

ABSTRACT
The adeno-associated viruses (AAVs), which are being developed as gene delivery vectors, display differential cell surface glycan binding and subsequent tissue tropisms. For AAV serotype 1 (AAV1), the first viral vector approved as a gene therapy treatment, and its closely related AAV6, sialic acid (SIA) serves as their primary cellular surface receptor. Toward characterizing the SIA binding site(s), the structure of the AAV1-SIA complex was determined by X-ray crystallography to 3.0 Å. Density consistent with SIA was observed in a pocket located at the base of capsid protrusions surrounding icosahedral 3-fold axes. Site-directed mutagenesis substitution of the amino acids forming this pocket with structurally equivalent residues from AAV2, a heparan sulfate binding serotype, followed by cell binding and transduction assays, further mapped the critical residues conferring SIA binding to AAV1 and AAV6. For both viruses five of the six binding pocket residues mutated (N447S, V473D, N500E, T502S, and W503A) abolished SIA binding, whereas S472R increased binding. All six mutations abolished or decreased transduction by at least 50% in AAV1. Surprisingly, the T502S substitution did not affect transduction efficiency of wild-type AAV6. Furthermore, three of the AAV1 SIA binding site mutants—S472R, V473D, and N500E—escaped recognition by the anti-AAV1 capsid antibody ADK1a. These observations demonstrate that common key capsid surface residues dictate both virus binding and entry processes, as well as antigenic reactivity. This study identifies an important functional capsid surface “hot spot” dictating receptor attachment, transduction efficiency, and antigenicity which could prove useful for vector engineering.
IMPORTANCE The adeno-associated virus (AAV) vector gene delivery system has shown promise in several clinical trials and an AAV1-based vector has been approved as the first gene therapy treatment. However, limitations still exist with respect to transduction efficiency and the detrimental effects of preexisting host antibodies. This study aimed to identify key capsid regions which can be engineered to overcome these limitations. A sialic glycan receptor recognition pocket was identified in AAV1 and its closely related AAV6, using X-ray crystallography. The site was confirmed by mutagenesis followed by cell binding and transduction assays. Significantly, residues controlling gene expression efficiency, as well as antibody escape variants, were also identified. This study thus provides, at the amino acid level, information for rational structural engineering of AAV vectors with improved therapeutic efficacy.
INTRODUCTION
Adeno-associated viruses (AAVs) are small, nonenveloped, single-stranded DNA packaging viruses that belong to the Dependoparvovirus genus of the Parvoviridae. There are more than 150 naturally occurring variants, and 13 nonhuman and human serotypes have been described (1–6). Due to their nonpathogenicity, ability to package recombinant DNA, long-term transgene expression, and ability to transduce dividing and nondividing cells, the AAVs are being developed as gene delivery vectors (7, 8). The most studied serotype, AAV2, exhibits broad tissue tropism, while several other serotypes exhibit better specific tissue transduction (9–18). Examples include AAV1 transduction of smooth muscle, the central nervous system, and retina at 1,000-, 35-, and 2-fold better, respectively (13–15), while AAV6 has superior smooth muscle, heart, and lung transduction by 500-, 10-, and 10-fold, respectively, compared to AAV2 (16–18). Diversity in the tissue tropism of different AAV serotypes extends their utility for the delivery of a large number of different genes for the treatment of a variety of genetic disorders.
However, despite the promise of the AAV as efficient gene delivery systems, several challenges need to be overcome for full clinical realization. This includes targeting specificity which is affected by (i) tissue tropism, with broad recognition resulting in less effective tissue specific application, and (ii) administration route, with direct or systemic injection affecting transgene expression profiles (19). Second, preexisting anti-AAV antibodies against different serotypes are found in 40 to 70% of the human population because of natural exposure to the viruses (20, 21). This leads to decreased therapeutic efficacy and reduces the population eligible for AAV vector treatment, with only immunologically privileged organs, such as the eye, being more amenable to viral vector delivery (22, 23). Capsid modifications such as mutations at critical antigenic sites (24) and rational capsid residue substitutions and insertion (25) have been used to generate antibody escape variants, as well as vectors with enhanced transduction. Since single residue mutations can result in dramatic effects on virus phenotypes (25, 26), it is important to understand how host cell molecules and antibodies interact with virus capsid.
The T=1 icosahedral capsid of AAVs are assembled from 60 copies of three overlapping viral proteins (VPs), VP1, VP2, and VP3 in a ratio of ∼1:1:10 (27, 28). The entire sequence of VP3 is contained within VP2, which is in turn contained within VP1. The VP1 thus contains a unique region, VP1u, and shares a common VP1/2 region with VP2. Superposition of the structurally ordered VP3 of available AAV serotype structures shows a core eight-stranded antiparallel β-barrel (βBIDG-βCHEF) motif and a conserved α-helix (αA), with variable loops inserted between these secondary structure elements (29). A total of nine variable regions (VRs) were defined within the loop insertions based on the comparison of two serotypes, AAV2 and AAV4, which differ in sequence and structure (30). The VR loops constitute the exterior capsid surface and dictate serotype diversity, such as capsid topology, tissue tropism, antibody recognition, and transduction properties (30–33). The capsid surface is characterized by a depression at the icosahedral 2-fold axis, protrusions surrounding a depression at the 3-fold axis, and a cylindrical channel at the 5-fold axis surrounded by a canyon-like depression (29).
Glycan receptors on cell surfaces facilitate the first step of many virus-host interactions and dictate virus host tissue tropism. Different AAV serotypes have evolved to use different glycan receptors for cell entry. These can be classified into three broad groups (34): (i) sialic acid (SIA) binders (AAV1, AAV4, AAV5, and AAV6) (35, 36), (ii) heparan sulfate proteoglycan (HSPG) binders (AAV2, AAV3, AAV6, and AAV13) (4, 26, 37, 38), and (iii) galactose binders (AAV9) (39, 40). AAV1 and AAV6, in group i, are closely related, differ by 6/736 amino acids (99.2% sequence identity), and recognize α2-3 and α2-6 N-linked SIA (35). In addition to SIA, AAV6 also binds HSPG dictated by the single K531E amino acid difference to AAV1 (26).
In this study, X-ray crystallography was used to identify the SIA binding site for AAV1, located on the base of the protrusions surrounding the icosahedral 3-fold axis of the capsid. Mutagenesis combined with cell binding and transduction assays confirmed N447, S472, V473, N500, T502, and W503 as being important for AAV1 and AAV6 SIA interaction. In addition, an overlap between the SIA binding site and the AAV1 ADK1a antigenic epitope (41) was confirmed by native dot blot analysis, enzyme-linked immunosorbent assay (ELISA), and antibody neutralization assay, suggesting steric hindrance of receptor attachment as the neutralization mechanism of this antibody. This study thus provides information for AAV1 and AAV6 determinants of cellular recognition, as well as for their engineering to escape from antibody recognition.
MATERIALS AND METHODS
Cell Culture.
Human embryonic kidney 293 (HEK293) cells were cultured in Dulbecco modified Eagle medium (Gibco, catalog no. 12100-046) supplemented with 10% fetal bovine serum and 1% antibiotic-antimycotic (Gibco, catalog no. 15240-052). The Chinese hamster ovary (CHO) cell lines Pro-5, Lec-2, Lec-8, and Lec-1 were obtained from the American Type Culture Collection (www.atcc.org). All of the CHO cells were cultured in α-minimum essential (α-MEM) medium (Gibco, catalog no. 32571-036) supplemented with 10% fetal bovine serum and 1% antibiotic-antimycotic. Cells were grown at 37°C in a 5% CO2 atmosphere.
Structure determination of the AAV1-SIA complex.
Expression and crystallization of the AAV1 virus-like particles (VLPs) have been previously described (42). Briefly, crystals of the AAV1 VLPs were grown at room temperature using the hanging-drop vapor diffusion method in buffer containing 0.1 M HEPES-NaOH (pH 7.3), 50 mM MgCl2, 0.03% NaN3, 1.0 M NaCl, 7% polyethylene glycol 6000, and 25% glycerol. Crystals were soaked into cryoprotectant solution (25% glycerol) containing a 10-fold molar excess (relative to VP binding sites) of a trisaccharide, 3′SLDN (Neu5Acα2-3GalNAcβ1-4GlcNAc) for 48 h and flash cooled in liquid nitrogen vapor prior to X-ray diffraction data collection. A total of 229 X-ray diffraction images were collected from one crystal at the A1 beamline at the Cornell High Energy Synchrotron Source (CHESS; Cornell University, Ithaca, NY). The data collection parameters were as follows: crystal-to-detector distance of 300 nm and an oscillation angle of 0.3° per image, with an exposure time of 70 s per image at a wavelength of λ = 0.979 Å.
The images were indexed, integrated, scaled, and merged using the HKL2000 suite (43). The crystals diffracted X-rays to 3.0-Å resolution. The crystals were shown to be monoclinic C2 with unit cell dimensions of a = 455.6, b = 261.8, c = 451.0 Å, α = γ = 90°, and β = 111.0°. The Rmerge and completeness values were 16.3 and 61.6%, respectively. The data processing statistics are provided in Table 1.
TABLE 1.
Data collection, processing, and refinement statistics of AAV1-SIA
| Parameter | AAV1-SIA |
|---|---|
| Data collection and processing | |
| Wavelength (λ, Å) | 0.979 (CHESS) |
| No. of films | 229 |
| Space group | C2 |
| Unit cell parameters | |
| a, b, c (Å) | 455.6, 261.8, 451.0 |
| β (°) | 111.0 |
| Resolution (Å) | 50.0–3.0 |
| Total no. of reflections | 1,170,740 |
| No. of unique reflections | 593,352 |
| Rmergea (%) | 16.3 |
| Completeness (%) | 61.6 |
| I/σ | 5.3 |
| Refinement | |
| Rfactorb (%) | 26.8 |
| Rfreec (%) | 27.6 |
| No. of atoms | 249,660 |
| RMSD | |
| Bond (Å) | 0.008 |
| Angle (°) | 1.065 |
| Ramachandran statistic (%) | |
| Favored | 92.9 |
| Allowed | 100 |
| Outliers | 0 |
| Avg B factor (Å) | |
| Main chain | 21.8 |
| Side chain | 23.2 |
| Ligand | 32.3 |
Rmerge = (Σ|Ihkl − 〈Ihkl〉 |/Σ|Ihkl|) × 100, where Ihkl is the intensity of an individual “hkl” reflection, and <Ihkl> is the mean intensity for all measured values of this reflection.
Rfactor = (Σ||Fobs| − |Fcalc||/Σ|Fobs|) × 100, where Fobs and Fcalc are the amplitudes for the observed and calculated reflections, respectively.
Rfree was calculated with 5% of the reflections excluded from the data set during refinement.
Based on the unit-cell volume and packing considerations, it was determined that there were four VLPs in the unit cell. The particle orientations were determined by self-rotation function searches using the GLRF program (44). Two particle orientations were observed, each with a noncrystallographic symmetry (NCS) 2-fold axis coincident with the crystallographic 2-fold axis, resulting in two independent half-capsids per asymmetric unit. The particles were positioned at (0.0,0.0,0.0) and (0.5,0.5,0.0) for the first orientation (Wyckoff notation a) and at (0.0,0.5,0.5) and (0.5,0.0,0.5) for the second orientation (Wyckoff notation b). Initial phases were calculated using the automr subroutine (molecular replacement) in the PHENIX program (45) with a 30-mer Cα model of the AAV1 VP3 monomer structure (PDB accession no. 3NG9).
After the automr procedure with the Cα model, which confirmed the presence of two half capsids (2 × 30-mers) in the crystallographic asymmetric unit, a cycle of refinement was carried to improve the initial phases using the phenix.refine subroutine in the PHENIX program. The reference VP3 Cα monomer was extracted from the starting 30-mer model, a polyalanine model of the AAV1 VP3 was superposed onto it, and the structure was converted to the wild-type (WT) AAV1 sequence by interactive substitution based on the resulting electron density map using the COOT program (46–48). The two half-capsids were then generated from the new VP3 model using NCS. Phases were improved by alternative interactive model building into averaged density maps using the COOT program (46–48), followed by coordinate refinement using the PHENIX program (49–51). After three rounds of this process, unassigned electron density on the exterior surface of the capsid that was not part of the VP3 and at a sigma (σ) threshold of 1.8 in an averaged “Fo−Fc” (where Fo and Fc represent the observed and calculated structure factor amplitudes, respectively) difference map, was interpretable as a SIA molecule associated with ∼30/60 VP molecules being used in the refinement process. The coordinate of a SIA was imported from the ligand library in the COOT program and manually docked into the averaged Fo−Fc difference map using the COOT program (46). The fitting of the docked SIA was refined against an averaged Fo−Fc difference map using “Real Space Refine Zone” in COOT and the coordinates of the AAV1-SIA complex were refined in PHENIX. The potential amino acids that interact with the SIA molecule were determined to be those with distances of ≤4 Å. The quality of the refined structure was analyzed by the MolProbity application (76). The data collection, processing, and refinement results are presented in Table 1. The figures were generated using the PyMOL program (52). The “Roadmap” image, depicting capsid surface accessible amino acid residues, was generated using the RIVEM program (53).
Generation of recombinant AAV (rAAV) mutant plasmid constructs.
SIA binding site mutants were designed based on the observed structural contacts between the AAV1 capsid and SIA (see above) and generated using the QuikChange Lightning site-directed mutagenesis kit (Agilent Technologies, catalog no. 210518-5) as described in the manufacturer's protocol. For each residue mutated, two complementary primers were used to introduce changes into the pXR1 (AAV1) or pXR6 (AAV6) plasmids with a single amino acid mutated to its structurally corresponding residue on AAV2, a HSPG binding serotype. The primers used to generate all these mutants are listed in Table 2. All of the mutants were sequenced before further analysis, and the plasmids were purified by the CsCl DNA extraction method.
TABLE 2.
Primer for generating SIA binding defective mutants and titer quantification
| Primer (VP aa)a | Sequence (5′–3′) |
|---|---|
| N447S-F | CCAATACCTGTATTACCTGAGCAGAACTCAAAATCAG |
| N447S-R | CTGATTTTGAGTTCTGCTCAGGTAATACAGGTATTGG |
| S472R-F | CTCCAGCTGGCATGCGGGTTCAGCCCAAAAACTG |
| S472R-R | GGGCTGAACACGCATGCCAGCTG |
| V473D-F | GCTGGCATGTCTGACCAGCCCAAAAACTGGC |
| V473D-R | CAGTTTTTGGGCTGATCAGACATGCCAG |
| N500E-F | CAGACAACAACAACAGCGAATTTACCTGGACTGGTGCTTC |
| N500E-R | GCACCAGTCCAGGTAAATTCGCTGTTGTTGTTG |
| T502S-F | CAACAACAGCAATTTTTCGTGGACTGGTGCTTC |
| T502S-R | TTTTGAAGCACCAGTCCACGAAAAATTGCTGTTGTTGTTG |
| W503A-F | CAACAGCAATTTTACCGCTACTGGTGCTTCAAAATATAACC |
| W503A-R | GGTTATATTTTGAAGCACCAGTAGCGGTAAAATTGCTGTTG |
| Luciferase-F | ATTACACCCGAGGGGGATGA |
| Luciferase-R | CTCTCACACACAGTTCGCCT |
aa, amino acid.
Expression and purification of WT and mutant rAAV vectors.
The rAAV vectors were generated in HEK293 cells by triple plasmid transfection (54). The plasmids used were the WT pXR1 or pXR6 or mutant capsid plasmids, pTR-Flag-Luciferase (Luc) reporter plasmid, and pXX6 helper plasmid (54). Ten 15-cm2 plates, seeded with HEK293 cells, were grown to 90% confluency and transfected by polyethyleneimine with the plasmids in a molar ratio of 1:1:3 (pXR:pTR-Flag-Luciferase:pXX6) for a total of 90 μg of plasmid DNA per plate. After 72 h, the cells were harvested and resuspended in TNET (500 mM Tris-HCl [pH 8.0], 300 mM NaCl, 5 mM EDTA, 0.06% Triton X-100), subjected to three rounds of freeze-thawing, and treated with 100 U/ml Benzonase (Millipore, catalog no. 71205-3) at 37°C for 30 min after the last thaw. The cell debris was removed by centrifugation at 3,500 × g for 10 min, and the supernatant was purified by iodixanol gradient ultracentrifugation and ion exchange using a HiTrap Q HP column (GE Healthcare, catalog no. 17-5158-01) (55). Fractions containing purified rAAV-Luc vectors were combined, concentrated, and dialyzed into phosphate-buffered saline (PBS) prior to use.
Negative-stain EM.
Portions (5 μl) of purified samples were loaded onto carbon-coated copper electron microscopy (EM) grids (Ted Pella) and incubated at room temperature for 5 min. The grids were blotted dry using filter paper (Whatman) and stained with Nano-W (Nanoprobes, catalog no. 2018) for 10 s. The grids were blotted with filter paper again to remove the staining solution and left at room temperature until completely dry. Micrographs were collected by imaging the grids on a Tecnai G2 Spirit transmission EM (FEI) microscope operated at an accelerating voltage of 120 keV.
Virus genome titer and capsid assembly quantification.
The genome titer of rAAV-Luc vectors was determined based on the packaged luciferase gene. To remove unpackaged DNA prior to packaged genome quantification, 10 μl of each purified rAAV vector was first treated with 1 U of benzonase in a total 200-μl reaction mixture containing 20 mM Tris-HCl (pH 8.5), 15 mM NaCl, and 2 mM MgCl2 at 37°C for 30 min. The packaged reporter gene was then released by adding 22 μl of proteinase K buffer (100 mM Tris HCl [pH 8.0], 100 mM EDTA, 10% sodium dodecyl sulfate) to the reaction mixture, followed by 2 μl of proteinase K (18.6 mg/ml; Fisher, catalog no. BP1700-05). The reaction was carried out at 37°C for 30 min, and the reporter gene was twice extracted by using phenol-chloroform-isoamyl alcohol, followed by ethanol precipitation. The genome titer of each rAAV-Luc vector was determined by quantitative-PCR (q-PCR) using primers (Table 2) targeting the pTR-Flag-Luciferase gene as previously described (55).
The capsid titer of the rAAV-Luc vectors was determined by an ELISA. Fourteen nanograms of cell lysate protein were used for each vector preparation. The assay was conducted as described in the AAV1 ELISA kit according to the manufacturer's protocol (Progen, catalog no. PRAAV1).
Cell binding assay.
Pro-5 cells were trypsinized from 10-cm2 plates and washed twice with cold serum-free α-MEM medium (Gibco, catalog no. 32571-036). The resuspended cells were added to 1.5-ml centrifuge tubes at 2.5 × 105 cells/tube in 50 μl of cold serum-free α-MEM medium. The cells were cooled to 4°C for 15 min before vector was added. Each rAAV-Luc vector was resuspended in 50 μl of cold serum-free α-MEM and added to the cells at a titer of 2.5 × 1010 vector genomes (vg)/tube (multiplicity of infection = 105). After incubation at 4°C for 90 min, the cells were collected and washed three times with cold PBS. The luciferase gene in bound vectors was extracted and quantified by q-PCR as described above.
Transduction assay.
CHO cell lines (Pro-5, Lec-2, Lec-8, and Lec-1) were seeded overnight at 5 × 104 cells/well into a 96-well plate. The cells were then washed with cold PBS, and 50 μl of cold serum-free α-MEM was added. After cooling down to 4°C for 15 min, rAAV-Luc vectors, at 5 × 109 vg in 50 μl of serum-free α-MEM, were added to each well, followed by incubation at 4°C for another 90 min. The cells were washed three times with cold PBS, warm complete α-MEM was added, and they were incubated in a CO2 culture incubator for 48 h. Luciferase expression was determined by a luciferase assay (Promega, catalog no. E1500) as described in the manufacturer's protocol.
Native dot blot assay.
To check for potential overlap of the SIA binding site with antigenic epitopes, the WT and mutant rAAV-Luc vectors were tested for reactivity against four different mouse monoclonal antibodies (MAbs)—ADK1a, ADK1b, 5H7, and 4E4—directed against WT AAV1 capsids (56, 75) and the B1 MAb that reacts against denatured AAV VPs (57). One hundred nanograms of the rAAV-Luc vectors in TNET buffer was used for native dot blot experiments. Each rAAV-Luc sample was loaded onto a nitrocellulose membrane (Amersham, catalog no. 10600007) using the Spot-Blot system (GE Healthcare). The unboiled vectors were blotted with hybridoma supernatant for ADK1b and 5H7 and purified antibodies for ADK1a and 4E4 produced as previously described (41, 58). Briefly, hybridoma supernatant isolated from cells producing the MAb was purified by Hi-Trap protein G HP columns (GE Healthcare). The intact IgG antibodies were eluted with glycine-HCl (pH 2.7) directly into 1 M Tris-HCl (pH 9.0) to achieve a neutral pH. The boiled samples were blotted with B1 (ARP, catalog no. 365158). Horseradish peroxidase-conjugated anti-mouse IgG (GE Healthcare, catalog no. NA931V) was used as secondary antibody, and the signal was detected by chemiluminescent reagent (Millipore, catalog no. WBKLS0500).
Neutralization assay.
To determine the ability of the AAV1 S472R mutant, which showed reduced binding by ADK1a, to escape neutralization by the IgG, purified ADK1a MAb was used. Briefly, Pro-5 cells were seeded at 5 × 104 cells/well in a 96-well plate and incubated at 37°C for 24 h. The rAAV-Luc vectors were preincubated with ADK1a at five different IgG/capsid ratios (6:1, 12:1, 30:1, 60:1, and 120:1) at 37°C for 30 min. The vector and antibody complexes were added to the cells in the appropriate volumes (based on the titer) required to achieve 5 × 109 vg/well. The expression of luciferase was assessed 48 h postinfection according to the manufacturer's protocol.
Coordinates.
The coordinates and structure factors for the AAV1-SIA complex structure have been deposited in the PDB database (http://www.rcsb.org/) under accession number 5EGC.
RESULTS
The base of the 3-fold protrusions serves as the AAV1 SIA binding pocket.
The AAV1-SIA crystal diffracted X-rays to 3.0-Å resolution. The data set was 61.6% complete and the overall Rmerge was 16.3% after data processing and reduction. After refinement, the R-work, indicating the quality of the data by measuring the agreement between the experimentally observed data and the model built into the electron density, is 26.8% (Table 1). These values are within acceptable range as shown in previously published virus structures at comparable resolution, including other AAV structures (30, 59). As reported for other AAV structures, the VP was ordered from residue 217 within the VP3 common region, with the VP1u and VP1/2 common regions being disordered (29). The ordered VP3 region exhibited the secondary structure topology of the AAVs, including the nine VRs previously described when superposed onto other AAV VP structures (not shown). The AAV1 capsid structure exhibited the structural characteristics previously reported for other AAVs, including the depressions at the icosahedral 2-fold and surrounding the 5-fold axes, protrusions surrounding the 3-fold axis, and channel at the 5-fold axis (Fig. 1A) (29, 59). Density for the single nucleotide residue ordered inside a pocket in most AAV structures determined to date (for example, see reference 59) was also observed in this complex structure (not shown).
FIG 1.
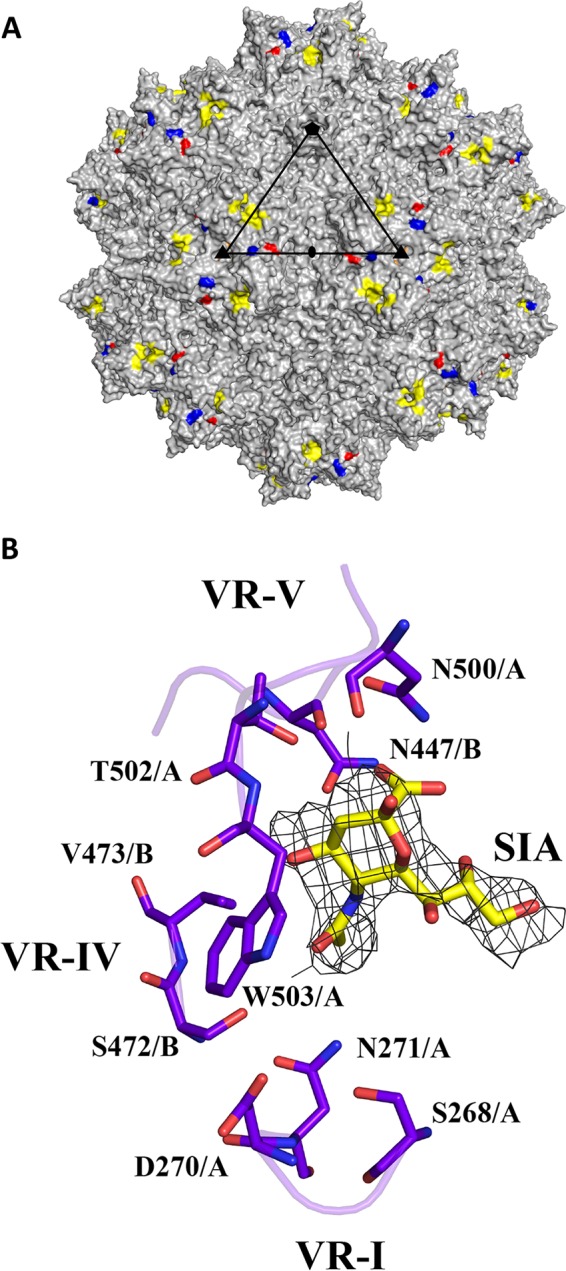
SIA binding site on the AAV1 capsid. (A) Capsid surface image of AAV1 with the location of observed SIA density (yellow) and three surface-exposed residues that differ between AAV1 and AAV6: E531K (red), F584L (blue), and A598V (orange, not visible in the orientation shown). A viral asymmetric unit is depicted by a triangle (in black) bounded by two 3-fold (filled triangle) axes, divided by a line drawn through the 2-fold (filled oval) and a 5-fold (filled pentagon) axis. (B) Stick representation of a section of the AAV1-SIA crystal structure and predicted SIA interacting residues. The binding pocket is formed by two monomers with chains designated A and B. Some of the residues are located at two variable regions: (i) VR-I for S268, D270, and N271; (ii) VR-IV for N447, S472, and V473; (iii) VR-V for N500, T502, and W503. The electron density map of SIA is an averaged 2Fo−Fc map shown at a sigma threshold of 2.0 with radius cutoff (carve) of 1.9 Å. This figure was generated using the PyMOL program (52).
The AAV1 crystal was soaked with the trisaccharide, Neu5Acα2-3GalNAcβ1-4GlcNAc (3′SLDN), which was identified as a top hit glycan interacting with AAV1 using a glycan microarray (35). However, only ordered electron density interpretable as the terminal SIA molecule was observed at the base of the protrusions surrounding icosahedral 3-fold axis (Fig. 1A and B). This indicates that the remaining GalNAcβ1-4GlcNAc moiety is flexible and/or not ordered. The terminal SIA was easily identifiable at a σ threshold of 1.8 and 0.5 in the averaged Fo−Fc and 2Fo−Fc density maps, respectively. Refinement of the SIA with a 0.5 occupancy resulted in SIA atom temperature factors (B-factors) within the range of those for the surrounding VP residue atoms with occupancy of 1.0 (Table 1). The SIA binding pocket is formed by two 3-fold symmetry related VP3 monomers contributed by three VRs: VR-I, VR-IV, and VR-V. Nine amino acids were identified as potential SIA binding contacts, based on a distance contact criteria of ≤4 Å between the SIA atoms and the capsid. These residues are as follows: S268, D270, and N271 in VR-I; N447, S472, and V473 in VR-IV; and N500, T502, and W503 in VR-V (Fig. 1B).
AAV1 and AAV6 utilize the same SIA binding pocket.
To confirm the structurally predicted AAV1 SIA binding residues, a structural alignment of the AAV1 VP3 with that of the HS binding AAV2 was performed. The five (out of the nine) residues which differed—N447, S472, V473, N500, and T502 (Table 3)—were mutated to their structurally corresponding amino acids in AAV2 in the AAV1 background for characterization. Tryptophan 503, conserved in AAV1, AAV2, and AAV9, was mutated to an alanine (Table 3).
TABLE 3.
SIA binding pocket residue alignment (in VP1 numbering)a
| AAV1 | AAV2 | AAV6 | AAV9 |
|---|---|---|---|
| S268 | S268 | S268 | S269 |
| D270 | D271 | D270 | D271 |
| N271 | N272 | N271 | N272 |
| Y445 | Y445 | Y445 | Y446 |
| N447 | S446 | N447 | S448 |
| G470 | G470 | G470 | N470 |
| S472 | R471 | S472 | A472 |
| V473 | D472 | V473 | V473 |
| N500 | E499 | N500 | E500 |
| T502 | S501 | T502 | A502 |
| W503 | W502 | W503 | W503 |
| SIA | HS | SIA | GAL |
Residues in boldface indicate amino acids that play a role in binding the glycan indicated for each serotype. Underlined residues for AAV2 are the type to which structurally equivalent AAV1 and AAV6 residues were substituted to eliminate SIA binding.
The amino acid composition of the SIA binding pocket is identical between AAV1 and AAV6 (Table 3). The 5/6 residues (out of 736) which differ between the two viruses that are ordered in their structures are E531K, F584L, and A598V (AAV1/AAV6), located around the 3-fold axis on the exterior capsid surface (Fig. 1A), and E418D and N642H located on the interior capsid surface, also close to the 3-fold axis (59). Residue L129F located within the VP1u is disordered in all AAV structures determined to date. The AAV1/6 differing residues ordered in the crystal structure do not overlap with those in the AAV1 SIA binding pocket (Fig. 1A); thus, it was rationalized that the dual glycan binding AAV6 may also utilize the same pocket as AAV1 to interact with SIA. Thus, the role of the pocket residues in SIA binding and cell transduction was tested for AAV6 alongside AAV1.
Negative-stain EM showed that all SIA site mutants formed intact capsids (Fig. 2A and C). Quantification of the luciferase reporter gene packaged inside the capsids by q-PCR showed no DNA packaging defects (Fig. 2B and D), except for mutant rAAV6-N447S, which showed a 100-fold decrease in packaged genome copy number compared to WT rAAV6. This defect in AAV6, but not in AAV1, is difficult to explain because the six residues which differ between AAV1 and AAV6 are not proximal to the SIA binding pocket. Thus, how this residue is affecting the packaging phenotype for AAV6 is unknown. Due to its packaging defect, the rAAV6-N447S mutant was excluded from analysis of cell binding and transduction efficiency of the binding pocket mutants compared to the WT virus.
FIG 2.
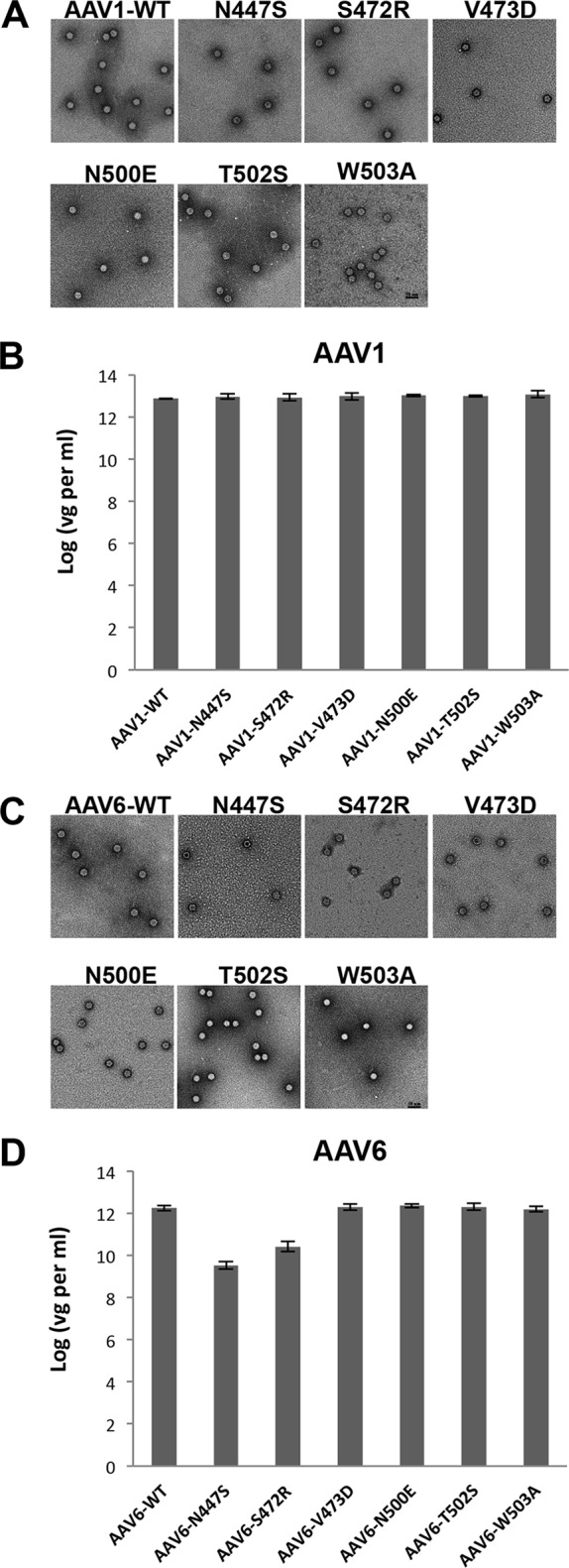
WT AAV and SIA-binding site mutant characterization. (A and C) Negative stain micrographs and (B and D) genome packaging titers of WT AAV and SIA binding site mutants. Panels A and B show results for AAV1, and panels C and D show results for AAV6. The genome packaging titers were determined by q-PCR. The values are means from three experiments displayed in a log scale; the error bars represent standard deviations. vg, vector genome.
Structurally predicted SIA binding residues are important in SIA-dependent cell attachment.
Cell binding assays with the parental CHO cell Pro-5, which displays SIA as a terminal glycan, showed a decrease for 5/6 AAV1/6 mutants: N447S, V473D, N500E, T502S, and W503A (Fig. 3). This confirmed the importance of these residues in SIA binding for both serotypes. However, unexpectedly, the mutant S472R showed an increased/comparable Pro-5 cell binding in AAV1 and AAV6 (Fig. 3). To test the possibility that the polar to basic residue change in S472R conferred a new binding phenotype for negatively charged molecules on the cell surface, such as the HS utilized by AAV2, a heparin column was used to test the affinity of this variant compared to AAV2. The result showed that S472R does not bind to the HS (data not shown). These observations indicate that this AAV1 and AAV6 residue is involved in initial cell engagement during infection and that the serine-to-arginine change alters the SIA interaction, possibly increasing affinity.
FIG 3.
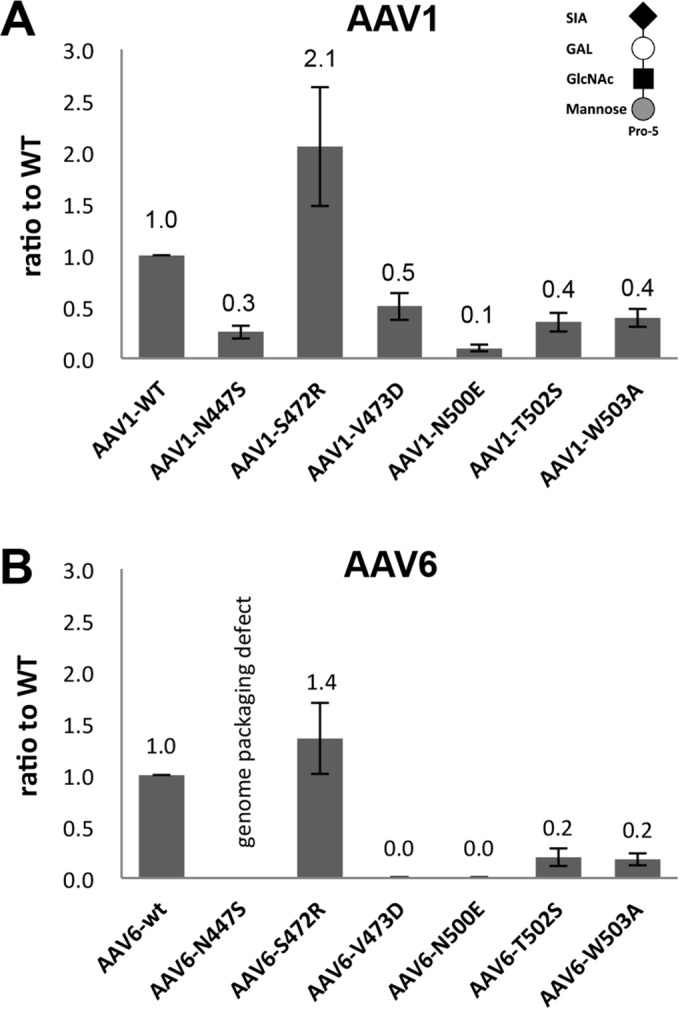
Pro-5 cell binding. The binding of AAV1 (A) and AAV6 (B) WT and SIA binding site mutants to Pro-5 cells. Data are normalized to the WT (1.0) in the y axis. The values are means from three experiments; the error bars represent standard deviations.
Structurally predicted SIA binding residues are involved in SIA-mediated cell transduction.
The infectivity of the AAV1 and AAV6 SIA binding mutants were tested in CHO cells with different levels of surface glycosylation to further assess the role of different cell surface glycans (Fig. 4). In addition to the Pro-5 cells, Lec-2 (deficient in the CMP-sialic acid Golgi transporter and exposes terminal galactose on surface glycans), Lec-8 (deficient in the UDP-galactose Golgi transporter and displays terminal N-acetylglucosamine on surface glycans), and Lec-1 (deficient in the N-acetylglucosaminyltransferase and displays terminal mannose on surface glycans) were tested. The transduction by WT rAAV1 was significantly decreased (∼72%) on the terminal SIA devoid cell line Lec-2 compared to the parental Pro-5 cell line (Fig. 4A). Transduction by rAAV1 mutants, N447S, V473D, N500E, T502S, and W503A, defective in Pro-5 binding also showed a >63% reduction in transduction in Pro-5 cells compared to WT rAAV1. Interestingly, although rAAV1-S472R showed a 2-fold increase in Pro-5 cell binding compared to the WT virus, the transduction ability of this mutant was also reduced by ∼68% in these cells. The transduction efficiency of WT rAAV1 was reduced to ∼30, 40, and 60% in the Lec-2, Lec-8, and Lec-1, respectively; while the mutants had negligible transduction levels (Fig. 4A).
FIG 4.
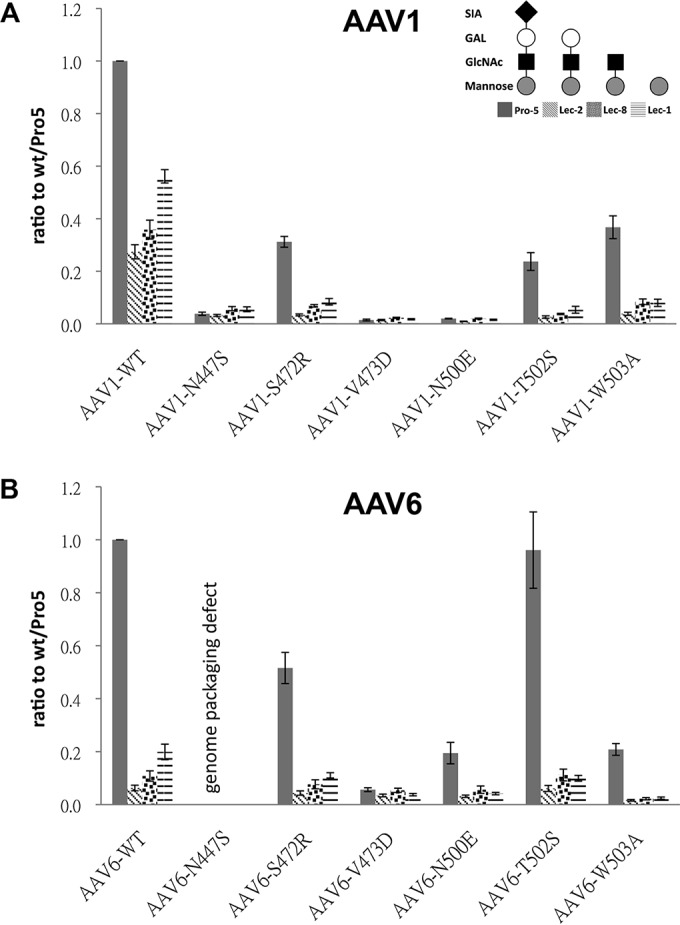
Transduction assays in CHO cells. Transduction by AAV1 (A) and AAV6 (B) WT and SIA binding mutants in Pro-5, Lec-2, Lec-8, and Lec-1, as indicated in panel A. Data are normalized to WT transduction in Pro-5 cells set to 1 in the y axis. The values are means from three experiments; the error bars represent standard deviations.
The transduction level of WT rAAV6 is also decreased in Lec-2, Lec-8, and Lec-1 cells (Fig. 4B). Four mutants—S472R, V473D, N500E, and W503A—showed a >50% reduction in transduction of Pro-5 cells. These same four mutants did not transduce the Lec2, Lec8, and Lec1 cells, an observation consistent with their lack of binding to Pro-5 cells. Surprisingly, mutant T502S transduced Pro-5 cell at the same level as WT AAV6 despite an ∼80% decrease in cell binding compared to the WT virus. This suggests a gain of function related to postentry events for this mutant that is not related to SIA binding.
SIA binding site overlaps with AAV1 ADK1a epitope.
Several residues in the SIA binding site overlap amino acids reported to form the antigenic footprint for the ADK1a MAb directed against the AAV1 capsid (Fig. 5A) (41). Thus, the possibility that this MAb neutralizes infection due to steric hindrance of the receptor attachment was explored. A native dot blot showed that three of the SIA binding mutants, S472R, V473D, and N500E, can escape from ADK1a recognition but not from MAbs ADK1b, 5H7, and 4E4, which are also directed against the AAV1 capsid but have different epitopes (Fig. 5B). This observation is consistent with the overlap of the SIA binding site with only the mapped ADK1a footprint (Fig. 5A). This escape from ADK1 was further confirmed by an ELISA using ADK1a as primary antibody (Fig. 5C). The rAAV1-S472R that still retained a reduced level of Pro-5 transduction (∼30% compared to WT) was thus tested for its ability to escape neutralization during infection in these cells. WT rAAV1 preincubated with ADK1a IgG prior to infection showed a dose-dependent decrease in transduction level in Pro5 cells with increasing ADK1a-to-capsid ratios (Fig. 6A). In contrast, the rAAV1-S472R transduced Pro5 cells in the presence of 120 IgG molecules per capsid, which is equivalent to two MAb molecules per VP (Fig. 6B). These data indicate that ADK1a neutralizes AAV1 infection at the viral entry stage by blocking attachment.
FIG 5.
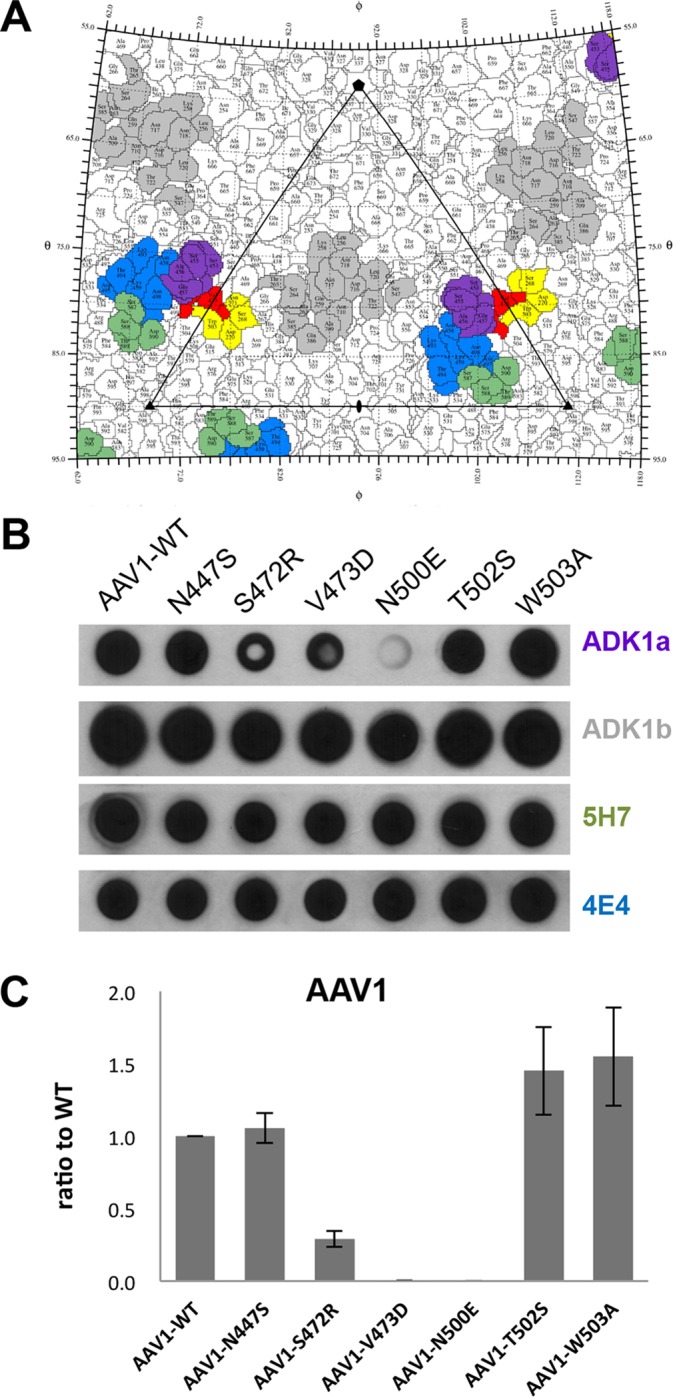
Antigenic reactivity of SIA binding mutants. (A) Stereographic Roadmap projection of the epitopes mapped for AAV1 specific antibodies: ADK1a (purple), ADK1b (gray), 5H7 (green), and 4E4 (blue), along with SIA binding site (yellow). Residues that overlap between the ADK1a epitope and the SIA binding site are colored in red. (B) Dot blot analysis of SIA binding mutants against AAV1 antibodies: ADK1a, ADK1b, 5H7, and 4E4. (C) ELISA results for AAV1 SIA mutants using ADK1a as primary antibody. Data are normalized to WT set to 1 in the y axis. The values are means from three experiments; the error bars represent standard deviations. Panel A was generated using the RIVEM program (53).
FIG 6.
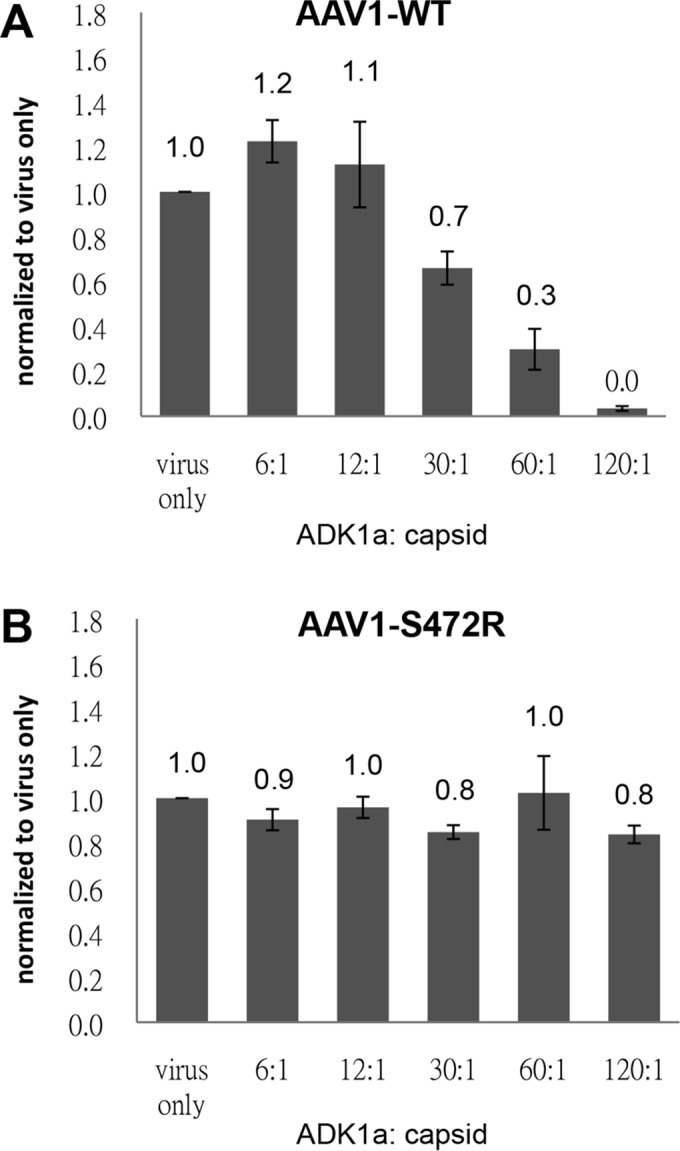
Neutralization of WT AAV1 and the AAV1-S472R infection of Pro-5 cells by ADK1a. Cell transduction by WT AAV1 (A) and AAV1-S472R (B), respectively, preincubated with ADK1a antibody was assessed. The ratios shown are ADK1a IgG molecules per capsid. Data are normalized to virus only set to 1 in the y axis. The values are means from three experiments; the error bars represent standard deviations.
DISCUSSION
Capsid modification via rational design is a tested strategy for overcoming challenges still remaining for the AAV delivery system. This approach can be used to generate new chimeric viruses by swapping or mutating specific capsid residues or regions between different serotypes based on the sequence and structural information to achieve desired functional phenotypes. An example is the liver detargeting and muscle tropic chimera, AAV2i8, generated by substituting a hexapeptide from AAV8 into AAV2's HS binding region based on structural information (60). Another example is the enhanced muscle-tropic chimera, AAV2.5, generated by introducing five residues from muscle tropic serotype AAV1 into the AAV2 background (61). Both of these chimeras showed altered transduction and antigenic profiles, including escape from antibody recognition compared to the WT parental vectors. Therefore, knowledge of capsid structures and mapped capsid regions that engage in cellular interaction is important for capsid manipulation for improved vector efficacy.
The structure of the AAV1-SIA complex was determined in an effort to identify the capsid region utilized for cell entry interactions. This region was mapped to a capsid surface pocket at the base of the 3-fold protrusion on AAV1 capsid and confirmed to play an essential role in cell binding and transduction for AAV1 and the closely related AAV6. Significantly, a structurally analogous pocket serves as the galactose (GAL) binding pocket in AAV9 (40). Residues dictating AAV9 GAL binding are D271, N272, Y446, N470, A472, V473, and W503 identified by mutagenesis and ligand docking (40). Structural alignment of the glycan binding pockets showed that 5/11 residues are different between AAV1/6 and AAV9 (Table 3). Among these residues, the long side chain of AAV9 N470, shown to be critical for its interaction with GAL, clashes with the modeled SIA molecule in the AAV1-SIA complex and would likely hinder an interaction (Fig. 7). This residue is a glycine in AAV1. Therefore, residue 470 may be the glycan usage determinant for AAV1 and AAV9.
FIG 7.
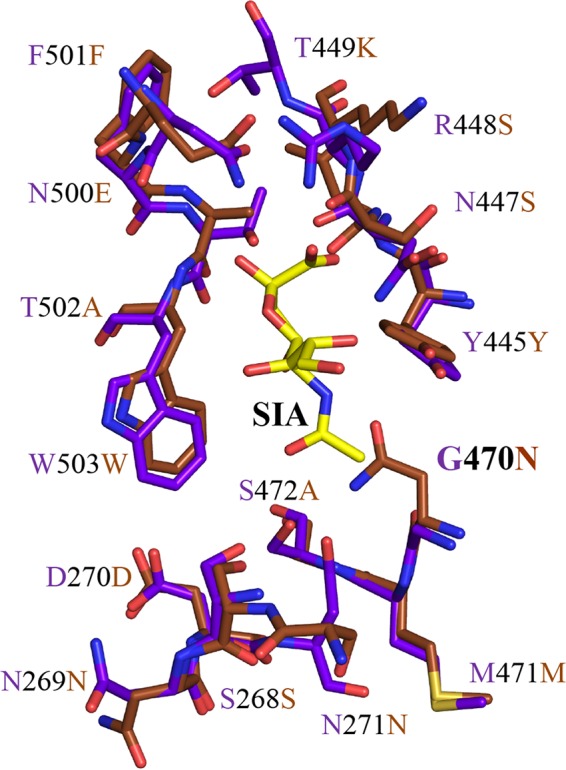
SIA and GAL binding pocket. A superposition of the stick representation of the AAV1 SIA binding pocket (in purple) and AAV9 GAL binding site (in brown) is shown. The predicted glycan binding determinant, residue 470, is glycine in AAV1 and asparagine in AAV9. This figure was generated using the PyMOL program (52).
Site-directed mutagenesis of the amino acids forming the SIA binding pocket in AAV1 and AAV6 to their structurally equivalent residue types in AAV2 confirmed the role of 5/6 residues mutated in cell binding and transduction. The observed increase in cell binding by the S472R mutation for both viruses did not translate to an increase in transduction. A three-dimensional (3D) model of the S472R variant indicates that substitution of the polar serine residue with the positively charged and larger arginine side chain conferred the possibility of forming five hydrogen bonds between the amino group of the arginine and the backbone carbonyl oxygen atoms of residues S268, D270, and N271 and the SIA molecule. These extra interactions may stabilize the capsid to increase SIA binding affinity. The same stabilizing interactions may prevent post-cell-entry processing events during infection resulting in the lack of correlation between increased cell binding and transduction.
Given that the residue composition of the SIA binding pocket is identical between AAV1 and AAV6, the difference in transduction levels for the T502S mutation in the background of the two viruses was unexpected (Fig. 4). This observation suggested that the residues which differ between AAV1 and AAV6 are affecting this mutant's phenotype. The variant is defective for cell binding, suggesting that the surface differences are not playing a role. The difference in transduction may thus be due to postentry events associated with the differences at positions 129, 418, and 642. Residues 129, 418, and 642 have been reported to play a role in AAV1 and AAV6 transduction (62, 63). Residue 129 is not ordered in the AAV structures, while T502 is at a distance of 38.9 and 35.4 Å from 418 and 642, respectively, located on the interior surface of the capsid. However, how these residues are exerting an effect is difficult to understand. Regardless of the types of glycan utilized by different AAV serotypes, the 3-fold protrusions and surrounding regions have been identified as common glycan binding sites (Fig. 8) (34). At the top of 3-fold protrusion, AAV4 binds to SIA via K492, K503, M523, G581, Q583, and N585 (64). Structural alignment showed that AAV4 K492 and K503, which superpose with AAV1 S499 and T504, are adjacent to the SIA binding residue N500 and W503 on AAV1. AAV5 binds to SIA by M569, A570, T571, A581, G583, T584, Y585, N586, and L587 (65). However, none of these AAV5 residues is close to the SIA binding site on AAV1. Rather the residues are located in the depression at the 3-fold axis. AAV6 interacts with HS using K459 and K493, which are located on the wall of the 3-fold protrusions facing the 5-fold axis (66). AAV6 also uses K531 and R576, at the base of 3-fold protrusion facing the 3-fold axis, to interact with HS (26, 66). K531 is structurally equivalent to K528 on AAV13 and dictates its HS binding. The residues utilized by AAV2, R484, R487, K532, R585, and R588, and AAV3B, R594, to interact with HS are also located at the inner wall of the 3-fold protrusions facing the 3-fold axis (67–69, 77, 78). These observations, along with the data from this study, suggest that AAVs have evolved glycan recognition niches, within the 3-fold region, instead of using the same residues, to engage their respective glycans during infection.
FIG 8.

AAV glycan receptor binding footprints. (A) Available glycan binding sites for AAV1, AAV2, AAV3B, AAV4, AAV5, AAV6, AAV9, and AAV13 projected onto the surface density of the AAV1 capsid. (B) Closeup of the receptor binding footprints viewed looking down the icosahedral 3-fold axis. Residues involved in glycan interactions for the different AAV serotypes are listed in the color key. Residues in parentheses indicate AAV1 numbering. The figures were generated using the PyMOL program (52).
The prevalence of preexisting antibodies against AAV vectors limits their therapeutic efficacy (70, 71). To overcome this challenge, capsid modification based on capsid antigenic epitopes can generate variants with the ability to escape from antibody recognition (25, 72). The antigenic structures of AAV1, AAV2, AAV5, AAV6, and AAV8 have been characterized by cryo-electron microscopy and image reconstruction (41, 58, 73, 74). Despite sequence diversity (∼60 to 99%) between these serotypes, two common antigenic regions were identified which are located on the 2/5-fold wall and 3-fold protrusion on the capsid surface (41). Although their mechanisms of neutralization are mostly unknown, some of these antigenic footprints coincided with regions determining receptor binding and regions controlling transduction phenotype. For example, the HS binding site for AAV2 overlaps with the epitope described for the C37-B MAb specific for this serotype which binds to the top of 3-fold protrusions (57, 67, 68, 74). This MAb blocks virus entry. On the other hand, A20 MAb, also directed against AAV2, binds to the 2/5-fold wall described as a transduction determinant (33, 73). This MAb inhibits infection at a postnuclear entry step. For AAV5, mutation of its SIA binding site has also been shown to create variants able to escape from antibody recognition (65), again pointing to a neutralization mechanism involving receptor attachment blockage. In the example of AAV8, its ADK8 MAb blocks infection at a post-cell/pre-nuclear-entry step, suggesting a block in cellular trafficking (58). In this study, the ADK1a was shown to escape recognition by SIA site mutants S472R, V473D, and N500E, pointing to a receptor blockage mechanism for neutralization. Information on receptor attachment sites, as presented here and their potential overlap with antigenic regions provides an important 3D guide for engineering AAV capsids that can escape from antibody recognition while retaining their transduction ability.
ACKNOWLEDGMENTS
We thank the UF Interdisciplinary Center for Biotechnology Research Electron Microscopy core electron microscopy access (funded by the UF College of Medicine and UF Division of Sponsored Programs). We also thank R. Jude Samulski (UNC Chapel Hill) for his generous gift of the pXR1 and pXR6 plasmids.
This project was funded in part by a UF Alumni Fellowship to L.-Y.H, and NIH R01 grants GM082946 (M.A.-M.) and HL089221 (M.A.-M. and A.A.).
REFERENCES
- 1.Gao G, Vandenberghe LH, Alvira MR, Lu Y, Calcedo R, Zhou X, Wilson JM. 2004. Clades of adeno-associated viruses are widely disseminated in human tissues. J Virol 78:6381–6388. doi: 10.1128/JVI.78.12.6381-6388.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schmidt M, Katano H, Bossis I, Chiorini JA. 2004. Cloning and characterization of a bovine adeno-associated virus. J Virol 78:6509–6516. doi: 10.1128/JVI.78.12.6509-6516.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schmidt M, Voutetakis A, Afione S, Zheng C, Mandikian D, Chiorini JA. 2008. Adeno-associated virus type 12 (AAV12): a novel AAV serotype with sialic acid- and heparan sulfate proteoglycan-independent transduction activity. J Virol 82:1399–1406. doi: 10.1128/JVI.02012-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schmidt M, Govindasamy L, Afione S, Kaludov N, Agbandje-McKenna M, Chiorini JA. 2008. Molecular characterization of the heparin-dependent transduction domain on the capsid of a novel adeno-associated virus isolate, AAV(VR-942). J Virol 82:8911–8916. doi: 10.1128/JVI.00672-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mori S, Wang L, Takeuchi T, Kanda T. 2004. Two novel adeno-associated viruses from cynomolgus monkey: pseudotyping characterization of capsid protein. Virology 330:375–383. doi: 10.1016/j.virol.2004.10.012. [DOI] [PubMed] [Google Scholar]
- 6.Bello A, Tran K, Chand A, Doria M, Allocca M, Hildinger M, Beniac D, Kranendonk C, Auricchio A, Kobinger GP. 2009. Isolation and evaluation of novel adeno-associated virus sequences from porcine tissues. Gene Ther 16:1320–1328. doi: 10.1038/gt.2009.82. [DOI] [PubMed] [Google Scholar]
- 7.Kotterman MA, Schaffer DV. 2014. Engineering adeno-associated viruses for clinical gene therapy. Nat Rev Genet 15:445–451. doi: 10.1038/nrg3742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mueller C, Flotte TR. 2008. Clinical gene therapy using recombinant adeno-associated virus vectors. Gene Ther 15:858–863. doi: 10.1038/gt.2008.68. [DOI] [PubMed] [Google Scholar]
- 9.Manno CS, Chew AJ, Hutchison S, Larson PJ, Herzog RW, Arruda VR, Tai SJ, Ragni MV, Thompson A, Ozelo M, Couto LB, Leonard DG, Johnson FA, McClelland A, Scallan C, Skarsgard E, Flake AW, Kay MA, High KA, Glader B. 2003. AAV-mediated factor IX gene transfer to skeletal muscle in patients with severe hemophilia B. Blood 101:2963–2972. doi: 10.1182/blood-2002-10-3296. [DOI] [PubMed] [Google Scholar]
- 10.Bartlett JS, Samulski RJ, McCown TJ. 1998. Selective and rapid uptake of adeno-associated virus type 2 in brain. Hum Gene Ther 9:1181–1186. doi: 10.1089/hum.1998.9.8-1181. [DOI] [PubMed] [Google Scholar]
- 11.Koeberl DD, Alexander IE, Halbert CL, Russell DW, Miller AD. 1997. Persistent expression of human clotting factor IX from mouse liver after intravenous injection of adeno-associated virus vectors. Proc Natl Acad Sci U S A 94:1426–1431. doi: 10.1073/pnas.94.4.1426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Takeda S, Takahashi M, Mizukami H, Kobayashi E, Takeuchi K, Hakamata Y, Kaneko T, Yamamoto H, Ito C, Ozawa K, Ishibashi K, Matsuzaki T, Takata K, Asano Y, Kusano E. 2004. Successful gene transfer using adeno-associated virus vectors into the kidney: comparison among adeno-associated virus serotype 1 to 5 vectors in vitro and in vivo. Nephron Exp Nephrol 96:e119–126. doi: 10.1159/000077378. [DOI] [PubMed] [Google Scholar]
- 13.Chao H, Liu Y, Rabinowitz J, Li C, Samulski RJ, Walsh CE. 2000. Several log increase in therapeutic transgene delivery by distinct adeno-associated viral serotype vectors. Mol Ther 2:619–623. doi: 10.1006/mthe.2000.0219. [DOI] [PubMed] [Google Scholar]
- 14.Wang C, Wang CM, Clark KR, Sferra TJ. 2003. Recombinant AAV serotype 1 transduction efficiency and tropism in the murine brain. Gene Ther 10:1528–1534. doi: 10.1038/sj.gt.3302011. [DOI] [PubMed] [Google Scholar]
- 15.Lebherz C, Maguire A, Tang W, Bennett J, Wilson JM. 2008. Novel AAV serotypes for improved ocular gene transfer. J Gene Med 10:375–382. doi: 10.1002/jgm.1126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Blankinship MJ, Gregorevic P, Allen JM, Harper SQ, Harper H, Halbert CL, Miller AD, Chamberlain JS. 2004. Efficient transduction of skeletal muscle using vectors based on adeno-associated virus serotype 6. Mol Ther 10:671–678. doi: 10.1016/j.ymthe.2004.07.016. [DOI] [PubMed] [Google Scholar]
- 17.Zincarelli C, Soltys S, Rengo G, Koch WJ, Rabinowitz JE. 2010. Comparative cardiac gene delivery of adeno-associated virus serotypes 1 to 9 reveals that AAV6 mediates the most efficient transduction in mouse heart. Clin Transl Sci 3:81–89. doi: 10.1111/j.1752-8062.2010.00190.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Halbert CL, Allen JM, Miller AD. 2001. Adeno-associated virus type 6 (AAV6) vectors mediate efficient transduction of airway epithelial cells in mouse lungs compared to that of AAV2 vectors. J Virol 75:6615–6624. doi: 10.1128/JVI.75.14.6615-6624.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Huda F, Konno A, Matsuzaki Y, Goenawan H, Miyake K, Shimada T, Hirai H. 2014. Distinct transduction profiles in the CNS via three injection routes of AAV9 and the application to generation of a neurodegenerative mouse model. Mol Ther Methods Clin Dev 1:14032. doi: 10.1038/mtm.2014.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Calcedo R, Morizono H, Wang L, McCarter R, He J, Jones D, Batshaw ML, Wilson JM. 2011. Adeno-associated virus antibody profiles in newborns, children, and adolescents. Clin Vaccine Immunol 18:1586–1588. doi: 10.1128/CVI.05107-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Boutin S, Monteilhet V, Veron P, Leborgne C, Benveniste O, Montus MF, Masurier C. 2010. Prevalence of serum IgG and neutralizing factors against adeno-associated virus (AAV) types 1, 2, 5, 6, 8, and 9 in the healthy population: implications for gene therapy using AAV vectors. Hum Gene Ther 21:704–712. doi: 10.1089/hum.2009.182. [DOI] [PubMed] [Google Scholar]
- 22.Dinculescu A, Glushakova L, Min SH, Hauswirth WW. 2005. Adeno-associated virus-vectored gene therapy for retinal disease. Hum Gene Ther 16:649–663. doi: 10.1089/hum.2005.16.649. [DOI] [PubMed] [Google Scholar]
- 23.Bennett J, Maguire AM, Cideciyan AV, Schnell M, Glover E, Anand V, Aleman TS, Chirmule N, Gupta AR, Huang Y, Gao GP, Nyberg WC, Tazelaar J, Hughes J, Wilson JM, Jacobson SG. 1999. Stable transgene expression in rod photoreceptors after recombinant adeno-associated virus-mediated gene transfer to monkey retina. Proc Natl Acad Sci U S A 96:9920–9925. doi: 10.1073/pnas.96.17.9920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Maheshri N, Koerber JT, Kaspar BK, Schaffer DV. 2006. Directed evolution of adeno-associated virus yields enhanced gene delivery vectors. Nat Biotechnol 24:198–204. doi: 10.1038/nbt1182. [DOI] [PubMed] [Google Scholar]
- 25.Li C, Diprimio N, Bowles DE, Hirsch ML, Monahan PE, Asokan A, Rabinowitz J, Agbandje-McKenna M, Samulski RJ. 2012. Single amino acid modification of adeno-associated virus capsid changes transduction and humoral immune profiles. J Virol 86:7752–7759. doi: 10.1128/JVI.00675-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wu Z, Asokan A, Grieger JC, Govindasamy L, Agbandje-McKenna M, Samulski RJ. 2006. Single amino acid changes can influence titer, heparin binding, and tissue tropism in different adeno-associated virus serotypes. J Virol 80:11393–11397. doi: 10.1128/JVI.01288-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Snijder J, van de Waterbeemd M, Damoc E, Denisov E, Grinfeld D, Bennett A, Agbandje-McKenna M, Makarov A, Heck AJ. 2014. Defining the stoichiometry and cargo load of viral and bacterial nanoparticles by Orbitrap mass spectrometry. J Am Chem Soc 136:7295–7299. doi: 10.1021/ja502616y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rose JA, Maizel JV, Inman JK, Shatkin AJ. 1971. Structural proteins of adenovirus-associated viruses. J Virol 8:766–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sujata H, Robert N, Mavis A-M. 2012. Parvoviruses: structure and infection. Future Virol 7:253–278. doi: 10.2217/fvl.12.12. [DOI] [Google Scholar]
- 30.Govindasamy L, Padron E, McKenna R, Muzyczka N, Kaludov N, Chiorini JA, Agbandje-McKenna M. 2006. Structurally mapping the diverse phenotype of adeno-associated virus serotype 4. J Virol 80:11556–11570. doi: 10.1128/JVI.01536-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Agbandje-McKenna M, Kleinschmidt J. 2011. AAV capsid structure and cell interactions. Methods Mol Biol 807:47–92. doi: 10.1007/978-1-61779-370-7_3. [DOI] [PubMed] [Google Scholar]
- 32.Wu P, Xiao W, Conlon T, Hughes J, Agbandje-McKenna M, Ferkol T, Flotte T, Muzyczka N. 2000. Mutational analysis of the adeno-associated virus type 2 (AAV2) capsid gene and construction of AAV2 vectors with altered tropism. J Virol 74:8635–8647. doi: 10.1128/JVI.74.18.8635-8647.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lochrie MA, Tatsuno GP, Christie B, McDonnell JW, Zhou S, Surosky R, Pierce GF, Colosi P. 2006. Mutations on the external surfaces of adeno-associated virus type 2 capsids that affect transduction and neutralization. J Virol 80:821–834. doi: 10.1128/JVI.80.2.821-834.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Huang LY, Halder S, Agbandje-McKenna M. 2014. Parvovirus glycan interactions. Curr Opin Virol 7:108–118. doi: 10.1016/j.coviro.2014.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wu Z, Miller E, Agbandje-McKenna M, Samulski RJ. 2006. α2,3 and α2,6 N-linked sialic acids facilitate efficient binding and transduction by adeno-associated virus types 1 and 6. J Virol 80:9093–9103. doi: 10.1128/JVI.00895-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kaludov N, Brown KE, Walters RW, Zabner J, Chiorini JA. 2001. Adeno-associated virus serotype 4 (AAV4) and AAV5 both require sialic acid binding for hemagglutination and efficient transduction but differ in sialic acid linkage specificity. J Virol 75:6884–6893. doi: 10.1128/JVI.75.15.6884-6893.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Summerford C, Samulski RJ. 1998. Membrane-associated heparan sulfate proteoglycan is a receptor for adeno-associated virus type 2 virions. J Virol 72:1438–1445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Handa A, Muramatsu S, Qiu J, Mizukami H, Brown KE. 2000. Adeno-associated virus (AAV)-3-based vectors transduce hematopoietic cells not susceptible to transduction with AAV-2-based vectors. J Gen Virol 81:2077–2084. doi: 10.1099/0022-1317-81-8-2077. [DOI] [PubMed] [Google Scholar]
- 39.Shen S, Bryant KD, Brown SM, Randell SH, Asokan A. 2011. Terminal N-linked galactose is the primary receptor for adeno-associated virus 9. J Biol Chem 286:13532–13540. doi: 10.1074/jbc.M110.210922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bell CL, Gurda BL, Van Vliet K, Agbandje-McKenna M, Wilson JM. 2012. Identification of the galactose binding domain of the adeno-associated virus serotype 9 capsid. J Virol 86:7326–7333. doi: 10.1128/JVI.00448-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tseng YS, Gurda BL, Chipman P, McKenna R, Afione S, Chiorini JA, Muzyczka N, Olson NH, Baker TS, Kleinschmidt J, Agbandje-McKenna M. 2015. Adeno-associated virus serotype 1 (AAV1)- and AAV5-antibody complex structures reveal evolutionary commonalities in parvovirus antigenic reactivity. J Virol 89:1794–1808. doi: 10.1128/JVI.02710-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Miller EB, Gurda-Whitaker B, Govindasamy L, McKenna R, Zolotukhin S, Muzyczka N, Agbandje-McKenna M. 2006. Production, purification and preliminary X-ray crystallographic studies of adeno-associated virus serotype 1. Acta Crystallogr Sect F Struct Biol Cryst Commun 62:1271–1274. doi: 10.1107/S1744309106048184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Otwinowski Z, Minor W. 1997. Processing of X-ray Diffraction data in oscillation mode. Methods Enzymol 276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 44.Tong L, Rossmann MG. 1997. Rotation function calculations with GLRF program. Methods Enzymol 276:594–611. doi: 10.1016/S0076-6879(97)76080-4. [DOI] [PubMed] [Google Scholar]
- 45.Adams PD, Afonine PV, Bunkóczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH. 2010. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr 66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Debreczeni JE, Emsley P. 2012. Handling ligands with Coot. Acta Crystallogr D Biol Crystallogr 68:425–430. doi: 10.1107/S0907444912000200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Emsley P, Cowtan K. 2004. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr 60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 48.Emsley P, Lohkamp B, Scott WG, Cowtan K. 2010. Features and development of Coot. Acta Crystallogr D Biol Crystallogr 66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Afonine PV, Grosse-Kunstleve RW, Urzhumtsev A, Adams PD. 2009. Automatic multiple-zone rigid-body refinement with a large convergence radius. J Appl Crystallogr 42:607–615. doi: 10.1107/S0021889809023528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Afonine PV, Grosse-Kunstleve RW, Echols N, Headd JJ, Moriarty NW, Mustyakimov M, Terwilliger TC, Urzhumtsev A, Zwart PH, Adams PD. 2012. Towards automated crystallographic structure refinement with phenix.refine. Acta Crystallogr D Biol Crystallogr 68:352–367. doi: 10.1107/S0907444912001308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Headd JJ, Echols N, Afonine PV, Grosse-Kunstleve RW, Chen VB, Moriarty NW, Richardson DC, Richardson JS, Adams PD. 2012. Use of knowledge-based restraints in phenix.refine to improve macromolecular refinement at low resolution. Acta Crystallogr D Biol Crystallogr 68:381–390. doi: 10.1107/S0907444911047834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Schrödinger, LLC. The PyMol. Molecular Graphics System, version 1.7.1.3. Schrödinger, LLC; http://www.pymol.org. [Google Scholar]
- 53.Xiao C, Rossmann MG. 2007. Interpretation of electron density with stereographic roadmap projections. J Struct Biol 158:182–187. doi: 10.1016/j.jsb.2006.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Xiao X, Li J, Samulski RJ. 1998. Production of high-titer recombinant adeno-associated virus vectors in the absence of helper adenovirus. J Virol 72:2224–2232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zolotukhin S, Byrne BJ, Mason E, Zolotukhin I, Potter M, Chesnut K, Summerford C, Samulski RJ, Muzyczka N. 1999. Recombinant adeno-associated virus purification using novel methods improves infectious titer and yield. Gene Ther 6:973–985. doi: 10.1038/sj.gt.3300938. [DOI] [PubMed] [Google Scholar]
- 56.Kuck D, Kern A, Kleinschmidt JA. 2007. Development of AAV serotype-specific ELISAs using novel monoclonal antibodies. J Virol Methods 140:17–24. doi: 10.1016/j.jviromet.2006.10.005. [DOI] [PubMed] [Google Scholar]
- 57.Wobus CE, Hügle-Dörr B, Girod A, Petersen G, Hallek M, Kleinschmidt JA. 2000. Monoclonal antibodies against the adeno-associated virus type 2 (AAV-2) capsid: epitope mapping and identification of capsid domains involved in AAV-2-cell interaction and neutralization of AAV-2 infection. J Virol 74:9281–9293. doi: 10.1128/JVI.74.19.9281-9293.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gurda BL, Raupp C, Popa-Wagner R, Naumer M, Olson NH, Ng R, McKenna R, Baker TS, Kleinschmidt JA, Agbandje-McKenna M. 2012. Mapping a neutralizing epitope onto the capsid of adeno-associated virus serotype 8. J Virol 86:7739–7751. doi: 10.1128/JVI.00218-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ng R, Govindasamy L, Gurda BL, McKenna R, Kozyreva OG, Samulski RJ, Parent KN, Baker TS, Agbandje-McKenna M. 2010. Structural characterization of the dual glycan binding adeno-associated virus serotype 6. J Virol 84:12945–12957. doi: 10.1128/JVI.01235-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Asokan A, Conway JC, Phillips JL, Li C, Hegge J, Sinnott R, Yadav S, DiPrimio N, Nam HJ, Agbandje-McKenna M, McPhee S, Wolff J, Samulski RJ. 2010. Reengineering a receptor footprint of adeno-associated virus enables selective and systemic gene transfer to muscle. Nat Biotechnol 28:79–82. doi: 10.1038/nbt.1599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bowles DE, McPhee SW, Li C, Gray SJ, Samulski JJ, Camp AS, Li J, Wang B, Monahan PE, Rabinowitz JE, Grieger JC, Govindasamy L, Agbandje-McKenna M, Xiao X, Samulski RJ. 2012. Phase 1 gene therapy for Duchenne muscular dystrophy using a translational optimized AAV vector. Mol Ther 20:443–455. doi: 10.1038/mt.2011.237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Limberis MP, Vandenberghe LH, Zhang L, Pickles RJ, Wilson JM. 2009. Transduction efficiencies of novel AAV vectors in mouse airway epithelium in vivo and human ciliated airway epithelium in vitro. Mol Ther 17:294–301. doi: 10.1038/mt.2008.261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Yan Z, Lei-Butters DC, Keiser NW, Engelhardt JF. 2013. Distinct transduction difference between adeno-associated virus type 1 and type 6 vectors in human polarized airway epithelia. Gene Ther 20:328–337. doi: 10.1038/gt.2012.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Shen S, Troupes AN, Pulicherla N, Asokan A. 2013. Multiple roles for sialylated glycans in determining the cardiopulmonary tropism of adeno-associated virus 4. J Virol 87:13206–13213. doi: 10.1128/JVI.02109-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Afione S, DiMattia MA, Halder S, Di Pasquale G, Agbandje-McKenna M, Chiorini JA. 2015. Identification and mutagenesis of the adeno-associated virus 5 sialic acid binding region. J Virol 89:1660–1672. doi: 10.1128/JVI.02503-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Xie Q, Lerch TF, Meyer NL, Chapman MS. 2011. Structure-function analysis of receptor-binding in adeno-associated virus serotype 6 (AAV-6). Virology 420:10–19. doi: 10.1016/j.virol.2011.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kern A, Schmidt K, Leder C, Müller OJ, Wobus CE, Bettinger K, Von der Lieth CW, King JA, Kleinschmidt JA. 2003. Identification of a heparin-binding motif on adeno-associated virus type 2 capsids. J Virol 77:11072–11081. doi: 10.1128/JVI.77.20.11072-11081.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Opie SR, Warrington KH, Agbandje-McKenna M, Zolotukhin S, Muzyczka N. 2003. Identification of amino acid residues in the capsid proteins of adeno-associated virus type 2 that contribute to heparan sulfate proteoglycan binding. J Virol 77:6995–7006. doi: 10.1128/JVI.77.12.6995-7006.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lerch TF, Xie Q, Chapman MS. 2010. The structure of adeno-associated virus serotype 3B (AAV-3B): insights into receptor binding and immune evasion. Virology 403:26–36. doi: 10.1016/j.virol.2010.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Manno CS, Pierce GF, Arruda VR, Glader B, Ragni M, Rasko JJ, Rasko J, Ozelo MC, Hoots K, Blatt P, Konkle B, Dake M, Kaye R, Razavi M, Zajko A, Zehnder J, Rustagi PK, Nakai H, Chew A, Leonard D, Wright JF, Lessard RR, Sommer JM, Tigges M, Sabatino D, Luk A, Jiang H, Mingozzi F, Couto L, Ertl HC, High KA, Kay MA. 2006. Successful transduction of liver in hemophilia by AAV-Factor IX and limitations imposed by the host immune response. Nat Med 12:342–347. doi: 10.1038/nm1358. [DOI] [PubMed] [Google Scholar]
- 71.Hurlbut GD, Ziegler RJ, Nietupski JB, Foley JW, Woodworth LA, Meyers E, Bercury SD, Pande NN, Souza DW, Bree MP, Lukason MJ, Marshall J, Cheng SH, Scheule RK. 2010. Preexisting immunity and low expression in primates highlight translational challenges for liver-directed AAV8-mediated gene therapy. Mol Ther 18:1983–1994. doi: 10.1038/mt.2010.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Huttner NA, Girod A, Perabo L, Edbauer D, Kleinschmidt JA, Büning H, Hallek M. 2003. Genetic modifications of the adeno-associated virus type 2 capsid reduce the affinity and the neutralizing effects of human serum antibodies. Gene Ther 10:2139–2147. doi: 10.1038/sj.gt.3302123. [DOI] [PubMed] [Google Scholar]
- 73.McCraw DM, O'Donnell JK, Taylor KA, Stagg SM, Chapman MS. 2012. Structure of adeno-associated virus-2 in complex with neutralizing monoclonal antibody A20. Virology 431:40–49. doi: 10.1016/j.virol.2012.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Gurda BL, DiMattia MA, Miller EB, Bennett A, McKenna R, Weichert WS, Nelson CD, Chen WJ, Muzyczka N, Olson NH, Sinkovits RS, Chiorini JA, Zolotutkhin S, Kozyreva OG, Samulski RJ, Baker TS, Parrish CR, Agbandje-McKenna M. 2013. Capsid antibodies to different adeno-associated virus serotypes bind common regions. J Virol 87:9111–9124. doi: 10.1128/JVI.00622-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Harbison CE, Weichert WS, Gurda BL, Chiorini JA, Agbandje-McKenna M, Parrish CR. 2012. Examining the cross-reactivity and neutralization mechanisms of a panel of mAbs against adeno-associated virus serotypes 1 and 5. J Gen Virol 93(Pt 2):347–355. doi: 10.1099/vir.0.035113-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Lovell SC, Davis IW, Arendall WB III, de Bakker PI, Word JM, Prisant MG, Richardson JS, Richardson DC. 2003. Structure validation by Calpha geometry: phi, psi and Cbeta deviation. Proteins. 50(3):437–450. doi: 10.1002/prot.10286. [DOI] [PubMed] [Google Scholar]
- 77.O'Donnell J, Taylor KA, Chapman MS. 2009. Adeno-associated virus-2 and its primary cellular receptor—cryo-EM structure of a heparin complex. Virology 385(2):434–443. doi: 10.1016/j.virol.2008.11.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Levy HC, Bowman VD, Govindasamy L, McKenna R, Nash K, Warrington K, Chen W, Muzyczka N, Yan X, Baker TS, Agbandje-McKenna M. 2009. Heparin binding induces conformational changes in adeno-associated virus serotype 2. J Struct Biol 165(3):146–156. doi: 10.1016/j.jsb.2008.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]