

ABSTRACT
Dissociation of imaginal disc cells has been carried out previously to enable flow cytometry and cell sorting to analyze cell cycle progression, cell size, gene expression, and other aspects of imaginal tissues. However, the lengthy dissociation protocols employed may alter gene expression, cell behavior and overall viability. Here we describe a new rapid and gentle method of dissociating the cells of wing imaginal discs that significantly enhances cell viability and reduces the likelihood of gene expression changes. Furthermore, this method is scalable, enabling collection of large amounts of sample for high-throughput experiments without the need for data-distorting amplifications.
KEYWORDS: Dissociation, Drosophila, FACS, imaginal disc, mRNA-seq
Introduction
Drosophila imaginal tissues, the larval epithelial structures that give rise to adult structures upon metamorphosis, have been used extensively to study patterning, growth and cell cycle, signal transduction, cell fate specification, and many other aspects of developmental biology.1 We use imaginal tissues to study how a specified and patterned epithelium carries out tissue regeneration after ablation of a significant portion of that tissue.2,3 To identify changes in gene expression at specific time points after tissue ablation, we sought to label and sort regeneration blastema cells from damaged wing imaginal discs for mRNA sequencing. This isolation of blastema cells was challenging because they are only about 2–5% of the cells in the wing disc. Therefore, we needed a cell dissociation protocol that would maximize cell viability and dissociation to enable collection of the small number of desired cells, while at the same time minimize the time taken to achieve dissociation to prevent changes in gene expression induced by the manipulations.
Dissociation of imaginal discs followed by flow cytometry and/or fluorescence-activated cell sorting (FACS) has been carried out using enzymes such as trypsin4 and collagenase5 with lengthy incubation times ranging from 1 to 4 h, raising the concern that the time in the enzyme incubation could alter cell physiology and gene expression. Furthermore, our initial trials using such protocols resulted in high levels of cell death, significantly reducing our yield of usable cells and mRNA after cell sorting and RNA preparation. Therefore, we examined many protocols for cell dissociation of imaginal discs,4-6 other Drosophila tissues,7,8 and other organisms,9 and tested numerous modifications of those protocols to identify the shortest protocol that produced the most live cells.
Here we present a rapid, gentle and scalable method to isolate fluorescently labeled cells from imaginal discs. This method uses a commercially available enzyme preparation in a glass dish to dissociate the discs in as little as 15 min, with significantly improved cell viability and mRNA yield.
Results
Identification of an appropriate cell marker
For our purposes, we isolated cells that expressed the gene nubbin10 (FBgn0085424) in the wing imaginal disc. We identified a line containing a MiMIC transposable element11 inserted in the nubbin (nub) locus that expressed GFP in the nubbin-expressing cells of the wing disc (Fig. 1)(nubMI05126, FBti0146996, Bloomington Stock Center line #37920). Similar enhancer trap and protein trap lines that express GFP in other patterns would enable use of this protocol for a wide variety of purposes.
Figure 1.
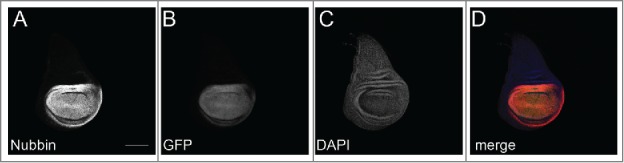
(A) MiMIC insertion into the nubbin locus marks wing pouch cells for sorting after dissociation. Third instar wing imaginal disc. A) Anti-Nubbin.10 B) nub-MiMIC-GFP. Image is native GFP fluorescence. C) DAPI. D) Merge. Scale bar is 100 μm.
For the optimization below we used undamaged wing discs, in which the number of GFP-labeled cells is significantly higher than it would be in our experimental system. Isolation and transcriptional profiling of regeneration blastema cells will be presented in a separate publication.
Dissociation optimization
To identify the optimal enzymes, buffers, length of time and temperature for disc dissociation, we tested multiple concentrations, combinations and variations of each (for examples see Table 1). Variations on the protocol were initially assessed qualitatively and visually for relative amount of dissociation and cell viability using Sytox Green or Red (Invitrogen #S7020, #S34859) to stain the dying cells, which were visualized under a compound microscope (Leica DM RXA2). After this initial optimization we tested different dissociation conditions by flow cytometry to quantify numbers of viable GFP+ and GFP- cells (Table 1 and Fig. 2).
Table 1.
Assessment of selected dissociation and sort trials.
| Enzyme | Buffer | Incubation | Method to stop dissociation | Cell dissociation | Cell viability (Sytox Red) | Percentage of flow cytometry “events” that were live cells | Percentage of live single cells that were GFP+ |
|---|---|---|---|---|---|---|---|
| Collagenase 1 mg/mL + Papain 1 mg/mL | Calcium Magnesium Free Buffer (CMF) | 30 min at 30°C | add CMF | ** | * | 25% | 37% |
| Collagenase 0.3 mg/mL | Calcium Magnesium Free Buffer (CMF) | 30 min at 30°C | add CMF | ** | * | 12% | 23% |
| Collagenase 1mg/mL | Rinaldini Solution | 30 min at 30°C | add SSM | ** | * | 8% | 10% |
| TrypLE 10x | Dulbecco's PBS | 15 min at 37°C | add SSM | *** | *** | 48% | 27% |
Figure 2.

Comparison of different dissociation protocols. A, B) Initial protocol using 160 imaginal discs, 1 mg/mL collagenase and Rinaldini solution. C, D) Final protocol using 120 imaginal discs, 10x TrypLE and Dulbecco's PBS. A, C) Separation by GFP content showing peak of GFP-positive cells (arrows). B, D) Gating to sort GFP- (blue) and GFP+ (green) cells. Gates were manually set to maximize separation of GFP+ and GFP- cells. E) RT-qPCR showing enrichment of nubbin and dMyc in the GFP+ cells relative to the GFP- cells, and enrichment of the notum marker teashirt and the hinge marker unpaired in the GFP- cells relative to the GFP+ cells, demonstrating the efficiency of the sort. F) Wing disc in which most of the wing pouch has undergone ablation, leaving a small population of nub-GFP+ cells remaining to be sorted (arrow), as well as GFP+ debris (arrowhead). G) FACS sorting of the GFP+ cells remaining after tissue ablation, using 400 imaginal discs, 10x TrypLE and Dulbecco's PBS. FACS data are presented in graphs generated using the FCS Express 4 software.
Handling of the dissected imaginal discs
While existing protocols called for collection of discs in microfuge tubes and centrifuging the tubes to pellet the discs between washes,5 we found these steps led to loss of tissue. Therefore, we collected discs in a glass dish (Carolina Biological Supply #742300), eliminated these washing steps and proceeded straight to dissociation in the glass dish.
Buffer
Our starting protocol called for using Rinaldini Solution7 (100 mL H2O, 800 mg NaCl, 20 mg KCl, 5 mg NaH2PO4, 100 mg NaHCO3, 100 mg glucose, pH 7.35). We also tested a Calcium and Magnesium free buffer (15 mM HEPES, 400 mg/L NaH2PO4, 800 mg/L NaCl, 1200 mg/L KCl, 800 mg/L NaHCO3, 240 mg/L glucose, 1% BSA, pH 7.3 ) used for cell dissociation in other organisms9, as well as Dulbecco's PBS (Sigma #D8537-500ML) as recommended for the TrypLE enzyme. The protocols compared in Figure 2 used Rinaldini Solution and Dulbecco's PBS.
Enzymes
We tested collagenase (Sigma #C0130-100MG)5 at a variety of concentrations, including 0.3 mg/mL, 0.5 mg/mL, and 1 mg/mL, collagenase with papain (Sigma #P4762-50MG)7 at a concentration of 1 mg/mL each, and the commercially available purified recombinant trypsin TrypLE (ThermoFisher #A12177-01) at the recommended 10x concentration. The protocols compared in Figure 2 used Collagenase 1 mg/mL and TrypLE 10x. We found the best dissociation and viability with the TrypLE.
Time and temperature
We tested incubation with the enzymes for 5, 10, 15, and 30 min at 30°C and 37°C (Table 1 and data not shown). Our qualitative assessment determined that the optimal dissociation and viability occurred using the TrypLE at 37°C for 15 min. These experiments were carried out with lot #1408696 of TrypLE, which had a reported activity of 0.40 recombinant protease units per mL (rPu/mL). Because activity varies by lot, the time required for dissociation should be optimized for each batch, taking the rPu/mL value into consideration. We also experimented with different methods of incubating the discs at the desired temperature in the glass dish, developing the method using metal beads described in the protocol (Fig. 3).
Figure 3.
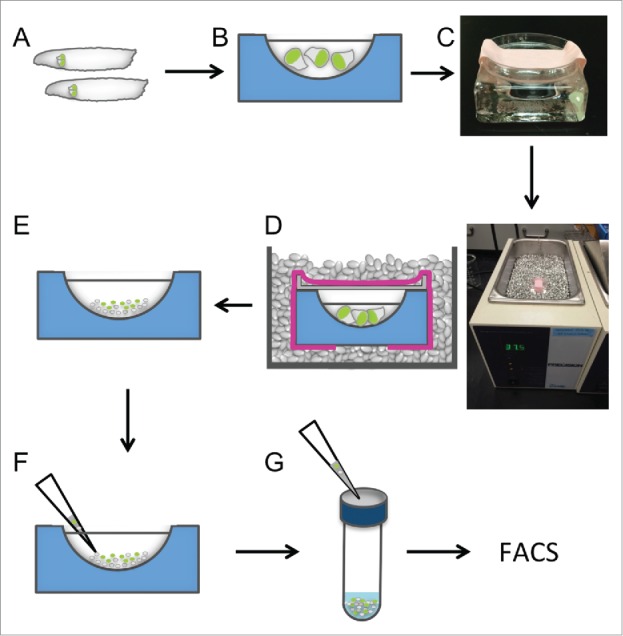
Schematic of dissociation protocol. A) Wash larvae and dissect in SSM. B) Collect imaginal discs in Dulbecco's PBS in a glass dish. C) Replace PBS with TrypLE, cover with Petri dish lid and secure with tape. D) Incubate in metal bead bath for 15 min at 37°C. E) Stop enzyme action by flooding with SSM. F) Further dissociate cells by pipetting 2–3 times with a 1000 μL pipet tip. F) Pass cells through a cell strainer to remove any remaining aggregates.
Halting the enzymatic reaction
Different protocols recommended halting the enzymatic reaction either with a series of washes in Rinaldini solution and SSM 7 or adding excess buffer to the reaction.5 Our optimization ensured that the discs were sufficiently dissociated such that they could not be centrifuged and washed. When testing collagenase we found that flooding the sample with SSM caused significantly less cell death than flooding with buffer (Table 1). In our final protocol we added excess SSM to stop the TrypLE reaction.
Mechanical dissociation
We tested mechanical dissociation after the enzyme incubation by pipetting through different sized pipet tips several times. We found that using a 200 μL pipet tip 5 caused significant cell death, whereas a 1000 μL pipet tip was sufficiently gentle (Fisherbrand Sureone #02-707-403) (data not shown). We also passed the cell suspension through a cell strainer (VWR #352235) to remove cells that remained clumped.7 While we have not compared our method to that described by Liang et al.,12 which uses mechanical dissociation by repeated passage through a small needle, we expect that, similar to the 200 μL pipet tip, the needle would induce levels of cell death that would be too high for our purposes.
FACS sorting
Cells were sorted in a BD FACS ARIA II sorter, using gentle conditions (85 μm nozzle and low pressure of 20 psi). Under these conditions, sorting cells from 400 imaginal wing discs took approximately one hour. We sorted directly into RNA Lysis buffer (Qiagen RNA isolation kit #74104), limited the number of cells sorted into each tube so as not to dilute the lysis buffer, and added β-mercaptoethanol immediately. We also found that how quickly we progressed through the dissociation protocol and started the sort affected the number of live cells at the time of the sort. To demonstrate this, we used a BD LSR II Flow Cytometry Analyzer to determine preparation quality at various times after dissociation, and found that a wait of as little as 30 min between dissociation and sorting led to a 26% decrease in the number of viable cells (data not shown).
RNA yield
For the 2 protocols in Figure 2, RNA was prepared using a Qiagen kit (#74104). Protocol 1 (Collagenase) started with 160 imaginal discs and yielded 3.2 ng/μL RNA for a total of 96 ng RNA, while protocol 2 (TrypLE) started with 120 imaginal discs and yielded 13.4 ng/μL RNA for a total of 402 ng RNA (Fig. 2). RNA concentrations were measured using a Nanodrop Spectrophotometer 2000C (ThermoScientific) for these optimizations. Prior to proceeding with high-throughput sequencing we recommend confirming RNA concentrations with a Qubit Fluorometer (ThermoFisher) and assessing RNA quality with an Agilent 2100 BioAnalyzer or similar machine.
Verification of sorting
To confirm separation of the nub-GFP expressing wing pouch cells from the remainder of the wing imaginal disc, we carried out RT-qPCR on the GFP+ and GFP- cells for the wing pouch marker nub,10 the growth driver dMyc, which is ubiquitously expressed in the wing disc but is higher in the pouch,13 as well as the notum marker teashirt (tsh) and the hinge marker unpaired (upd)14 (Fig. 2E). Transcripts for nub were highly enriched in the GFP+ cells relative to the GFP- cells, while transcripts for dMyc were slightly elevated in the GFP+ cells as expected. Conversely, transcripts for tsh and upd were highly enriched in the GFP- cells relative to the GFP+ cells. Three or 4 biological replicates of 100 discs each were used. Primers: tsh F5′ AGGGGTAAAAGCTCCAGAAGAA, R5′ GCTCAATGGCACTTAAAACAGA; nub F5′ GCTACTCATTCGGCATTCAAGT, R5′ TTTGAAAATTGTGCAAAGAGTG, upd F5′ AGCGTCCCAGGCAGAGCTTCA, R5′ TACTCCCGAAAGGCGTGGCG; dMyc F5′ CAACTTGCTGGCGGCACGG, R5′ TCCTGAGTTATATGCCTGTCTCGCT. For these experiments we used TrypLE from lot 1755986, which had an rPu/mL of 0.22. Consequently, we increased dissociation time to 30 min and did not see any decrease in viability.
Sorting rare cells
To demonstrate that this protocol can be used to sort small populations of cells, we dissociated and sorted regeneration blastema cells. We have previously described our method of ablating most of the wing pouch in the early third larval instar using rotund-GAL4, UAS-reaper, and GAL80ts 15. Twenty-four hours after ablation, the regeneration blastema is marked via nub-GFP (Fig. 2F). Although they represent only 5.86% of the total dissociated cell population, these cells can be sorted via FACS (Fig. 2G).
Scaling the preparation
The number of cells isolated by sorting and used for each mRNA sample could be increased at several points in the protocol. To increase the number of discs used per sort we increased the number of people dissecting at once, rather than the length of time a single researcher was dissecting. We found that 4 researchers dissecting discs simultaneously could isolate at least 400 discs in about 1 h. We limited the number of discs per glass dish to 100, processing multiple glass dishes in parallel.
If more RNA is desired for each sample beyond what can be achieved by dissections in parallel, multiple rounds of dissections and sorts could be pooled. When such pooling was necessary, we chose to pool samples after isolating RNA, rather than at other points in the protocol, because the RNA was more stable than frozen sorted or unsorted cells. Thus, the protocol would proceed directly from dissection through dissociation and sort to RNA isolation as quickly as possible before storage and pooling.
Dissociation protocol
Before starting the protocol, autoclave microfuge tubes (ThermoFisher #Am12400), close the tubes, and use a razor to cut the connector between the cap and the tube.
Select 50 third-instar larvae using appropriate markers. Place the larvae into a glass dish (Carolina Biological Supply #742300) containing water to wash off residual food.
Remove the water with a transfer pipet, leaving the larvae in the dish, and replace with enough 1x PBS (Gold Biotechnology #P-271-200) to submerge and wash the larvae. Repeat with 70% ethanol to sterilize the larval cuticles.
Wash in the same manner with Supplemented Schnieder's Medium (SSM) (Schnieder's Medium [Sigma #S0146], Fetal bovine serum 10% [VWR #1300500], penicillin/streptomycin 2% [VWR 17602E], Insulin 0.02 mg/mL [Sigma #I2643-50MG]) before keeping in SSM.
Dissect larvae with forceps (Dumont no. 5, Ted Pella #505) in SSM to isolate the wing imaginal discs. Collect the discs in a second glass dish containing Dulbecco's PBS (Sigma #D8537-500ML) placed on ice, up to 100 discs per dish.
Pipet out the Dulbecco's PBS using a 200 μL pipet tip (VWR ultrafine #89079-444). Add 100 μL TrypLE undiluted from its 10x working concentration.
Cover the glass dish with a petri dish lid (95 × 15 mm, Fisher #FB0875714G) flat side down and wrap with lab tape to keep secure (Fig. 3). Place the dish in a heated bath filled with metallic beads (ThermoFisher #A12543-02) at 37°C and cover with beads (Fig. 3).
Incubate the dish for 15 min and remove from the metal bead bath.
Stop the reaction by adding 500 μL SSM.
Gently pipet the suspension up and down using a 1000 μL pipet tip 2–3 times for mechanical dissociation.
Pass the cells through a cell strainer (VWR #352235) to eliminate clumps. Cell concentration can be quantified using a hemocytometer. Cell viability can be assessed using Sytox staining.
Seal the cell strainer tube with parafilm, place on ice, and transport for FACS sorting. Keep all samples on ice before, after, and during the sort.
Adjust the FACS machine to use an 85 μm nozzle and low pressure of 20 psi.
Sort directly into the autoclaved tubes from step 1 containing 500 μL Lysis Buffer (Qiagen RNA isolation kit #74104), and keep the samples on ice.
As soon as a tube contains 20,000 cells, remove it and immediately add 5μL β-mercaptoethanol (Sigma #M6250-100ML) and close with an autoclaved cap. Use a new tube with Lysis Buffer to continue to collect cells. Repeat as needed.
Once sorting is complete, transport the samples back to the laboratory and continue the RNA isolation (Qiagen #74104) immediately using sterile and RNAse-free equipment (RNase Zap ThermoFisher #AM9780). To pool the RNA from the same sample collected in multiple tubes, run the sample from each tube through a single RNA column.
To assess RNA yield use a Nanodrop spectrophotometer (ThermoScientific) or a Qubit Fluorometer (ThermoFisher). To assess RNA quality, use an Agilent 2100 Bioanalyzer or similar machine.
Store RNA at −80°C before use for qPCR or deep sequencing.
Discussion
Here we detail the optimization and final protocol for dissociating imaginal discs and sorting fluorescently labeled cells for RNA extraction. Our aim was to develop a protocol that was rapid and gentle so as to avoid changes in gene expression and cell physiology that would mask the phenomena we sought to study. We also needed a protocol that maximized cell viability, because in our studies of tissue damage 15 very few of the GFP-labeled cells remain in the disc after damage. Thus the protocol presented here enables isolation of cells that comprise less than 5% of the imaginal disc in sufficient quantities for high-throughput RNA sequencing.
While it is possible that the 15 min incubation in the 37°C metal bead bath was sufficient to activate the heat-shock stress response, the brief incubation followed quickly by a sort into lysis buffer likely minimized the changes in gene expression induced. Furthermore, these changes would occur in both experimental and control samples and thus not be identified as differential expression. Indeed, in our unpublished transcriptional profile of our experimental sorted cells compared with control sorted cells, 11 heat-shock protein (Hsp) genes, including the Hsp90 homolog Hsp83, were either not differentially expressed or not detected (data not shown). The six Hsp proteins that were differentially expressed showed very low fold increases in expression such that they would not be deemed of interest.
The ability to scale the protocol by dissecting in parallel and pooling collections enables analysis of cells that make up a small portion of the imaginal disc. While protocols and kits exist to prepare RNA for high-throughput sequencing from small numbers of cells,16 the ability to collect sufficient numbers of cells eliminates the need for amplifications that might alter the proportion of transcripts to be sequenced.
Given the genetic tools available in Drosophila, cells of interest might be positively or negatively fluorescently labeled using enhancer traps and protein traps, or generation of mitotic clones, MARCM clones, or flip-out clones.11,17-19 These techniques enable labeling of cells from different regions of the discs, of different cell-fate specifications, or different genetic makeup. Therefore, this protocol will be widely applicable for a variety of studies using imaginal discs.
Abbreviations
- FACS
fluorescence-activated cell sorting
- mRNA-seq
deep sequencing of messenger ribonucleic acids
- PBS
phosphate buffered saline
- SSM
Supplemented Schnieder's Medium
- nub
nubbin
- tsh
teashirt
- upd
unpaired
- rPu/mL
recombinant protease units per milliliter
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
Sumbul Jawed Khan and Syeda Nayab Fatima Abidi contributed equally to this work. Dr. Barbara Pilas, director of the flow cytometry facility in the University of Illinois Roy J. Carver Biotechnology Center, provided technical support throughout the optimization of the cell sorting. A.R. Brock provided helpful comments on the manuscript. The anti-Nubbin antibody was a gift of S. Cohen. Stocks obtained from the Bloomington Drosophila Stock Center (NIH P40OD018537) were used in this study.
Funding
This work was supported by a Young Investigator Award from the Roy J. Carver Charitable Trust (#12-4041) and a grant from NIH (R01GM107140).
References
- [1].Held LI., Jr Imaginal Discs. Cambridge, UK: Cambridge University Press; 2005. [Google Scholar]
- [2].Schuster KJ, Smith-Bolton RK. Taranis Protects Regenerating Tissue from Fate Changes Induced by the Wound Response in Drosophila. Dev Cell 2015; 34:119-28; PMID:26096735; http://dx.doi.org/ 10.1016/j.devcel.2015.04.017 [DOI] [PubMed] [Google Scholar]
- [3].Skinner A, Khan SJ, Smith-Bolton RK. Trithorax regulates systemic signaling during Drosophila imaginal disc regeneration. Development 2015; 142:3500-11; PMID:26487779; http://dx.doi.org/ 10.1242/dev.122564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Neufeld TP, de la Cruz AF, Johnston LA, Edgar BA. Coordination of growth and cell division in the Drosophila wing. Cell 1998; 93:1183-93; PMID:9657151; http://dx.doi.org/ 10.1016/S0092-8674(00)81462-2 [DOI] [PubMed] [Google Scholar]
- [5].Collesano M, Corona DFV. Flow cytometry and karyotype analysis of D.melanogaster eye disc cells. Fly (Austin) 2007; 1:242-4; PMID:18820443; http://dx.doi.org/ 10.4161/fly.4766 [DOI] [PubMed] [Google Scholar]
- [6].de la Cruz AFA, Edgar BA. Flow cytometric analysis of Drosophila cells. Methods Mol Biol 2008; 420:373-89; PMID:18641961; http://dx.doi.org/ 10.1007/978-1-59745-583-1_24 [DOI] [PubMed] [Google Scholar]
- [7].Berger C, Harzer H, Burkard TR, Steinmann J, van der Horst S, Laurenson A-S, Novatchkova M, Reichert H, Knoblich JA. FACS purification and transcriptome analysis of drosophila neural stem cells reveals a role for Klumpfuss in self-renewal. Cell Rep 2012; 2:407-18; PMID:22884370; http://dx.doi.org/ 10.1016/j.celrep.2012.07.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Dutta D, Xiang J, Edgar BA. RNA expression profiling from FACS-isolated cells of the Drosophila intestine. Curr Protoc Stem Cell Biol 2013; 27:Unit2F.2; PMID:24510286 [DOI] [PubMed] [Google Scholar]
- [9].Reddien PW, Oviedo NJ, Jennings JR, Jenkin JC, Sánchez Alvarado A. SMEDWI-2 is a PIWI-like protein that regulates planarian stem cells. Science 2005; 310:1327-30; PMID:16311336; http://dx.doi.org/ 10.1126/science.1116110 [DOI] [PubMed] [Google Scholar]
- [10].Ng M, Diaz-Benjumea FJ, Cohen SM. Nubbin encodes a POU-domain protein required for proximal-distal patterning in the Drosophila wing. Development 1995; 121:589-99; PMID:7768195 [DOI] [PubMed] [Google Scholar]
- [11].Venken KJT, Schulze KL, Haelterman NA, Pan H, He Y, Evans-Holm M, Carlson JW, Levis RW, Spradling AC, Hoskins RA, et al.. MiMIC: a highly versatile transposon insertion resource for engineering Drosophila melanogaster genes. Nat Methods 2011; 8:737-43; PMID:21985007; http://dx.doi.org/ 10.1038/nmeth.1662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Liang L, Haug JS, Seidel CW, Gibson MC. Functional genomic analysis of the periodic transcriptome in the developing Drosophila wing. Dev Cell 2014; 29:112-27; PMID:24684830; http://dx.doi.org/ 10.1016/j.devcel.2014.02.018 [DOI] [PubMed] [Google Scholar]
- [13].Johnston LA, Prober DA, Edgar BA, Eisenman RN, Gallant P. Drosophila myc regulates cellular growth during development. Cell 1999; 98:779-90; PMID:10499795; http://dx.doi.org/ 10.1016/S0092-8674(00)81512-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Ayala-Camargo A, Anderson AM, Amoyel M, Rodrigues AB, Flaherty MS, Bach EA. JAK/STAT signaling is required for hinge growth and patterning in the Drosophila wing disc. Dev Biol 2013; 382:413-26; PMID:23978534; http://dx.doi.org/ 10.1016/j.ydbio.2013.08.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Smith-Bolton RK, Worley MI, Kanda H, Hariharan IK. Regenerative growth in Drosophila imaginal discs is regulated by Wingless and Myc. Dev Cell 2009; 16:797-809; PMID:19531351; http://dx.doi.org/ 10.1016/j.devcel.2009.04.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Combs PA, Eisen MB. Low-cost, low-input RNA-seq protocols perform nearly as well as high-input protocols. PeerJ 2015; 3:e869; PMID:25834775; http://dx.doi.org/ 10.7717/peerj.869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Kelso RJ, Buszczak M, Quiñones AT, Castiblanco C, Mazzalupo S, Cooley L. Flytrap, a database documenting a GFP protein-trap insertion screen in Drosophila melanogaster. Nucleic Acids Res 2004; 32:D418-20; PMID:14681446; http://dx.doi.org/ 10.1093/nar/gkh014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Lee T, Luo L. Mosaic analysis with a repressible cell marker (MARCM) for Drosophila neural development. Trends Neurosci 2001; 24:251-4; PMID:11311363; http://dx.doi.org/ 10.1016/S0166-2236(00)01791-4 [DOI] [PubMed] [Google Scholar]
- [19].Struhl G, Basler K. Organizing activity of wingless protein in Drosophila. Cell 1993; 72:527-40; PMID:8440019; http://dx.doi.org/ 10.1016/0092-8674(93)90072-X [DOI] [PubMed] [Google Scholar]