

Abstract
A new type of Pd-catalyzed alkene carboamination reaction that provides direct access to enantioenriched 2-aminoindanes from 2-allylphenyltriflate derivatives and aliphatic amines is described. A catalyst generated in situ from Pd(OAc)2 and (S)-tert-ButylPHOX provides the functionalized carbocycles in good yield with up to >99:1 er. The transformations occur via a key anti-aminopalladation that involves intermolecular attack of an amine nucleophile on an arylpalladium alkene complex.
TOC Graphic
Over the past several years our group has developed an efficient and practical protocol for the generation of heterocycles via Pd-catalyzed alkene carboamination reactions between aryl or alkenyl-X species (X = Br, I, Cl, OTf) and alkenes bearing pendant nucleophiles.1 This method has been used to prepare a number of heterocyclic motifs, and has been applied to enantioselective syntheses of pyrrolidines2, imidazoldin-2-ones3, tetrahydroquinolines, tetrahydroquinoxalines, and tetrahydroisoquinolines4 with excellent yields and stereoselectivity. Although the scope of these transformations is quite broad, in all cases the reactions involve the coupling of an alkene bearing a tethered nucleophile with an exogenous electrophile such as an aryl bromide or triflate. To date, no examples of Pd-catalyzed alkene carboamination reactions have been described in which the coupling partners are reversed such that a free amine nucleophile is coupled with an alkene bearing a pendant electrophile. In this communication we describe the first examples of Pd-catalyzed alkene carboamination reactions between amine nucleophiles and 2-allylphenyltriflate derivatives, which afford substituted carbocycles in good yield with excellent enantioselectivity.
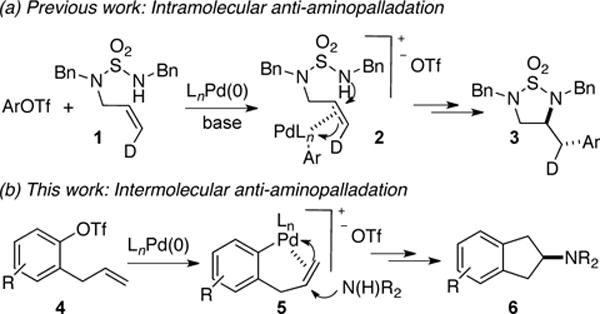
We envisioned that a new synthesis of amino-substituted carbocycles could potentially be achieved through a Pd-catalyzed carboamination reaction between an aryl electrophile bearing a pendant alkene and a free amine nucleophile (Scheme 1b). Most known alkene carboamination reactions between aminoalkenes and aryl/alkenyl electrophiles proceed via intramolecular syn-aminopalladation of an intermediate palladium amido complex, and provide products resulting from an exocyclization process.1 However, it seemed un-likely that the syn-aminopalladation pathway would provide access to the desired carbocycles, as this pathway would require an endocyclization through a highly strained transition state.
Scheme 1.
anti-Aminopalladation Mechanisms
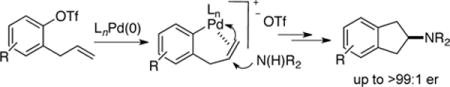
We recently reported a new synthesis of cyclic sulfamides via Pd-catalyzed alkene carboamination reactions.5 During the course of those studies we discovered that transformations could be induced to proceed via anti-aminopalladation6 under appropriate reaction conditions (Scheme 1a, 1 → 3). In general, conditions leading to a more electrophilic metal center (aryl triflate substrates and relatively polar PhCF3 solvent) favored the formation of anti-addition products. It seemed that the anti-aminopalladation pathway could provide access to the desired carbocycles in this new type of alkene carboamination reaction. As shown in Scheme 1b, oxidative addition of aryl triflate 4 to Pd(0) followed by alkene coordination would afford 5, which could undergo intermolecular anti-aminopalladation followed by reductive elimination to generate the desired product 6.
In our initial studies we elected to explore intermolecular carboamination reactions between 2-allylphenyltriflate derivatives 4 and amine nucleophiles. These transformations would give rise to 2-aminoindane derivatives 6, which have not only attracted attention due to their pronounced CNS activities,7 but also appear as motifs in pharmaceutically relevant molecules including autotaxin inhibitors,8 and cathepsin d inhibitors.9 In addition, the required substrates could be prepared from readily available phenols in three steps via O-allylation, aromatic Claisen rearrangement, and installation of the triflate moiety. Optimization was initiated by employing conditions similar to those used in our prior syntheses of cyclic sulfamides (which proceed via anti-aminopalladation). Efforts to couple relatively electron-poor nitrogen nucleophiles such as sulfonamides, amides or imides with 4a were unsuccessful, and unreacted starting material was recovered (Table 1, entries 1–3). However, we were delighted to see that the coupling of pyrrolidine with 4a led to the formation of significant amounts of desired product 7 (entry 4). To further improve reactivity a series of Buchwald-type biarylphosphines were surveyed,10 and we found BrettPhos outperformed other ligands and provided a 68% isolated yield of the desired product (entry 7). Efforts to improve yield by decreasing reaction temperature were unsuccessful (entries 8 and 9). After continued experimentation we found that performing the reaction with slight excess of pyrrolidine led to a cleaner reaction with no observed by-products by crude 1H NMR (entry 10), and provide the desired product in essentially quantitative yield. Interestingly, despite the utility of the Buchwald ligands for Pd-catalyzed N-arylation reactions, in this system side products resulting from N-arylation were not observed.
Table 1.
Influence of Nucleophile and Ligand on Reactivitya
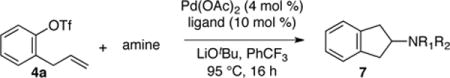
| |||||
|---|---|---|---|---|---|
|
| |||||
| entry | amine | ligand | T (°C) | conv (%)a,b | yield (%)a |
| 1 |
|
RuPhos | 95 | 0 | – |
| 2 |
|
CPhos | 95 | 0 | – |
| 3 |
|
RuPhos | 95 | 0 | – |
| 4 |
|
RuPhos | 95 | 80 | 45 |
| 5 |
|
CPhos | 95 | 85 | 62 |
| 6 |
|
JackiePhos | 95 | 100 | 45 |
| 7 |
|
BrettPhos | 95 | 100 | 73 (68) |
| 8 |
|
BrettPhos | 40 | 32 | 5 |
| 9 |
|
BrettPhos | 70 | 65 | 70 |
| 10c |
|
BrettPhos | 95 | 100 | quant. |
Conditions: 1.0 equiv 4a, 1.0 equiv amine, 1.4 equiv LiOtBu, 4 mol % Pd(OAc)2, 10 mol % ligand, PhCF3 (0.1 M), 95 °C, 16 h.
Determined by 1H NMR using phenanthrene as an internal standard. Isolated yields are given in parentheses.
1.0 equiv of 2-allylphenyl triflate and 1.2 equiv of pyrrolidine were used.
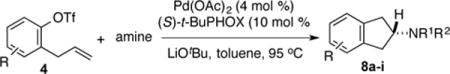
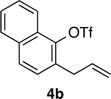
Having successfully optimized the coupling of pyrrolidine with 2-allylphenyl triflate to generate the achiral 2-aminoindane derivative 7, we turned our attention to the development of an asymmetric variant of this reaction to generate chiral aminoindane products. In our optimization studies we examined the coupling of 2-allyl-1-naphthyl triflate 4b with pyrrolidine. A series of chiral phosphine ligands were examined that have similar steric and electronic properties as the BrettPhos ligand, which provided good results in our achiral aminoindane synthesis. Initial studies with MOP-type ligands (L1–L5) did provide some asymmetric induction, but efforts to improve enantioselectivity by modifying the alkoxy substituent, the phosphine moiety, or use of a partially hydrogenated backbone did not lead to satisfactory results. We subsequently turned our attention to PHOX-type ligands L8-L10,11 as these ligands have previously shown utility in other asymmetric Pd-catalyzed reactions.11,12 Ligand L8 afforded the product in 86:14 er, which was improved to 88:12 er by conducting the reaction in toluene. Replacement of the diphenylphosphino group with a dicyclohexylphosphino group (L9) provided lower selectivity. However, use of a bulky tert-butyl group rather than an isopropyl group on the oxazoline (L10) led to significant improvement, and the desired product (R)-8a was generated with >99:1 er in 98% isolated yield.13
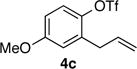
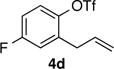
With optimized conditions in hand we proceeded to explore the scope of this transformation. The coupling reactions of 2-allylnaphthyl triflate 4b with several different amines proceeded in good to excellent yield and enantioselectivity. The best results were obtained with cyclic amines (entries 1–4), however, the use of an acyclic secondary amine (diethylamine) proceeded in lower yield and selectivity (entry 5). Both linear and α-branched 1° amines (entries 6 and 7) afforded the desired product in good to excellent yields with enantioselectivities of >99:1 and 97:3 er respectively. Transformations of 4-substituted 2-allyphenyltriflate derivatives were also successful. The coupling of electron-rich 2-allyl-4-methoxyphenyl triflate 4c with pyrrolidine afforded a moderate yield of 56% but excellent 97:3 er (entry 8). Similar results were obtained with n-octylamine, (entry 9). The coupling of the less-electron-rich 2-allyl-4-fluorophenyl triflate 4d with pyrrolidine proceeded in 77% yield and 98:2 er. The modest yields obtained with electron-rich substrate 4c could be due to the reduced electrophilicity of cationic Pd-intermediate 5 (Scheme 1b), which could inhibit alkene coordination and decrease the facility of nucleophilic attack on the alkene. The coupling of 4b with pyrrolidine on a 1.0 mmol scale provided essentially identical results to the small scale (0.1 mmol) coupling reaction (entry 2).
To further probe the mechanism of this transformation we examined the coupling of deuterated substrate d-4a with pyrrolidine. As shown in equation 1, this reaction provided d-7 with a cis-relationship between the D-atom and the proton adjacent to the amino group. This outcome confirmed the anti-addition pathway by way of intermediate Pd-alkene complex 9.
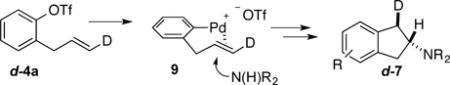 |
(1) |
The stereochemical outcome of these reactions is likely determined in the C–N bond-forming aminopalladation step. As shown in Scheme 2, attack of the amine nucleophile on the Pd-bound alkene in complex 10 leads to the observed major enantiomer. In contrast, nucleophilic addition to complex 11 is likely disfavored due to steric repulsion between the bulky tert-butyl substituent on the ligand and both the alkene and the incoming nucleophile.14
Scheme 2.
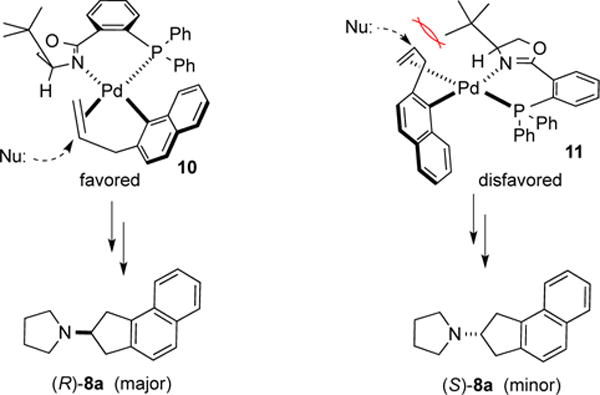
Stereochemical Model
In conclusion, we have developed a new class of alkene carboamination reactions that generate carbocyclic products bearing amino groups via coupling of amine nucleophiles with 2-allylphenyltriflate derivatives. These are the first examples of Pd-catalyzed alkene carboamination reactions in which the key aminopalladation step is an intermolecular process. These reactions illustrate the potential to employ Pd-catalyzed alkene carboamination reactions for the generation of carbocycles rather than heterocycles, and provide straightforward access to potentially useful 2-aminoindane derivatives.
Supplementary Material
Table 2.
Optimization of Asymmetric Reactionsa
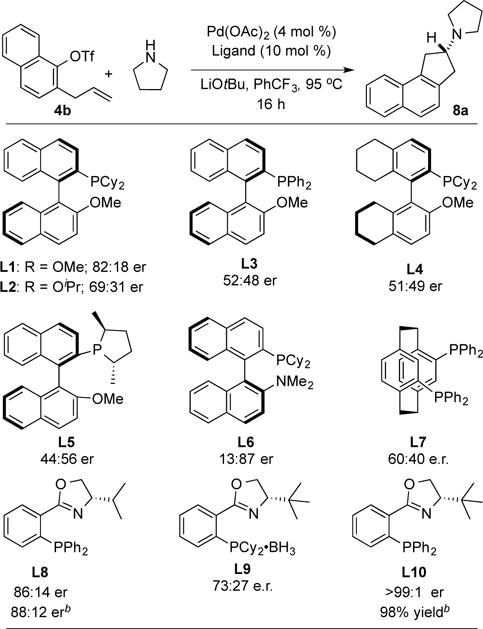
|
Conditions: 1.0 equiv 4b, 1.2 equiv amine, 1.4 equiv LiOtBu, 4 mol % Pd(OAc)2, 10 mol % ligand, PhCF3 (0.1 M), 95 °C, 16 h.
The reaction was conducted in toluene solvent.
Table 3.
Asymmetric Synthesis of Aminoindanesa
| ||||
|---|---|---|---|---|
|
| ||||
| entry | substrate | amine | % yieldb | er |
| 1 2c |
|
|
98 95 |
>99:1 99:1 |
| 3 | 4b |
|
88 | 95:5 |
| 4 | 4b |
|
90 | 95:5 |
| 5 | 4b |
|
47 | 89:11 |
| 6 | 4b |
|
63 | >99:1 |
| 7 | 4b |
|
82 | 97:3 |
| 8 |
|
|
56 | 97:3 |
| 9 | 4c |
|
59 | >99:1 |
| 10 |
|
|
77 | 98:2 |
Conditions: Reactions were conducted on a 0.1 mmol scale using 1.0 equiv 4, 1.2 equiv amine, 1.4 equiv LiOtBu, 4 mol % Pd(OAc)2, 10 mol % (S)-tBu-PHOX, toluene (0.1 M), 95 °C, 3–16 h.
Isolated yields (average of two or more experiments).
The reaction was conducted on a 1.0 mmol scale.
Acknowledgments
The authors thank the NIH-NIGMS (GM098314) for financial support of this work, and Dr. Nicholas Babij and Dr. Brett Hopkins for helpful discussions.
Footnotes
Supporting Information
Experimental procedures, compound characterization data, and copies of 1H and 13C NMR spectra. “This material is available free of charge via the Internet at http://pubs.acs.org.”
Notes
The authors declare no competing financial interests.
References
- 1.For reviews on Pd-catalyzed alkene carboamination reactions for the synthesis of nitrogen heterocycles, see:; (a) Schultz DM, Wolfe JP. Synthesis. 2012;44:351. doi: 10.1055/s-0031-1289668. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Wolfe JP. Top Heterocycl Chem. 2013;32:1–38. [Google Scholar]
- 2.Mai DN, Wolfe JP. J Am Chem Soc. 2010;132:12157. doi: 10.1021/ja106989h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hopkins BA, Wolfe JP. Angew Chem Int Ed. 2012;51:9886. doi: 10.1002/anie.201205233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hopkins BA, Wolfe JP. Chem Sci. 2014;5:4840. doi: 10.1039/C4SC01327A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fornwald RM, Fritz JA, Wolfe JP. Chem Eur J. 2014;20:8782. doi: 10.1002/chem.201402258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.For reviews on stereochemical pathways in alkene aminopalladation reactions, see:; (a) McDonald RI, Liu G, Stahl SS. Chem Rev. 2011;111:2981. doi: 10.1021/cr100371y. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Jensen KH, Sigman MS. Org Biomol Chem. 2008;6:4083. doi: 10.1039/b813246a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.(a) Simmler LD, Rickli A, Schramm Y, Hoener MC, Liechti ME. Biochem Pharmacol. 2014;88:237. doi: 10.1016/j.bcp.2014.01.024. [DOI] [PubMed] [Google Scholar]; (b) Brandt SD, Braithwaite RA, Evans-Brown M, Kicman AT. In: Novel Psychoactive Substances. Dargan PI, Wood DM, editors. Elsevier; London: 2013. pp. 261–283. [Google Scholar]
- 8.Jones SB, Norman BH, Pfeifer LA. WO2013133583A1. PCT Int Appl. 2014 Sep 18;161:505176. Scifinder Scholar.
- 9.Klein M, Tsaklakidis C. WO2014127881A1. PCT Int Appl. 2014 Aug 28;161:390014. Scifinder Scholar.
- 10.(a) Surry DS, Buchwald SL. Chem Sci. 2011;2:27. doi: 10.1039/C0SC00331J. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Surry DS, Buchwald SL. Angew Chem, Int Ed. 2008;47:6338. doi: 10.1002/anie.200800497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Helmchen G, Pfaltz A. Acc Chem Res. 2000;33:336. doi: 10.1021/ar9900865. [DOI] [PubMed] [Google Scholar]
- 12.(a) Behenna DC, Stoltz BM. J Am Chem Soc. 2004;126:15044. doi: 10.1021/ja044812x. [DOI] [PubMed] [Google Scholar]; (b) Doran R, Guiry PJ. J Org Chem. 2014;79:9112. doi: 10.1021/jo5014806. [DOI] [PubMed] [Google Scholar]; (c) Linton EC, Kozlowski MC. J Am Chem Soc. 2008;130:16162. doi: 10.1021/ja807026z. [DOI] [PubMed] [Google Scholar]
- 13.The absolute configuration of (R)-8a was established through x-ray crystallographic analysis of the corresponding HBr salt. The absolute configuration of all other products was assigned based on analogy to 8a.
- 14.For a related model of asymmetric induction in asymmetric Heck reactions, see:; Kilroy TG, Hennessy AJ, Connolly DJ, Malone YM, Farrell A, Guiry PJ. J Mol Catal A. 2003;196:65. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.