain maximal responsiveness? This has great clinical relevance for those patients too medically ill to start intense rehabilitation immediately.
In a recent influential randomized placebo-controlled clinical trial, the selective serotonin reuptake inhibitor (SSRI), fluoxetine, was given within 10 days after stroke and then continued for 3 months.18 At 3 months, the group receiving fluoxetine had significantly enhanced recovery from motor impairment compared to the control group.18–20 A recent meta-analysis showed that post-stroke patients treated with SSRIs were less likely to be dependent, disabled, and/or neurologically impaired.21 The exact mechanism of how fluoxetine potentiates recovery is not known but is likely related to its ability to potentiate synaptic plasticity. For example, fluoxetine can restore time-dependent visual cortical plasticity in the adult rodent,22 likely through a decrease in the inhibition/excitation ratio,22, 23 and by stimulating gene expression important for plasticity.22, 24
Using a mouse model of focal motor stroke and a skilled prehension (reach-to-grasp) task,25 we first sought to demonstrate that there is diminished responsiveness to training when it is delayed by a week compared to when it is initiated 24 hours after stroke. We then tested the hypothesis that fluoxetine, when administered 24h after stroke, would maintain a maximal response to training even after a delay of a week between stroke induction and training.
Material and Methods
Mice
Adult male C57bl/6 mice 100 to 140 days old were singly housed in custom-made chambers and kept on a 12/12-hour light/dark cycle. A total of 70 mice were used; two animals were excluded from the analysis due to death prior to completion of the study (one receiving saline and one receiving fluoxetine). The specific number of animals for each behavioral experiment was chosen based on previously published experiments5, 25, 26 and “n” is reported in each figure legend. Three to four days prior to training on the prehension task, the mice were placed on scheduled administration of daily 2.0–3.0 g Bio-serv dustless precision pellet mouse chow with water ad libitum, with food restriction to 85% of their starting weight. All animal handling and use was performed according to and with approval from the Johns Hopkins University Animal Care and Use Committee.
Skilled prehension task
Training was conducted in an identical manner to that described before.25 Briefly, mice were trained in modified cages to reach for 45 mg dustless precision pellets (Bio-serv). Prehension was scored as successful when the mouse reached its forelimb through the slit, grabbed the pellet, and ate it without knocking it from its resting space, dropping it, or in any other way losing control. The percent of successful prehension attempts was determined per pellet. A training block consisted of 30 pellets at a distance of 1 cm with each pellet presented one at a time. After familiarization and paw determination, the animals underwent 2 blocks of 30 reaching attempts per training day. The animals had one training day off per week (including the day after stroke induction). Mice not undergoing training (days off) were treated just like the trained mice (allowed to run free in their home cage, were food restricted, fed the same pellets, and maintained on the same light/dark cycle) but were never exposed to the prehension task. Training after stroke began after a 1-day or 7-day delay after stroke induction. All investigators were blinded to training condition after stroke induction.
Fluoxetine
Mice were randomized after stroke induction to receive either fluoxetine (Tocris; prepared in sterile normal saline) 10 mg/kg daily or normal saline 5 ml/kg daily via intraperitoneal injection beginning either 24 hours or 7 days after stroke induction, i.e., the injections were always given 24 hours prior to the commencement of training. Investigators were blinded to stroke vs. sham condition as well as to fluoxetine vs. saline injection.
Stroke Induction
The location of motor areas was identified based upon prior anatomic27 and functional28 data. These data also indicate that these areas are geographically consistent within a given strain. We have used these with prior success and followed our previously published protocol.25 A fiber optic bundle of a cold light source (Zeiss 1500 electronic, Jena, Germany) with a 20 gauge aperture was centered at 2 mm lateral and 0.5 mm anterior from bregma for caudal forelimb area (CFA) infarction. The brains were then illuminated through the intact skull for 15 minutes, starting 5 minutes after the intraperitoneal injection of 150 μl of a 10 mg/mL rose Bengal solution in sterile normal saline. Animals undergoing sham had an identical procedure performed except that no illumination occurred.
Tissue preparation and histology
On the day of sacrifice, the mice were placed under deep anethesia with 2.5% avertin and transcardially perfused with 4% paraformaldehyde (PFA) in 0.1 M sodium phosphate, pH7.4. The brains were dissected out and placed in 4% PFA for 24 hours. Brains were coronally sliced at 50 μm on a vibrating microtome. Free-floating sections were washed 3 × 5 minutes in PBS and subsequently stained in one of four different ways.
Cresyl violet staining: slices were stained in 0.1% Cresyl violet with 0.25% glacial acetic acid followed by dehydration in graded alcohols. Slices were mounted in Permount (Fischer Chemical).
Immunofluorescent preparation: slices were placed for 4 hours in block solution (10% normal goat serum and 0.04% triton X-100, in tris buffered saline) followed by overnight incubation at 4°C with primary antibody diluted in block solution. Sigma primary antibodies diluted at 1:1000 included anti-calbindin (monoclonal goat anti-mouse), anti-calretinin (polyclonal goat anti-rabbit), and anti-parvalbumin (monoclonal goat anti-mouse). Sections were subsequently washed 3 × 5 minutes in tris buffered saline with 0.04% triton and incubated at room temperature for 4 hours with secondary antibodies (Alexa goat-anti-rabbit 633 diluted 1:500; and goat-anti-mouse 488 diluted 1:250). Sections were washed 2 × 5minutes in tris buffered saline + triton ×100, 1 × 5 min tris buffered saline, and mounted in ProLong Gold reagent (Invitrogen).
Fluoro-Jade C preparation (Histo-Chem Inc): sections were mounted onto superfrost slides (Fischer Chemical) and allowed to dry overnight. Sections were the treated per Hisot-Chem Inc protocol.
Quantification of stroke volume
From brain slices prepared with Cresyl violet at the indicated times of sacrifice, the entire anterior-posterior extent of the CFA contralateral to the preferred paw was imaged and reconstructed in 3 dimensions using Imaris (Bitplane) imaging software. An investigator blinded to conditions demarcated the stroke pathology and volumes were calculated using Imaris (Bitplane) imaging software.
Inhibitory interneuron counts
Using coronal sections, we defined counting areas anatomically (and not based on visually perceived differences of marker expression) based on previous definitions of the boundaries of medial agranular cortex (AGm)25, 28 and medial frontal cortex (mFC).27, 29 For AGm, we defined a medial boundary from which we extended a 1.2 mm2 area slice from the medial and dorsal pial boundaries; for mFC we extended a 1.2 mm2 area slice from the midline pial boundary. These areas represent subareas of AGm and mFC, which prevented us from confounding our counts with cells from neighboring areas. The entire extent of a 50 μm slice was then imaged at 2 μm intervals using Zeiss Apotome technology to precisely localize cells30 and the resulting data was reconstructed in 3 dimensions using Imaris (Bitplane) imaging software to create a 60 mm3 volume obtained in an unbiased manner (please see supplementary figures). Such volumes were taken from each animal both ipsilesional and contralesional to the stroke. An investigator blinded to the experimental condition counted the number of cells immunofluorescently labeled with PV, CR, or CB within this volume. A cell was counted as positive if it had any immunofluorescent label for the indicated marker.
Analysis of cell death
Seven days after CFA stroke induction, tissue was prepared for Fluoro-Jade C analysis as described above. The entire anterior-posterior extent of the CFA stroke was analyzed and sections representing the largest medial-lateral area were imaged (thus representing the central core and largest area of stroke damage). The entire extent of 50 μm slices were then imaged at 2 μm intervals using Zeiss Apotome technology to precisely localize puncta30 and the resulting data was reconstructed in 3 dimensions using Imaris (Bitplane) imaging software. An investigator blinded to the experimental condition counted the number of Fluoro-Jade C positive cells. A cell was counted as positive if it had any immunofluorescent label for Fluoro-Jade C. The volume of the stroke was used to normalize the Fluoro-Jade C counts.
Statistics
Behavioral data were analyzed with both generalized and linear mixed effect models.31, 32 Separate models were fit before and after stroke induction. The basic linear model was Yit=β0+Ui+β1Gi+β2t+β3×Gi×t+εi, where the Yit is the percentage of correct grabs for mouse i on day t, G_i is group status (fluoxetine versus saline), Ui is a mouse-specific random intercept. These are assumed to be independent and identically distributed Gaussian random variables and εi are normally distributed random errors. Both natural scale and logit transformed versions of the model were considered. In addition to this model we considered a generalized linear mixed model Cit|Ui~Binomial(pit, ni) where Ci was the count of the correct grabs for mouse i on day t out of n_i trials. The model assumed that logit(pit)=γ0+Ui+γ1Gi+γ2t+γ3×Gi×t where logit(pit) is the natural logarithm of the odds of a correct reach for mouse i on day t. In both models the random intercept and intercept terms account for any mouse- and group-specific differences at either baseline or after training. As the results were in agreement, we report the results of the simpler linear mixed effect model with no transformations.
Stroke volume data were analyzed using GraphPad Prism’s 2-way ANOVA with correction for multiple comparisons. Immunofluorescent data were analyzed using GraphPad Prism’s 2-way ANOVA with Tukey’s posttest. Cell death data were analyzed using GraphPad Prism’s 2-way t-test (not assuming a Gaussian distribution).
Results
Recovery of prehension was incomplete when training was delayed
In a previous study we showed that focal stroke in CFA, contralateral to the preferred paw, led to a large decrement in skilled prehension, which recovered to normal pre-stroke levels if training was initiated after a one-day delay and continued for 7 days.25 Here, as a control, we reproduced this result in mice receiving daily saline injections (the fluoxetine results will be given in the next section). Specifically, wild-type mice were trained to perform the skilled prehension task and reached asymptotic performance after 6 to 8 training days. An approximate 0.25 mm3 focal stroke in CFA contralateral to the paw used for prehension led to a large decrement in prehension accuracy, which recovered fully to pre-stroke levels when training was started after a 1-day delay (Figure 1A).
Figure 1.
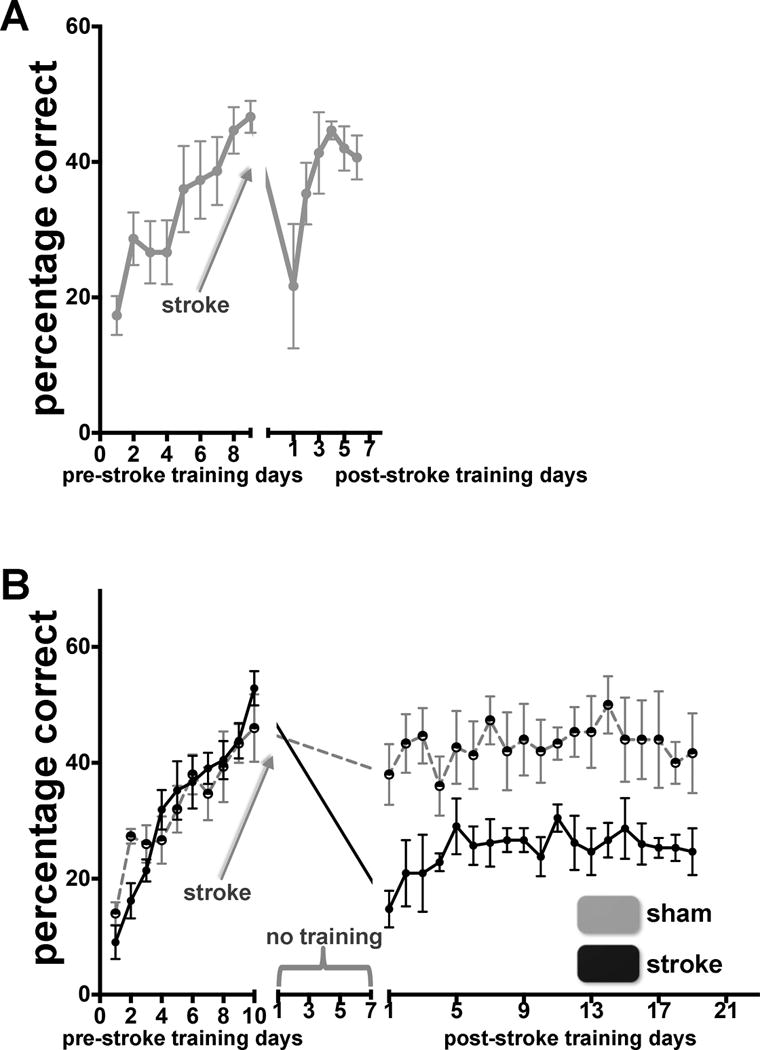
There is a temporal period sensitive to motor training after CFA stroke. (A) Skilled prehension recovers after CFA infarction with daily saline injection beginning 24 hours after stroke coupled with training beginning after a 1-day post-stroke delay. Prehension success (mean +/− SEM) for mice undergoing CFA stroke. n=5. (B) Skilled prehension displays suboptimal recovery after a 7-day delay. Reaching success (mean +/− SEM) for mice undergoing CFA stroke and training beginning after 7-day post-stroke delay (sham stroke – gray n = 5; stroke – black n = 7).
To examine the effect a training delay would have on motor recovery, we trained wild-type mice to perform the skilled prehension task, induced a focal CFA infarction, and then had the mice remain in their home-cages for 7 days, free to move about but without prehension training. Assessment on the prehension task on post-stroke day 8, revealed that there was little spontaneous recovery of performance (Figure 1B). Starting training on the prehension task on post-stroke day 8 did lead to performance gains but these were markedly lower than for mice that began training after only a 1-day delay (Figure 1A).25 Mice that underwent sham procedures had the same performance level before and after the delay, which indicates that the 7-day delay itself did not lead to a decrement in performance via forgetting. To summarize, there was a return to normal pre-stroke performance on the prehension task if training was started within 48 hours but this was not the case if initiation of training was delayed by a week; the mice improved by a small amount then hit a plateau.
Fluoxetine increased responsiveness to delayed training
The main hypothesis of this study was that fluoxetine would improve motor recovery by maintaining the post-stroke sensitive period. To test this, we administered daily IP injections of fluoxetine beginning 24 hours after CFA stroke. The experiments with fluoxetine duplicated those described above with saline. The group that began training after a 1-day post-stroke delay showed a return to pre-stroke levels of prehension, as was seen in the analogous saline group (Figure 2A). The critical difference between saline and fluoxetine groups became apparent when training was delayed by a week: only the fluoxetine group showed a return to pre-stroke levels of prehension (Figure 2B). Specifically, before stroke induction there was an estimated 3.4 percentage point increase per day in correct reaches (standard error 0.42 with a highly significant p-value 1e-13). The saline group was an estimated 0.7 percentage points lower at baseline (non-significant p-value of 0.839) but there was no significant difference in the slope between the saline and fluoxetine groups (p-value 0.6). After stroke induction, the saline group started an estimated 2 percentage points lower on correct reaches (p-value 0.6). The fluoxetine group gained an estimated 4.5 percentage points of correct reaches per day while the saline group gained an estimated 1.7 per day. The estimated difference in slopes (2.8 percentage points per day) was highly statistically significant (standard error 0.97, p = 0.005).
Figure 2.

Fluoxetine rescued the mice from the only minimal gains expected when training is delayed by 7 days. (A) Skilled prehension recovers after focal CFA infarction with daily fluoxetine injections beginning 24 hours after stroke coupled with training beginning after a 1-day post-stroke delay. Reaching success (mean +/− SEM) for mice undergoing CFA stroke. n = 5. Saline injected mice (re-graphed from Figure 1A) shown for comparison. (B) Fluoxetine administration beginning 24 hours after CFA stroke extends the time window for recovery. Reaching success (mean +/− SEM) for mice undergoing CFA stroke and training beginning after a 7-day post-stroke delay. Mice received daily injections of saline (gray n = 8) or fluoxetine (black n = 9). (C) Fluoxetine administration 7 days after CFA stroke does NOT lead to improved recovery. Reaching success (mean +/− SEM) for mice undergoing CFA stroke and training beginning after a 7-day post-stroke delay. Mice received daily injections of saline (gray n = 10) or fluoxetine (black n = 9) beginning 7 days after the stroke (24 hours prior to training). Behavioral statistics are reported in material and methods.
To test if the timing of fluoxetine administration itself has an effect on recovery, we performed the same 7-day post-stroke delay-experiment but with the difference that fluoxetine was only started on day 7 post-stroke (Figure 2C). The learning effect prior to stroke was a little lower for the fluoxetine group (1.77 increase in percent correct per day versus 2.41 increase per day for saline). After stroke the fluoxetine group learned at a rate of 0.98 percentage points per day while the saline group learned at 0.732 (p-value of 0.60 for the comparison in the groups). Thus the mice that received fluoxetine 7 days after stroke never returned to pre-stroke levels of prehension and looked similar to the saline group.
In summary, if fluoxetine was given 24 hours after stroke then recovery occurred to pre-stroke levels whether training was begun after a 1-day or 7-day post-stroke delay. This apparent fluoxetine-induced maintenance of maximal responsiveness to training over a delay only occurred if the drug was administered early.
Fluoxetine administration after stroke did not reduce stroke volume or neuronal death
Fluoxetine has been shown to be neuroprotective if given within hours after ischemic stroke.33–35 To determine if fluoxetine administration had an effect on neuronal death and/or infarct volume, which could have confounded our results, we performed two controls. First, to assess neuronal survival within the stroke core, we used Fluoro-Jade C staining, which makes use of a cationic fluorescent dye empirically demonstrated to bind to degenerating neuron cell bodies, dendrites, and axons after tissue fixation and provides a reliable and quantifiable index for assessing neuronal damage.36, 37 We assessed for neuronal death at 7 days after stroke. We chose this time point because 1) it would reflect a pure effect of the drug before the onset of training and 2) Fluoro-Jade C staining remains positive for at least 7 days. Quantification showed that there were increased Fluoro-jade C puncta in mice receiving daily fluoxetine administration beginning 24 hours after stroke compared to mice receiving daily saline (Figure 3A).
Figure 3.

Fluoxetine administration at 1 and 7 days does not reduce cell degeneration or infarct volume. (A) Mice receiving fluoxetine injections beginning 1-day after CFA stroke demonstrated increased neuronal degradation compared to mice receiving saline injections beginning 1-day after CFA stroke as indicated by Flouro-jade C quantification. n = 6 for both conditions; * p = 0.0043. (B) There is no significant difference between CFA infarct volumes in mice receiving training beginning after a 1-day (train early) or 7-day (train late) post-stroke delay coupled with either saline or fluoxetine injections beginning 1-day (early) or 7-days (late) after stroke or in mice with 7-day delayed retraining and no drug administration. Number (n) of mice used for volumetric analyses are listed.
As a second control, mice were sacrificed within 2 hours of their last training session and stroke volumes were measured. Regardless of timing, there were no significant stroke volume differences between animals receiving no drug (i.e. the animals graphed in Figure 1B), animals receiving daily fluoxetine, or animals receiving daily saline (Figure 3B). Combined, these data indicate that fluoxetine’s beneficial effect on post-stroke recovery mechanisms is not attributable to either a neuroprotective effect the first week after stroke (in fact, there was more neuronal death in the fluoxetine group) or stroke volume reduction.
Fluoxetine administration after stroke decreased inhibitory marker expression in a medial premotor area
We have previously demonstrated that medial premotor cortex (AGm) mediates recovery after a stroke in motor cortex and that this is associated with decreases in parvalbumin (PV), calbindin (CB), and calretinin (CR) expression in this area. This suggests that modulation of region-specific excitatory/inhibitory balance is important for recovery.25 To test if the observed effects of fluoxetine occur through a similar mechanism, we performed blinded quantification of cells expressing PV, CB, and CR in AGm seven days after CFA stroke. There was a statistically significant decrease in ipsilesional AGm PV expression (as well as a trend towards decreased CB expression) in animals that received daily fluoxetine beginning 24 hours after stroke as compared with contralesional AGm and compared to animals receiving daily saline injection beginning 24 hours after stroke (Figure 4). Alternatively, however, an increase in contralesional AGm PV expression could have just given the impression of decreased ipsilesional AGm PV expression. To rule out this possibility, we performed a comparison to historical controls in which PV expression was measured in mice who underwent sham stroke and subsequent prehension training.25 There was a significant decrease in ipsilesional AGm PV expression in animals with stroke (23.27 mm3 +/− 0.81) versus sham stroke (31.56 mm3 +/− 3.7), which indicates that PV expression ispsilesional to stroke in animals treated with fluoxetine was indeed lower than baseline. A potential objection, however, is that the animals in the sham group underwent post-stroke prehension training whereas as the animals in the stroke + fluoxetine group did not, raising the possibility that training alone may lead to a generalized increase in PV expression. However, we find this highly unlikely since 1) previous studies have shown decreases and not increases in PV expression after learning in normal animals. A training-induced decrease in PV expression in AGm would make our claim even stronger, as fluoxetine would have needed to decreased this expression even further. 2) Other studies have demonstrated a similar number of PV-positive neurons in normal motor cortex as we saw in the sham stroke group.38 Fluoro-Jade C staining of AGm revealed no immunofluorescence, which indicates that there was no death of PV interneurons (data not shown).
Figure 4.

fluoxetine administration after stroke is associated with decreased inhibitory interneuron marker expression in medial premotor cortex (AGm). (A) Schematic of CFA stroke and the imaged medial premotor cortex (AGm) volumes (box) of animals undergoing stroke with daily fluoxetine (A1) or saline (A2) injections beginning 24 hours after stroke; schematic of mFC (A3) which is approximately 1.3 mm anterior of the imaged motor regions. Representative immunofluorescent images labeled with PV (B1, B2), CB (C1, C2), or CR (D1, D2) in animals receiving fluoxetine (B1, C1, D1) or saline (B2, C2, D2) both contra- and ipsi-lesional relative to CFA stroke. (B3, C3, D3) Number of PV (B3), CB (C3), and CR (D3) positive neurons per 107 μm3 in the AGm. Ipsi refers to cortex ipsilesional to stroke and contralateral to the reaching paw; contra refers to cortex contralesional to stroke and ipsilateral to the reaching paw. (** p=0.001 compared relative to ipsilesional cortex in animals receiving fluoxetine; no other comparison was statistically significant). n = 6 for all conditions. Scale bars, 200 μm.
Prior work has shown that 3 weeks of fluoxetine administration leads to decreased PV expression in the mFC.29 However, seven days of fluoxetine or saline administration after stroke revealed no change in the number of cells expressing PV in the mFC either ipsilesional or contralesional to CFA stroke (data not shown). These data indicate that post-stroke fluoxetine administration alters PV expression in the ipsilesional premotor area and this change is anatomically specific. There was no motor training in any of the conditions tested.
Discussion
Using a mouse stroke model, we first showed that there is a gradient of diminishing responsiveness to training within the first week after CFA stroke. We then showed that daily fluoxetine administration post-stroke mitigated this gradient and was associated with full recovery of prehension accuracy even if training was delayed by a week. Fluoxetine was associated with decreased medial premotor inhibitory interneuron expression but did not reduce either stroke volume or neuronal death.
The post-stroke sensitive period
Here we demonstrated the existence of a post-stroke sensitive period in mice, although our data do not indicate when this sensitive period ends, as motor training after 7 days still led to a small increase in post-stroke motor performance that then reached plateau despite 19 days of training. This lack of responsiveness to further training is consistent with studies in the rat that show no benefit of later tune-ups despite a response to training earlier after stroke.39 That the impact of training falls off rapidly within one week post-stroke is also consistent with results in rats showing only a modest response to training and enrichment begun after 2 weeks post-stroke, compared to when begun early.5
It is possible that increasing the dose of training per day (intensity) could have led to further gains even with a delay of 7 days. The idea that there is a threshold of intensity for any given level of plasticity, below which there will be no behavioral response, is supported by the finding in rats that when training was begun 5 days post-stroke, 140 reaches per day led to no improvement but 240 reaches did.40 The existence of such a dose by time interaction is entirely consistent with the existence of a window within which there is a gradient of diminishing responsiveness to a fixed amount of training.
Human data also suggest a short-lived plasticity window after stroke, with most spontaneous recovery occurring in the first 3 months.2, 41 More recently, we have shown that the degree of recovery from impairment at 3 months follows a predictable proportionality rule: most patients, except a subset of patients with severe hemiparesis, regain approximately 70% of their maximal potential recovery from their initial impairment level.1, 42
There is increasing evidence that there are qualitative and quantitative differences in the cortical milieu when training occurs in the post-stroke sensitive period as compared to similar training in healthy subjects or in chronic stroke (reviewed in Zeiler and Krakauer).9 For example, multiple studies of the cortical environment early after stroke have demonstrated evidence for a shift in the cortical excitatory-inhibitory balance,43 either through an increase in excitability44–46 or a decrease in inhibition.25, 45, 47 Moreover, direct manipulation of excitatory-inhibitory balance in rodents promotes axonal and dendritic growth, and enhances recovery after stroke.11, 48 These data suggest that an initial reduction in inhibitory tone (especially phasic inhibition) enhances plasticity but then normalizes, which limits further large-scale cortical reorganization in response to sensory input or training.49, 50 Time-dependent changes in both glial scar and in molecules that regulate regrowth of axons through damaged tissue are also likely to be important.51, 52
The interaction between fluoxetine and the post-stroke sensitive period
Here we found that fluoxetine given 24 hours after stroke rescued post-stroke mice from the only minimal gains expected when training is delayed by 7 days. As in a previous report, this rescue did not occur if fluoxetine was only started at 7 days,53 which suggests that its effect is itself dependent on conditions in the sensitive period. Although earlier work has suggested a neuroprotective effect for SSRIs, especially within hours of an ischemic insult,33, 34, 54, 55 we found no evidence for either reduced infarct volume or reduced cell death.
Early after stroke, mice receiving daily fluoxetine performed worse compared to mice receiving saline (Figure 2A). This is similar to prior studies showing acute fluoxetine administration leads to poorer performance on motor and spatial learning tests56 and could be related to fluoxetine’s effects on non-hippocampal dependent learning mechanisms.57 It is also possible that the increased cell death in mice receiving fluoxetine (Figure 3A) played a role in the initial poorer performance. Although most studies have indicated that SSRIs tend to potentiate cell survival, a few have demonstrated fluoxetine-induced growth factor reduction58 as well as SSRI-induced apoptosis, especially of diseased cells,59–62 perhaps by potentiation of the immune system.63 Nevertheless, despite increased cell death, animals receiving fluoxetine eventually outperformed animals receiving saline. This is similar to prior reports in which rehabilitation-induced increases in stroke volume were associated with improved behavioral outcomes64, 65 reflecting a possible pruning effect, whereby energy-compromised neurons are eliminated early on as a result of use-dependent activation associated with rehabilitation.6
There is accumulating evidence that SSRIs enhance central nervous system plasticity mechanisms.66 An intriguing mechanism that could link fluoxetine and stroke recovery is SSRI-induced disinhibition.22, 23, 29, 67, 68 In prior work, we have shown a correlation between training-induced recovery from CFA stroke and a reduction of PV expression in a medial motor area.25 We conclude that decreased PV expression indicates a reduction in inhibition based on the following: 1) The changes were region specific, occurring only in relevant motor areas and not in mFC (which is a non-motor association cortex), 2) The changes occurred in the absence of cell death, and 3) PV immunoreactivity marks the activity of an inhibitory interneuron.69, 70 This conclusion, however, remains conjecture in the absence of direct physiological measurements.
Fluoxetine administration led to a decrease in post-stroke PV expression in the ipsilesional medial premotor cortex in the absence of training but the behavioral benefit only manifested with subsequent training. Prior studies suggest that pharmacological interventions must be paired with training to induce behavioral changes.18, 71–74 For example, fluoxetine enhances plasticity in fear circuits when combined with environmental experience, reshaping behavioral responses.75 The influence of fluoxetine on inhibition may operate directy or indirectly, for example via BDNF.76, 77
It is unlikely that the effect of fluoxetine reported here is attributable to an effect on mood, as this would predict a benefit regardless of when it was administered, which was not what was observed. Furthermore, our data are consistent with previous reports showing that fluoxetine administration for 7 days, and at dosages similar to those used in our study, is not associated with changes in mood in rodents.78–80
The lack of efficacy of fluoxetine when started at 7 days post-stroke is not likely attributable to ischemia’s effect on the blood brain barrier. This is because it appears that fluoxetine acts to promote reorganization in undamaged cortical areas (e.g. the medial premotor area); fluoxetine has good blood brain barrier penetration in the absence of injury81–83 and can produce a similar decrease in PV in uninjured wild-type mice29. Most importantly, our main hypothesis was that the lack of responsiveness to training at 7 days can be reversed by fluoxetine given on day 1 post-stroke. That this did not happen when it was given at 7 days does not detract from the principal demonstration that the temporal gradient of decreasing responsiveness to training was reversible.
Comparison to motor recovery in humans
It is of interest to compare and contrast our results in mice with the FLAME study in humans. Patients that received fluoxetine by post-stroke day 5–10 showed significantly greater recovery from motor impairment at 3 months than patients who received placebo.18 A key methodological difference between our mouse study versus the FLAME study is that we pre-trained our animals on the task with which their post-stroke recovery was subsequently assessed, whereas in FLAME, patients were assessed with the Fugl-Meyer Scale, on which they were not pre-trained. Why did the mice not spontaneously recover prehension with fluoxetine but the patients got better without training on the assessment task? There are a number of possibilities. First, rodents do spontaneously recover natural behaviors.5 It could be argued that prehension is overlearned in humans whereas it is a difficult and novel behavior for rodents. Second, the Fugl-Meyer scale can be contaminated by strength improvements.84 Third, patients in FLAME received rehabilitative therapy, which might overlap with components of the Fugl-Meyer Scale. It is of further interest that if one calculates expected proportional recovery1 of 70% of maximal potential Fugl-Meyer in the fluoxetine and placebo groups in the FLAME study, the fluoxetine group on average showed full proportional recovery, which suggests fluoxetine nudged them into full spontaneous recovery.
Equating the sensitive period in rodents and humans is difficult due to size, metabolism, and central nervous system differences. Nevertheless, comparison of human1, 2, 41 and rodent data5 suggest that the rodent sensitive period closes at least three times faster than that in humans, which is good news as it suggests that there is more time available to intervene in patients. Overall, existing data provide compelling evidence for a post-stroke sensitive period in both humans and in rodent models. Future electrophysiological studies will be required to demontrate decreased inhibition in premotor areas after a stroke in primary motor cortex. Prolonging the post-sensitive period pharmacologically, as was done here in mice, holds great promise for human neurorehabilitation.
Supplementary Material
Acknowledgments
Not applicable.
Sources of funding
Dr S.R.Z. is currently supported by the Richard S. Ross Clinician Scientist Award, a startup fund from the Johns Hopkins department of Neurology, as well as K08 NS085033-01. Dr J.W.K. is currently supported by R01 NS052804– 05, R01 120 86264, R01 HD073147, James S. McDonnell Foundation 220020220, and NSF 1358756. Imaris (Bitplane) imaging software was supported via grant NG050274.
Footnotes
Disclosures
The authors have no conflicts of interest to disclose.
References
- 1.Prabhakaran S, Zarahn E, Riley C, Speizer A, Chong JY, Lazar RM, et al. Inter-individual variability in the capacity for motor recovery after ischemic stroke. Neurorehabil Neural Repair. 2008;22:64–71. doi: 10.1177/1545968307305302. [DOI] [PubMed] [Google Scholar]
- 2.Jorgensen HS, Nakayama H, Raaschou HO, Olsen TS. Stroke. Neurologic and functional recovery the copenhagen stroke study. Phys Med Rehabil Clin N Am. 1999;10:887–906. [PubMed] [Google Scholar]
- 3.Hankey GJ, Spiesser J, Hakimi Z, Bego G, Carita P, Gabriel S. Rate, degree, and predictors of recovery from disability following ischemic stroke. Neurology. 2007;68:1583–1587. doi: 10.1212/01.wnl.0000260967.77422.97. [DOI] [PubMed] [Google Scholar]
- 4.Duncan PW, Goldstein LB, Matchar D, Divine GW, Feussner J. Measurement of motor recovery after stroke. Outcome assessment and sample size requirements. Stroke. 1992;23:1084–1089. doi: 10.1161/01.str.23.8.1084. [DOI] [PubMed] [Google Scholar]
- 5.Biernaskie J, Chernenko G, Corbett D. Efficacy of rehabilitative experience declines with time after focal ischemic brain injury. J Neurosci. 2004;24:1245–1254. doi: 10.1523/JNEUROSCI.3834-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Krakauer JW, Carmichael ST, Corbett D, Wittenberg GF. Getting neurorehabilitation right: What can be learned from animal models? Neurorehabil Neural Repair. 2012;26:923–931. doi: 10.1177/1545968312440745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Biernaskie J, Corbett D. Enriched rehabilitative training promotes improved forelimb motor function and enhanced dendritic growth after focal ischemic injury. J Neurosci. 2001;21:5272–5280. doi: 10.1523/JNEUROSCI.21-14-05272.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Li S, Overman JJ, Katsman D, Kozlov SV, Donnelly CJ, Twiss JL, et al. An age-related sprouting transcriptome provides molecular control of axonal sprouting after stroke. Nature neuroscience. 2010;13:1496–1504. doi: 10.1038/nn.2674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zeiler SR, Krakauer JW. The interaction between training and plasticity in the poststroke brain. Curr Opin Neurol. 2013;26:609–616. doi: 10.1097/WCO.0000000000000025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Urban ET, 3rd, Bury SD, Barbay HS, Guggenmos DJ, Dong Y, Nudo RJ. Gene expression changes of interconnected spared cortical neurons 7 days after ischemic infarct of the primary motor cortex in the rat. Molecular and cellular biochemistry. 2012;369:267–286. doi: 10.1007/s11010-012-1390-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Clarkson AN, Huang BS, Macisaac SE, Mody I, Carmichael ST. Reducing excessive gaba-mediated tonic inhibition promotes functional recovery after stroke. Nature. 2010;468:305–309. doi: 10.1038/nature09511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Manganotti P, Acler M, Zanette GP, Smania N, Fiaschi A. Motor cortical disinhibition during early and late recovery after stroke. Neurorehabil Neural Repair. 2008;22:396–403. doi: 10.1177/1545968307313505. [DOI] [PubMed] [Google Scholar]
- 13.Clarkson AN, Lopez-Valdes HE, Overman JJ, Charles AC, Brennan KC, Thomas Carmichael S. Multimodal examination of structural and functional remapping in the mouse photothrombotic stroke model. J Cereb Blood Flow Metab. 2013;33:716–723. doi: 10.1038/jcbfm.2013.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hinman JD, Rasband MN, Carmichael ST. Remodeling of the axon initial segment after focal cortical and white matter stroke. Stroke. 2013;44:182–189. doi: 10.1161/STROKEAHA.112.668749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Muller HD, Hanumanthiah KM, Diederich K, Schwab S, Schabitz WR, Sommer C. Brain-derived neurotrophic factor but not forced arm use improves long-term outcome after photothrombotic stroke and transiently upregulates binding densities of excitatory glutamate receptors in the rat brain. Stroke. 2008;39:1012–1021. doi: 10.1161/STROKEAHA.107.495069. [DOI] [PubMed] [Google Scholar]
- 16.Schabitz WR, Berger C, Kollmar R, Seitz M, Tanay E, Kiessling M, et al. Effect of brain-derived neurotrophic factor treatment and forced arm use on functional motor recovery after small cortical ischemia. Stroke. 2004;35:992–997. doi: 10.1161/01.STR.0000119754.85848.0D. [DOI] [PubMed] [Google Scholar]
- 17.Ploughman M, Windle V, MacLellan CL, White N, Dore JJ, Corbett D. Brain-derived neurotrophic factor contributes to recovery of skilled reaching after focal ischemia in rats. Stroke. 2009;40:1490–1495. doi: 10.1161/STROKEAHA.108.531806. [DOI] [PubMed] [Google Scholar]
- 18.Chollet F, Tardy J, Albucher JF, Thalamas C, Berard E, Lamy C, et al. Fluoxetine for motor recovery after acute ischaemic stroke (flame): A randomised placebo-controlled trial. Lancet Neurol. 2011;10:123–130. doi: 10.1016/S1474-4422(10)70314-8. [DOI] [PubMed] [Google Scholar]
- 19.Acler M, Robol E, Fiaschi A, Manganotti P. A double blind placebo rct to investigate the effects of serotonergic modulation on brain excitability and motor recovery in stroke patients. Journal of neurology. 2009;256:1152–1158. doi: 10.1007/s00415-009-5093-7. [DOI] [PubMed] [Google Scholar]
- 20.Mikami K, Jorge RE, Adams HP, Jr, Davis PH, Leira EC, Jang M, et al. Effect of antidepressants on the course of disability following stroke. The American journal of geriatric psychiatry: official journal of the American Association for Geriatric Psychiatry. 2011;19:1007–1015. doi: 10.1097/JGP.0b013e31821181b0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mead GE, Hsieh CF, Lee R, Kutlubaev M, Claxton A, Hankey GJ, et al. Selective serotonin reuptake inhibitors for stroke recovery: A systematic review and meta-analysis. Stroke. 2013;44:844–850. doi: 10.1161/STROKEAHA.112.673947. [DOI] [PubMed] [Google Scholar]
- 22.Maya Vetencourt JF, Sale A, Viegi A, Baroncelli L, De Pasquale R, O’Leary OF, et al. The antidepressant fluoxetine restores plasticity in the adult visual cortex. Science. 2008;320:385–388. doi: 10.1126/science.1150516. [DOI] [PubMed] [Google Scholar]
- 23.Mendez P, Pazienti A, Szabo G, Bacci A. Direct alteration of a specific inhibitory circuit of the hippocampus by antidepressants. J Neurosci. 2012;32:16616–16628. doi: 10.1523/JNEUROSCI.1720-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Maya-Vetencourt JF, Tiraboschi E, Greco D, Restani L, Cerri C, Auvinen P, et al. Experience-dependent expression of npas4 regulates plasticity in adult visual cortex. The Journal of physiology. 2012;590:4777–4787. doi: 10.1113/jphysiol.2012.234237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zeiler SR, Gibson EM, Hoesch RE, Li MY, Worley PF, O’Brien RJ, et al. Medial premotor cortex shows a reduction in inhibitory markers and mediates recovery in a mouse model of focal stroke. Stroke. 2013;44:483–489. doi: 10.1161/STROKEAHA.112.676940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Farr TD, Whishaw IQ. Quantitative and qualitative impairments in skilled reaching in the mouse (mus musculus) after a focal motor cortex stroke. Stroke. 2002;33:1869–1875. doi: 10.1161/01.str.0000020714.48349.4e. [DOI] [PubMed] [Google Scholar]
- 27.Paxinos G, Franklin KBJ. Plates. In: Paxinos G, Franklin KBJ, editors. The mouse brain in stereotaxic coordinates. San Diego: Academic Press; 2001. pp. 49–350. [Google Scholar]
- 28.Tennant KA, Adkins DL, Donlan NA, Asay AL, Thomas N, Kleim JA, et al. The organization of the forelimb representation of the c57bl/6 mouse motor cortex as defined by intracortical microstimulation and cytoarchitecture. Cereb Cortex. 2011;21:865–876. doi: 10.1093/cercor/bhq159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ohira K, Takeuchi R, Iwanaga T, Miyakawa T. Chronic fluoxetine treatment reduces parvalbumin expression and perineuronal nets in gamma-aminobutyric acidergic interneurons of the frontal cortex in adult mice. Molecular brain. 2013;6:43. doi: 10.1186/1756-6606-6-43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Das RK, Pal M, Barui A, Paul RR, Chakraborty C, Ray AK, et al. Apotome to visualize e-cadherin and p63 expression in oral pre-cancer. Biotechnology journal. 2012;7:602–607. doi: 10.1002/biot.201100013. [DOI] [PubMed] [Google Scholar]
- 31.Laird NM, Ware JH. Random-effects models for longitudinal data. Biometrics. 1982;38:963–974. [PubMed] [Google Scholar]
- 32.Verbeke G, Molenberghs Geert. Linear mixed models for longitudinal data. Springer Science & Business Media; 2009. pp. 1–484. (Springer series in statistics). [Google Scholar]
- 33.Shin TK, Kang MS, Lee HY, Seo MS, Kim SG, Kim CD, et al. Fluoxetine and sertraline attenuate postischemic brain injury in mice. The Korean journal of physiology & pharmacology: official journal of the Korean Physiological Society and the Korean Society of Pharmacology. 2009;13:257–263. doi: 10.4196/kjpp.2009.13.3.257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lim CM, Kim SW, Park JY, Kim C, Yoon SH, Lee JK. Fluoxetine affords robust neuroprotection in the postischemic brain via its anti-inflammatory effect. Journal of neuroscience research. 2009;87:1037–1045. doi: 10.1002/jnr.21899. [DOI] [PubMed] [Google Scholar]
- 35.Zhu BG, Sun Y, Sun ZQ, Yang G, Zhou CH, Zhu RS. Optimal dosages of fluoxetine in the treatment of hypoxic brain injury induced by 3-nitropropionic acid: Implications for the adjunctive treatment of patients after acute ischemic stroke. CNS neuroscience & therapeutics. 2012;18:530–535. doi: 10.1111/j.1755-5949.2012.00315.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Duckworth EA, Butler TL, De Mesquita D, Collier SN, Collier L, Pennypacker KR. Temporary focal ischemia in the mouse: Technical aspects and patterns of fluoro-jade evident neurodegeneration. Brain Res. 2005;1042:29–36. doi: 10.1016/j.brainres.2005.02.021. [DOI] [PubMed] [Google Scholar]
- 37.Bendel O, Alkass K, Bueters T, von Euler M, von Euler G. Reproducible loss of ca1 neurons following carotid artery occlusion combined with halothane-induced hypotension. Brain Res. 2005;1033:135–142. doi: 10.1016/j.brainres.2004.11.033. [DOI] [PubMed] [Google Scholar]
- 38.Falco A, Pennucci R, Brambilla E, de Curtis I. Reduction in parvalbumin-positive interneurons and inhibitory input in the cortex of mice with experimental autoimmune encephalomyelitis. Experimental brain research Experimentelle Hirnforschung Experimentation cerebrale. 2014;232:2439–2449. doi: 10.1007/s00221-014-3944-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Clarke J, Mala H, Windle V, Chernenko G, Corbett D. The effects of repeated rehabilitation “tune-ups” on functional recovery after focal ischemia in rats. Neurorehabil Neural Repair. 2009;23:886–894. doi: 10.1177/1545968309341067. [DOI] [PubMed] [Google Scholar]
- 40.MacLellan CL, Keough MB, Granter-Button S, Chernenko GA, Butt S, Corbett D. A critical threshold of rehabilitation involving brain-derived neurotrophic factor is required for poststroke recovery. Neurorehabil Neural Repair. 2011;25:740–748. doi: 10.1177/1545968311407517. [DOI] [PubMed] [Google Scholar]
- 41.Duncan PW, Lai SM, Keighley J. Defining post-stroke recovery: Implications for design and interpretation of drug trials. Neuropharmacology. 2000;39:835–841. doi: 10.1016/s0028-3908(00)00003-4. [DOI] [PubMed] [Google Scholar]
- 42.Winters C, van Wegen EE, Daffertshofer A, Kwakkel G. Generalizability of the proportional recovery model for the upper extremity after an ischemic stroke. Neurorehabil Neural Repair. 2014;11:2014. doi: 10.1177/1545968314562115. [DOI] [PubMed] [Google Scholar]
- 43.Carmichael ST. Brain excitability in stroke: The yin and yang of stroke progression. Arch Neurol. 2012;69:161–167. doi: 10.1001/archneurol.2011.1175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Centonze D, Rossi S, Tortiglione A, Picconi B, Prosperetti C, De Chiara V, et al. Synaptic plasticity during recovery from permanent occlusion of the middle cerebral artery. Neurobiol Dis. 2007;27:44–53. doi: 10.1016/j.nbd.2007.03.012. [DOI] [PubMed] [Google Scholar]
- 45.Manganotti P, Patuzzo S, Cortese F, Palermo A, Smania N, Fiaschi A. Motor disinhibition in affected and unaffected hemisphere in the early period of recovery after stroke. Clin Neurophysiol. 2002;113:936–943. doi: 10.1016/s1388-2457(02)00062-7. [DOI] [PubMed] [Google Scholar]
- 46.Laaksonen K, Kirveskari E, Makela JP, Kaste M, Mustanoja S, Nummenmaa L, et al. Effect of afferent input on motor cortex excitability during stroke recovery. Clin Neurophysiol. 2012;123:2429–2436. doi: 10.1016/j.clinph.2012.05.017. [DOI] [PubMed] [Google Scholar]
- 47.Schiene K, Bruehl C, Zilles K, Qu M, Hagemann G, Kraemer M, et al. Neuronal hyperexcitability and reduction of gabaa-receptor expression in the surround of cerebral photothrombosis. J Cereb Blood Flow Metab. 1996;16:906–914. doi: 10.1097/00004647-199609000-00014. [DOI] [PubMed] [Google Scholar]
- 48.Greifzu F, Pielecka-Fortuna J, Kalogeraki E, Krempler K, Favaro PD, Schluter OM, et al. Environmental enrichment extends ocular dominance plasticity into adulthood and protects from stroke-induced impairments of plasticity. Proc Natl Acad Sci U S A. 2014;111:1150–1155. doi: 10.1073/pnas.1313385111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hensch TK. Critical period plasticity in local cortical circuits. Nat Rev Neurosci. 2005;6:877–888. doi: 10.1038/nrn1787. [DOI] [PubMed] [Google Scholar]
- 50.Hensch TK. Critical period regulation. Annual review of neuroscience. 2004;27:549–579. doi: 10.1146/annurev.neuro.27.070203.144327. [DOI] [PubMed] [Google Scholar]
- 51.Wahl AS, Omlor W, Rubio JC, Chen JL, Zheng H, Schroter A, et al. Neuronal repair. Asynchronous therapy restores motor control by rewiring of the rat corticospinal tract after stroke. Science. 2014;344:1250–1255. doi: 10.1126/science.1253050. [DOI] [PubMed] [Google Scholar]
- 52.Wahl AS, Schwab ME. Finding an optimal rehabilitation paradigm after stroke: Enhancing fiber growth and training of the brain at the right moment. Frontiers in human neuroscience. 2014;8:381. doi: 10.3389/fnhum.2014.00381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Windle V, Corbett D. Fluoxetine and recovery of motor function after focal ischemia in rats. Brain Res. 2005;1044:25–32. doi: 10.1016/j.brainres.2005.02.060. [DOI] [PubMed] [Google Scholar]
- 54.Zhang F, Zhou H, Wilson BC, Shi JS, Hong JS, Gao HM. Fluoxetine protects neurons against microglial activation-mediated neurotoxicity. Parkinsonism & related disorders. 2012;18(Suppl 1):S213–217. doi: 10.1016/S1353-8020(11)70066-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Taguchi N, Nakayama S, Tanaka M. Fluoxetine has neuroprotective effects after cardiac arrest and cardiopulmonary resuscitation in mouse. Resuscitation. 2012;83:652–656. doi: 10.1016/j.resuscitation.2011.11.004. [DOI] [PubMed] [Google Scholar]
- 56.Majlessi N, Naghdi N. Impaired spatial learning in the morris water maze induced by serotonin reuptake inhibitors in rats. Behavioural pharmacology. 2002;13:237–242. doi: 10.1097/00008877-200205000-00007. [DOI] [PubMed] [Google Scholar]
- 57.Valluzzi JA, Chan K. Effects of fluoxetine on hippocampal-dependent and hippocampal-independent learning tasks. Behavioural pharmacology. 2007;18:507–513. doi: 10.1097/FBP.0b013e3282ee2a91. [DOI] [PubMed] [Google Scholar]
- 58.Dias BG, Banerjee SB, Duman RS, Vaidya VA. Differential regulation of brain derived neurotrophic factor transcripts by antidepressant treatments in the adult rat brain. Neuropharmacology. 2003;45:553–563. doi: 10.1016/s0028-3908(03)00198-9. [DOI] [PubMed] [Google Scholar]
- 59.Frick LR, Rapanelli M, Arcos ML, Cremaschi GA, Genaro AM. Oral administration of fluoxetine alters the proliferation/apoptosis balance of lymphoma cells and up-regulates t cell immunity in tumor-bearing mice. European journal of pharmacology. 2011;659:265–272. doi: 10.1016/j.ejphar.2011.03.037. [DOI] [PubMed] [Google Scholar]
- 60.Schaz U, Fohr KJ, Liebau S, Fulda S, Koelch M, Fegert JM, et al. Dose-dependent modulation of apoptotic processes by fluoxetine in maturing neuronal cells: An in vitro study. The world journal of biological psychiatry: the official journal of the World Federation of Societies of Biological Psychiatry. 2011;12:89–98. doi: 10.3109/15622975.2010.506927. [DOI] [PubMed] [Google Scholar]
- 61.Lee CS, Kim YJ, Jang ER, Kim W, Myung SC. Fluoxetine induces apoptosis in ovarian carcinoma cell line ovcar-3 through reactive oxygen species-dependent activation of nuclear factor-kappab. Basic & clinical pharmacology & toxicology. 2010;106:446–453. doi: 10.1111/j.1742-7843.2009.00509.x. [DOI] [PubMed] [Google Scholar]
- 62.Liu KH, Yang ST, Lin YK, Lin JW, Lee YH, Wang JY, et al. Fluoxetine, an antidepressant, suppresses glioblastoma by evoking ampar-mediated calcium-dependent apoptosis. Oncotarget. 2015;6:5088–5101. doi: 10.18632/oncotarget.3243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Nunez MJ, Balboa J, Rodrigo E, Brenlla J, Gonzalez-Peteiro M, Freire-Garabal M. Effects of fluoxetine on cellular immune response in stressed mice. Neurosci Lett. 2006;396:247–251. doi: 10.1016/j.neulet.2005.11.042. [DOI] [PubMed] [Google Scholar]
- 64.Risedal A, Zeng J, Johansson BB. Early training may exacerbate brain damage after focal brain ischemia in the rat. J Cereb Blood Flow Metab. 1999;19:997–1003. doi: 10.1097/00004647-199909000-00007. [DOI] [PubMed] [Google Scholar]
- 65.Farrell R, Evans S, Corbett D. Environmental enrichment enhances recovery of function but exacerbates ischemic cell death. Neuroscience. 2001;107:585–592. doi: 10.1016/s0306-4522(01)00386-4. [DOI] [PubMed] [Google Scholar]
- 66.Andrade C, Rao NS. How antidepressant drugs act: A primer on neuroplasticity as the eventual mediator of antidepressant efficacy. Indian journal of psychiatry. 2010;52:378–386. doi: 10.4103/0019-5545.74318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Caiati MD, Cherubini E. Fluoxetine impairs gabaergic signaling in hippocampal slices from neonatal rats. Frontiers in cellular neuroscience. 2013;7:63. doi: 10.3389/fncel.2013.00063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Guirado R, Perez-Rando M, Sanchez-Matarredona D, Castren E, Nacher J. Chronic fluoxetine treatment alters the structure, connectivity and plasticity of cortical interneurons. The international journal of neuropsychopharmacology/official scientific journal of the Collegium Internationale Neuropsychopharmacologicum. 2014:1–12. doi: 10.1017/S1461145714000406. [DOI] [PubMed] [Google Scholar]
- 69.Mainardi M, Landi S, Berardi N, Maffei L, Pizzorusso T. Reduced responsiveness to long-term monocular deprivation of parvalbumin neurons assessed by c-fos staining in rat visual cortex. PloS one. 2009;4:e4342. doi: 10.1371/journal.pone.0004342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Schwaller B. Cytosolic ca2+ buffers. Cold Spring Harbor perspectives in biology. 2010;2:a004051. doi: 10.1101/cshperspect.a004051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Gladstone DJ, Danells CJ, Armesto A, McIlroy WE, Staines WR, Graham SJ, et al. Physiotherapy coupled with dextroamphetamine for rehabilitation after hemiparetic stroke: A randomized, double-blind, placebo-controlled trial. Stroke. 2006;37:179–185. doi: 10.1161/01.STR.0000195169.42447.78. [DOI] [PubMed] [Google Scholar]
- 72.Walker-Batson D, Smith P, Curtis S, Unwin H, Greenlee R. Amphetamine paired with physical therapy accelerates motor recovery after stroke. Further evidence. Stroke. 1995;26:2254–2259. doi: 10.1161/01.str.26.12.2254. [DOI] [PubMed] [Google Scholar]
- 73.Feeney DM, Gonzalez A, Law WA. Amphetamine, haloperidol, and experience interact to affect rate of recovery after motor cortex injury. Science. 1982;217:855–857. doi: 10.1126/science.7100929. [DOI] [PubMed] [Google Scholar]
- 74.Hovda DA, Fenney DM. Amphetamine with experience promotes recovery of locomotor function after unilateral frontal cortex injury in the cat. Brain Res. 1984;298:358–361. doi: 10.1016/0006-8993(84)91437-9. [DOI] [PubMed] [Google Scholar]
- 75.Karpova NN, Pickenhagen A, Lindholm J, Tiraboschi E, Kulesskaya N, Agustsdottir A, et al. Fear erasure in mice requires synergy between antidepressant drugs and extinction training. Science. 2011;334:1731–1734. doi: 10.1126/science.1214592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Khundakar AA, Zetterstrom TS. Biphasic change in bdnf gene expression following antidepressant drug treatment explained by differential transcript regulation. Brain Res. 2006;1106:12–20. doi: 10.1016/j.brainres.2006.05.063. [DOI] [PubMed] [Google Scholar]
- 77.De Foubert G, Carney SL, Robinson CS, Destexhe EJ, Tomlinson R, Hicks CA, et al. Fluoxetine-induced change in rat brain expression of brain-derived neurotrophic factor varies depending on length of treatment. Neuroscience. 2004;128:597–604. doi: 10.1016/j.neuroscience.2004.06.054. [DOI] [PubMed] [Google Scholar]
- 78.Rogoz Z, Kabzinski M, Sadaj W, Rachwalska P, Gadek-Michalska A. Effect of co-treatment with fluoxetine or mirtazapine and risperidone on the active behaviors and plasma corticosterone concentration in rats subjected to the forced swim test. Pharmacological reports: PR. 2012;64:1391–1399. doi: 10.1016/s1734-1140(12)70936-2. [DOI] [PubMed] [Google Scholar]
- 79.Gomes KS, de Carvalho-Netto EF, Monte KC, Acco B, Nogueira PJ, Nunes-de-Souza RL. Contrasting effects of acute and chronic treatment with imipramine and fluoxetine on inhibitory avoidance and escape responses in mice exposed to the elevated t-maze. Brain research bulletin. 2009;78:323–327. doi: 10.1016/j.brainresbull.2008.11.003. [DOI] [PubMed] [Google Scholar]
- 80.Kelly JP, Wrynn AS, Leonard BE. The olfactory bulbectomized rat as a model of depression: An update. Pharmacology & therapeutics. 1997;74:299–316. doi: 10.1016/s0163-7258(97)00004-1. [DOI] [PubMed] [Google Scholar]
- 81.Uhr M, Steckler T, Yassouridis A, Holsboer F. Penetration of amitriptyline, but not of fluoxetine, into brain is enhanced in mice with blood-brain barrier deficiency due to mdr1a p-glycoprotein gene disruption. Neuropsychopharmacology: official publication of the American College of Neuropsychopharmacology. 2000;22:380–387. doi: 10.1016/S0893-133X(99)00095-0. [DOI] [PubMed] [Google Scholar]
- 82.Bolo NR, Hode Y, Nedelec JF, Laine E, Wagner G, Macher JP. Brain pharmacokinetics and tissue distribution in vivo of fluvoxamine and fluoxetine by fluorine magnetic resonance spectroscopy. Neuropsychopharmacology: official publication of the American College of Neuropsychopharmacology. 2000;23:428–438. doi: 10.1016/S0893-133X(00)00116-0. [DOI] [PubMed] [Google Scholar]
- 83.Henry ME, Moore CM, Kaufman MJ, Michelson D, Schmidt ME, Stoddard E, et al. Brain kinetics of paroxetine and fluoxetine on the third day of placebo substitution: A fluorine mrs study. The American journal of psychiatry. 2000;157:1506–1508. doi: 10.1176/appi.ajp.157.9.1506. [DOI] [PubMed] [Google Scholar]
- 84.Gladstone DJ, Danells CJ, Black SE. The fugl-meyer assessment of motor recovery after stroke: A critical review of its measurement properties. Neurorehabil Neural Repair. 2002;16:232–240. doi: 10.1177/154596802401105171. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.