

Abstract
The kidney is one of the primary organs targeted by cadmium (Cd), a widely distributed environmental pollutant. The glomerular endothelium is the major component of the glomerular filtration barrier. However, the effects of Cd on glomerular endothelial cells remain largely unknown. For this purpose, we aimed to determine the effects of low dose Cd on the survival of human renal glomerular endothelial cells (HRGECs). Cultured HRGECs were exposed to 4 µM cadmium chloride (CdCl2) and examined at different time-points. We found that Cd activates the nuclear factor-κB (NF-κB) pathway without inducing the apoptosis of HRGECs. Pre-treating the cells with pyrrolidine dithiocarbamate (PDTC), a potent NF-κB inhibitor, prior to Cd exposure triggered extensive cell death (73.5%). In addition, Cd activates the c-Jun N-terminal kinase (JNK) pathway, and inhibition of the NF-κB pathway significantly elevates Cd-induced JNK phosphorylation in HRGECs (p<0.01). The combination treatment of PDTC and SP600125, a JNK pathway inhibitor, increased the survival of Cd-stimulated HRGECs compared with those cells treated with PDTC alone (p<0.05). Taken together, these findings demonstrate that the NF-κB pathway plays an essential role in maintaining the survival of Cd-exposed HRGECs.
Keywords: cadmium, nuclear factor-κB signaling, renal glomerular endothelial cells, c-Jun N-terminal kinase signaling, apoptosis
Introduction
Cadmium (Cd) is an environmental pollutant which poses significant health risks to humans (1). After entering into human body through ingestion, inhalation or skin absorption, Cd is not biodegradable and persists in many human organs (2). The kidney is one of the primary organs targeted by Cd (3). With chronic exposure, Cd accumulates in the epithelial cells of the proximal tubule, resulting in tubular impairment with a loss of reabsorptive capacity and the development of proteinuria (4). Previous findings suggest that Cd also affects the vasculature in the kidney (3). In the human body, Cd ions enter the blood circulation and bind to plasma proteins (5,6). Through the circulation, Cd reaches the targeted organs and passes through the endothelium of each vascular bed (5). In the kidney, Cd directly interacts with glomerular endothelial cells (GECs), which comprise the inner layer of the glomerular filtration barrier. However, the effects of Cd on GECs remain largely unknown.
Cd induces cell death in a variety of cell types (7,8). Cell death is an active process that crucially maintains homeostasis in multicellular organisms (9). There are three major types of cell death: apoptosis, autophagy and necrosis. Apoptosis occurs primarily through two well known pathways, the intrinsic or mitochondrial-mediated pathway and the extrinsic or death receptor-mediated pathway (10). Both pathways are mediated by cell membrane receptors, such as tumor necrosis factor receptor 1/2 (TNFR1/2), Fas/CD95, death receptor 3 (DR3) and TRAIL-receptor (TRAIL-R)1 and TRAIL-R2 (11,12), and activate the caspase family of proteases, which ultimately dismantle the cell. Autophagy is a survival mechanism in which parts of the cytosol and specific organelles are engulfed by a double-membrane autophagosome and degraded (13). Necrosis is characterized by the rapid loss of membrane integrity (13). All three may be activated through distinct or overlapping signaling pathways in response to Cd (9). These signaling pathways include reactive oxygen species (ROS), nuclear factor-κB (NF-κB), B-cell lymphoma 2 (Bcl-2) and mitogen-activated protein kinas (MAPK) signaling (9).
NF-κB is a heterodimeric protein composed of different combinations of members of the Rel family of transcription factors including RelA (p65), RelB (p50), c-Rel, NF-κB1 (p59/p105), and NF-κB2 (p52/p100) (14). Usually, NF-κB dimers reside in the cytosol, bound to the inhibitory protein inhibitor of κB (IκB), and are activated by stimuli capable of inducing phosphorylation and proteolysis of IκB (15). Following the removal of IκB, NF-κB enters the nucleus to induce the expression of coordinate sets of targets genes for cell survival (15). Previous findings have reported that the activation of the NF-κB pathway antagonizes apoptosis by the ligand engagement of 'death receptors' such as TNFR1 (12). Other evidence suggests that the NF-κB pathway is required for cell survival (16). The NF-κB pathway regulates the expression of several inhibitors of apoptosis (IAPs), including c-IAP1, c-IAP2 and X chromosome-linked IAP (XIAP), which inhibits the caspase pathway (17,18). In addition, it has been demonstrated that the negative regulation of c-Jun N-terminal kinase (JNK) activation by NF-κB contributes to the inhibition of apoptosis (19).
JNK, a member of the MAPK superfamily, also mediates apoptosis-related pathways. The persistent activation of JNK correlates with apoptosis, whereas suppression of JNK correlates with cell survival (20). In the nuclear apoptosis pathway, activated JNK translocates to the nucleus and activates c-Jun (21), leading to the formation of activator protein 1 (AP-1) (22). The JNK-AP-1 pathway is involved in the increased expression of pro-apoptotic genes such as tumor necrosis factor-α (TNF-α), Fas-L and Bak (23). In the mitochondrial signaling of apoptosis, JNK is required for the release of cytochrome c from the inner membrane space of mitochondria and the activation of the caspase-9 cascade (24). JNK also promotes the activity of pro-apoptotic BH3, which is the only subgroup of the Bcl-2 family of proteins such as Bim and Bmf capable of activating Bax and/or Bak to initiate apoptosis (25).
In the present study, we evaluated the effects of low dose Cd on the apoptosis of human renal glomerular endothelial cells (HRGECs) and explored the underlying mechanisms. Although 4 µM Cd does not affect the survival of HRGECs, it significantly activates the NF-κB pathway. Treatment of the cells with pyrrolidine dithiocarbamate (PDTC), a potent NF-κB inhibitor, followed by Cd exposure induced extensive apoptosis of HRGECs. Inhibition of the NF-κB pathway significantly increased Cd-induced JNK phosphorylation. The addition of SP600125, a JNK pathway inhibitor, partially reversed the apoptosis of HRGECs induced by the combination treatment of Cd and PDTC. Our results indicate that Cd maintains the survival of Cd-exposed HRGECs through the activation of the NF-κB pathway.
Materials and methods
Cell culture
HRGECs were purchased from ScienCell Research Laboratories (Carlsbad, CA, USA). HRGECs were cultured in Dulbecco's modified Eagle's medium (DMEM)/F12, supplemented with 10% fetal bovine serum (Lonza, Basel, Switzerland), 100 IU/ml penicillin (Sigma-Aldrich,St. Louis, MO, USA) and 100 µg/ml streptomycin (Sigma-Aldrich). The cells were cultured in humidified air at 37°C with 5% CO2. Cadmium chloride (CdCl2) was purchased from Sigma-Aldrich and dissolved in phosphate-buffered saline (PBS) at a stock concentration of 1 mM. PDTC and SP600125 were purchased from Cell Signaling Technology (Danvers, MA, USA) and dissolved in dimethyl sulfoxide (DMSO). During the experiments, the cells were treated with various combinations of 4 µM CdCl2, 10 µM PDTC (for 1 h prior to Cd exposure) and 10 µM SP600125.
Cell viability assay
The viability of HRGECs was assessed following exposure to CdCl2, PDTC or combination treatment. The cell cultures were washed with PBS and incubated in 0.05% trypsin for 2 min at 37°C. After disaggregation with pipettes, the single cell suspension was diluted 1:1 in 0.4% trypan blue (w/v in 0.9% NaCl) (Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA). The number and percentage of dye-free cells was calculated.
Detection of apoptosis by Annexin V-fluorescein isothiocyanate (FITC) and propidium iodide (PI) analysis
The apoptosis of HRGECs following Cd exposure was detected by Annexin V-FITC and propidium iodide (PI) staining using an assay kit (Neobiosciences, Shenzhen, China) according to the manufacturer's instructions. Briefly, the cells were trypsinized, pelleted, washed twice with PBS and resuspended into single cells. Then, 1×106 cells were stained with Annexin V-FITC (0.25%) and PI (1 µg/ml) in 1X binding buffer (10 mM HEPES, pH 7.4, 140 mM NaOH, 2.5 mM CaCl2) for 15 min at room temperature in the dark. Positive staining of the cells was detected using a FACSAria II flow cytometer, and analyzed using the FACSDiva acquisition and analysis software (both from BD Biosciences, San Jose, CA, USA).
Western blot analysis
After treatment, HRGECs were lysed using RIPA buffer (20 mM Tris, pH 7.5, 150 mM NaCl, 50 mM NaF, 1% NP-40, 0.1% deoxycholate, 0.1% sodium dodecyl sulfate, 1 mM EDTA) supplemented with protease inhibitors aprotonin (1 µg/ml), leupeptin (10 µg/ml) and PMSF (1 mM). The protein concentration was determined using the bicinchoninic acid (BCA) assay (Bio-Rad Laboratories, Inc., Berkeley, CA, USA). Equal amounts of protein (40 µg) were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) (10% acrylamide gel) and transferred to a PVDF membrane. The membrane was blocked in TBST (20 mM Tris, 150 mM NaCl, 0.1% Tween-20) with 2.5% non-fat milk at 37°C for 1.5 h prior to incubation with primary antibodies overnight at 4°C. After washing three times with TBST, the membrane was incubated with secondary antibody at 37°C for 2 h. The following primary antibodies were used: rabbit anti-SAPK/JNK (9258), rabbit anti-phospho-SAPK/JNK (4668), mouse anti-IκBα (L35A5), rabbit anti-NF-κB p65 (6956), and rabbit anti-GAPDH (2118) (all from Cell Signaling Technology). The secondary antibody was HRP-linked anti-rabbit IgG (7074) and HRP-linked anti-mouse IgG (7076) (Cell Signaling Technology). The blots were developed with enhanced chemiluminescence reagents (Millipore, Billerica, MA, USA), and the relative intensities of the blots were quantified using ImageJ software (National Institutes of Health, Bethesda, MD, USA).
Immunofluorescence
HRGECs were allowed to grow to confluence on fibronectin-coated glass chamber slides and exposed to Cd for 24 h. The monolayers of HRGECs were then washed with PBS containing 100 mM glycine, fixed with 4% paraformaldehyde for 5 min, and washed three times with PBS for 10 min. Immunoreactivity was examined by staining with a rabbit polyclonal anti NF-κB p65 antibody (1:400; Cell Signaling Technology) overnight at 4°C and incubation with an Alexa 546-labeled anti-rabbit secondary antibody (1:200; Molecular Probes, Eugene, OR, USA) for 2 h. Nuclear staining was achieved using DAPI (1:1,000; Molecular Probes) and images of the the samples were captured using an Olympus LCX100 Imaging system (Olympus Corp., Tokyo, Japan) with an excitation wavelength of 546 nm.
Statistical analysis
The data are presented as the means ± standard error. All experiments were performed using at least three separate cell preparations. The differences between the groups were evaluated using a Student's t-test (two-tailed). A p-value <0.05 was considered to indicate a statistically significant difference.
Results
Cd activates NF-κB signaling in HRGECs
NF-κB signaling regulates apoptosis in response to oxidative stress, which may be induced by Cd (26). The activation of NF-κB requires the degradation of IκBa, which forms a cytoplasmic and inactive complex with the p65-p50 heterodimer (27). We examined the effects of Cd on NF-κB signaling in HRGECs. Western blot analysis revealed that the protein level of IκBα in the cytoplasm of HRGECs was significantly decreased by Cd exposure, whereas levels of the internal control GAPDH remained unchanged (Fig. 1A and B). Using immunofluorescence, we were able to demonstrate the nuclear translocation of the NF-κB/p65 subunit in HRGECs (Fig. 1C). Following exposure to Cd for 24 h, p65 in the nucleus markedly increased compared with that in the untreated cells. These results indicate that NF-κB signaling pathway is activated by Cd in HRGECs.
Figure 1.

Cadmium (Cd) activates nuclear factor-κB (NF-κB) signaling in human renal glomerular endothelial cells (HRGECs). (A) Representative blots of IκBα protein samples from HRGECs treated with 4 µM cadmium chloride (CdCl2) evaluated at different time-points. GAPDH was used as loading control. (B) The densitometry analysis of IκBα/GAPDH blots. n=4; NS, not significant; *p<0.05; **p<0.01 vs. control. (C) Immunofluoresence staining of p65 in an HRGEC monolayer treated with phosphate-buffered saline (PBS) (control) and 4 µM CdCl2 for 24 h.
Effects of Cd exposure on the apoptosis of HRGECs
Cd induces cytotoxicity in different types of cell depending on the concentration and the exposure time (28,29). We examined the effect of low dose Cd on the apoptosis of HRGECs. Using PI/Annexin V flow cytometric analysis, 4 µM Cd did not change the percentage of viable cells of HRGECs (86.8±1.6 vs. 84.7±2.9%, p=0.34). PDTC prevents IκBα degradation and the dissociation of NF-κB from IκBα thus inhibits the NF-κB signaling pathway (30). We pre-treated HRGECs with 10 µM PDTC for 1 h prior to Cd exposure and found that combination treatment with PDTC and Cd significantly decreased the percentage of viable cells of HRGECs compared to that of PDTC treatment alone (6.5±0.4 vs. 85.1±3.2%, p<0.01) (Fig. 2A and B). At a concentration of 4 µM, Cd did not significantly affect cell viability after 24 h in a trypan blue exclusion assay (93.4±0.6 vs. 91.1±0.7%, p=0.26) (Fig. 2C); however, combination treatment with PDTC and Cd significantly reduced the viability of HRGECs (91.1±0.7 vs. 33.4±0.8%, p<0.01) (Fig. 2C). Taken together, these findings demonstrated that NF-κB signaling pathway was required for the survival of Cd-stimulated HRGECs.
Figure 2.

Nuclear factor-κB (NF-κB) signaling is required for the survival of cadmium (Cd)-treated human renal glomerular endothelial cells (HRGECs). (A) Representative image of flow cytometry with Annexin V-fluorescein isothiocyanate (FITC)/propidium iodide (PI) double-staining for HRGECs treated with cadmium chloride (CdCl2), pyrrolidine dithiocarbamate (PDTC), or a combination of CdCl2 and PDTC for 24 h. (B) Bar graph of representative image of flow cytometry. n=3; NS, not significant; **P<0.05. (C) Trypan blue exclusion assay of HRGECs treated with CdCl2, PDTC or combination of CdCl2 and PDTC. n=3; **P<0.05.
Activation of NF-κB signaling inhibits JNK signaling in Cd-stimulated HRGECs
The JNK pathway mediates apoptosis and is directly activated by Cd in several cell lines (7,8). It has been reported that NF-κB signaling pathway negatively regulates the JNK pathway (31). We examined JNK activation in HRGECs exposed to Cd and PDTC. Fig. 3A showed that Cd increased phosphorylated (p-)JNK in HRGECs, whereas the total levels of JNK protein and the internal control GAPDH remain unchanged. In the presence of PDTC, Cd markedly increased the phosphorylation of JNK (Fig. 3B). Densitometry analyses showed that the combination of PDTC and Cd significantly elevated p-JNK compared with exposure to Cd alone (p<0.01) (Fig. 3C). Thus, the JNK pathway is suppressed by Cd-activated NF-κB signaling pathway.
Figure 3.

c-Jun N-terminal kinase (JNK) signaling is suppressed by nuclear factor-κB (NF-κB) activation in cadmium (Cd)-treated human renal glomerular endothelial cells (HRGECs). (A and B) Representative blots of phosphorylated (p-)JNK and total JNK from protein samples of HRGECs exposed to Cd in the absence or presence of pyrrolidine dithiocarbamate (PDTC) at different time-points. GAPDH was used as loading control. (C) The densitometry analysis of p-JNK/total JNK. n=3; **p<0.01.
SP600125 partially reverses the apoptosis of HRGECs induced by Cd and PDTC
To further confirm the role of the JNK pathway in Cd-stimulated HRGECs, 10 µM SP600125, an inhibitor of JNK pathway, was applied to cells in addition to Cd and PDTC. The addition of SP600125 partially reversed the apoptosis of HRGECs (76.8±3.5 vs. 62.4±3.7%, p<0.05) (Fig. 4). This suggests that NF-κB partially inhibits the Cd-induced apoptosis of HRGECs through the negative regulation of the JNK pathway.
Figure 4.
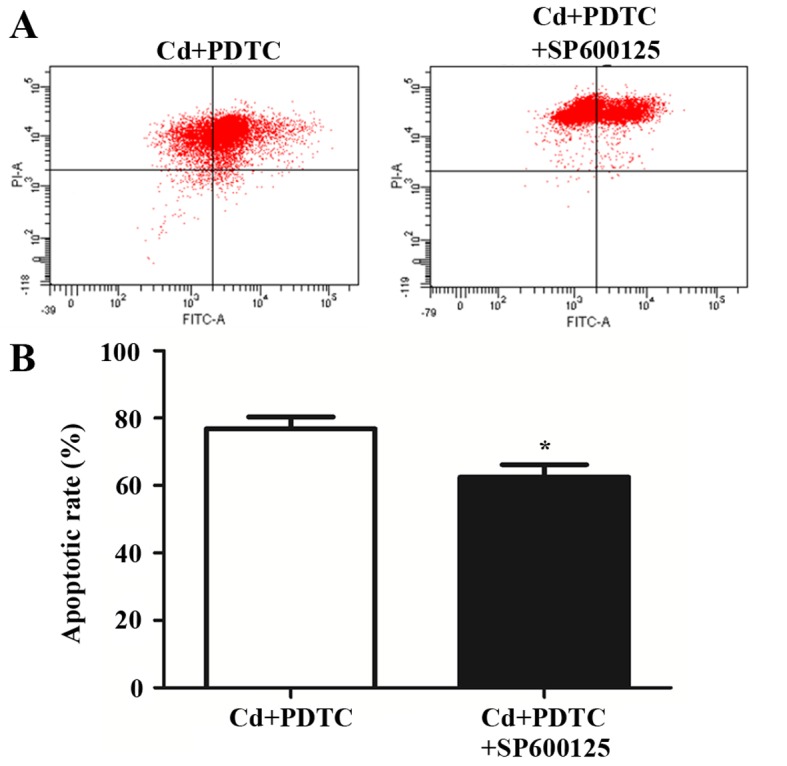
SP600125 partially reverses cadmium (Cd)-induced apoptosis of human renal glomerular endothelial cells (HRGECs) pre-treated with pyrrolidine dithiocarbamate (PDTC). (A) Representative image of flow cytometry with Annexin V-fluorescein isothiocyanate (FITC)/propidium iodide (PI) double-staining for HRGECs stimulated with Cd and PDTC in the absence or presence of SP600125 for 24 h. (B) Bar graph of flow cytometry. n=3; *p<0.05.
Discussion
Cd, one of the most widespread pollutants, induces a number of clinical symptoms of kidney damage (3). In the present study, we evaluated the effects of low dose Cd on the apoptosis of HRGECs, which comprise the primary layer of glomerular filtration. Our data indicated that 4 µM Cd does not induce the apotosis of HRGECs. Cd activates the NF-κB pathway and inhibition of NF-κB pathway significantly induces cell apoptosis in Cd-stimulated HRGECs. In addition, the NF-κB pathway maintains cell survival partially through the suppression of the JNK pathway. The present study indicates that the NF-κB pathway is essential for maintaining the survival of HRGECs exposed to Cd.
GECs are a specialized type of microvascular cell which form the inner layer of the glomerular filter barrier (32). Injury to GECs leads to nephrotoxicity characterized by proteinuria, polyuria and glucosuria (3). We have recently reported that 1 µM and 1 h exposure of Cd increases the permeability in HRGECs monolayers and redistributes the adherens junction proteins vascular endothelial-cadherin and β-catenin; however, this short-term, low dose of Cd does not induce the apoptosis of HRGECs (33). Morales et al have shown that after a 9-week sub-chronic exposure to Cd in rats, renal endothelial nitric oxide synthase (eNOS) was upregulated by 10–20-fold (34). It is recognized that Cd potentially causes damage to the glomeruli endothelium; however, the detailed mechanism responsible for thse effects remains unclear. In the present study, we determined that 4 µM and 24 h Cd exposure does not induce the apoptosis of HRGECs, by balancing the activation of pro-apoptotic and pro-survival signaling pathways.
The NF-κB pathway is closely associated with cell survival (35). Mice with a genetic disruption of RelA, a major activating subunit of NF-κB, died from massive apoptosis of hepatocytes in the liver (36). In the present study, we found that low dose Cd activates the NF-κB pathway in HRGECs through the degradation of IκBα and the translocation of the NF-κB p65 subunit from the cytosol to the nucleus. Cd induces the activation of NF-κB in several types of cell. Jeong et al proved that 3 µM Cd activates the NF-κB pathway in cerebrovascular endothelial cells (37). Moreover, high concentrations of Cd (20 or 30 µM) significantly upregulated the protein levels of p65 in the nuclei of bronchial epithelial cells (38). We also found that the inhibition of the NF-κB pathway by PDTC induces marked apoptosis of Cd-stimulated HRGECs. In another study, the inhibitory protein IκBα was modified to resist ubiquitin-mediated degradation; it consistently prevented NF-κB from entering into the nucleus, leading to the sensitization of the cells to apoptotic stimuli (39). Taken together, these findings suggest that Cd activates the NF-κB pathway, which maintains the survival of HRGECs.
To maintain the survival of HRGECs, it is likely that Cd activates pro-apoptotic pathways together with the NF-κB pathway. The JNK pathway is one of the initiators of apoptosis, and is regulated by the NF-κB pathway (20). Tang et al (19) and De Smaele et al (41) have independently demonstrated that TNF-α induces prolonged JNK activation in NF-κB activation-deficient cells, such as RelA and IKKβ knockouts. In the present study, we examined the role of the JNK pathway in Cd-induced apoptosis. We found that Cd activates the JNK pathway in HRGECs and Cd induced the activation of the NF-κB pathway which inhibits the JNK pathway. In Cd- and PDTC-treated HRGECs, the addition of a JNK inhibitor partially reversed the Cd-induced apoptosis of HRGECs. Thus, NF-κB promotes cell survival partially by inhibiting the JNK pathway. In addition to the JNK pathway, NF-κB is involved in multiple mechanisms which suppress apoptosis. The NF-κB pathway upregulates the Bcl-2 family members Bcl-xL and A1/Bfl-1, which reduce the effect of mitochondrial depolarization and inhibit cytochrome c release from mitochondria (42). NF-κB also increases the expression of TRAF1/2, c-IAP 1/2 and XIAP, which suppress apoptosis at the level of caspase-8 (40). Therefore, the downstream effectors of the NF-κB pathway which mediate the survival of HRGECs warrant further investigation.
In conclusion, we found that low dose Cd activates the NF-κB pathway through the degradation of IκBα, and promotes the translocation of NF-κB/p65 into the nucleus of HRGECs. The activation of NF-κB is required for the survival of Cd-stimulated HRGECs. In addition, the Cd-activated NF-κB pathway prevents cell death partially by inhibiting the JNK pathway. The present study provides important information for improving our understanding of the molecular mechanisms underlying Cd-induced nephrotoxicity.
Acknowledgments
The present study was supported by grants from the Science and Technology Development Plan of Shandong Province (no. 2013GSF11805), the National Natural Science Foundation of China (no. 81370269) and the Shandong Taishan Scholarship (Ju Liu).
References
- 1.Järup L, Berglund M, Elinder CG, Nordberg G, Vahter M. Health effects of cadmium exposure - a review of the literature and a risk estimate. Scand J Work Environ Health. 1998;24(Suppl 1):1–51. [PubMed] [Google Scholar]
- 2.Thévenod F, Lee WK. Toxicology of cadmium and its damage to mammalian organs. Met Ions Life Sci. 2013;11:415–490. doi: 10.1007/978-94-007-5179-8_14. [DOI] [PubMed] [Google Scholar]
- 3.Prozialeck WC, Edwards JR. Early biomarkers of cadmium exposure and nephrotoxicity. Biometals. 2010;23:793–809. doi: 10.1007/s10534-010-9288-2. [DOI] [PubMed] [Google Scholar]
- 4.Chakraborty PK, Lee WK, Molitor M, Wolff NA, Thévenod F. Cadmium induces Wnt signaling to upregulate proliferation and survival genes in sub-confluent kidney proximal tubule cells. Mol Cancer. 2010;9:102. doi: 10.1186/1476-4598-9-102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Prozialeck WC, Edwards JR, Woods JM. The vascular endothelium as a target of cadmium toxicity. Life Sci. 2006;79:1493–1506. doi: 10.1016/j.lfs.2006.05.007. [DOI] [PubMed] [Google Scholar]
- 6.Dong F, Guo F, Li L, Guo L, Hou Y, Hao E, Yan S, Allen TD, Liu J. Cadmium induces vascular permeability via activation of the p38 MAPK pathway. Biochem Biophys Res Commun. 2014;450:447–452. doi: 10.1016/j.bbrc.2014.05.140. [DOI] [PubMed] [Google Scholar]
- 7.Liu J, Kapron CM. Differential induction of MAP kinase signalling pathways by cadmium in primary cultures of mouse embryo limb bud cells. Reprod Toxicol. 2010;29:286–291. doi: 10.1016/j.reprotox.2010.02.001. [DOI] [PubMed] [Google Scholar]
- 8.Yuan Y, Jiang C, Hu F, Wang Q, Zhang K, Wang Y, Gu J, Liu X, Bian J, Liu Z. The role of mitogen-activated protein kinase in cadmium-induced primary rat cerebral cortical neurons apoptosis via a mitochondrial apoptotic pathway. J Trace Elem Med Biol. 2015;29:275–283. doi: 10.1016/j.jtemb.2014.06.006. [DOI] [PubMed] [Google Scholar]
- 9.Green DR, Llambi F. Cell Death Signaling. Cold Spring Harb Perspect Biol. 2015;7:7. doi: 10.1101/cshperspect.a006080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wyllie AH, Kerr JF, Currie AR. Cell death: The significance of apoptosis. Int Rev Cytol. 1980;68:251–306. doi: 10.1016/S0074-7696(08)62312-8. [DOI] [PubMed] [Google Scholar]
- 11.Kucharczak J, Simmons MJ, Fan Y, Gélinas C. To be, or not to be: NF-kappaB is the answer - role of Rel/NF-kappaB in the regulation of apoptosis. Oncogene. 2003;22:8961–8982. doi: 10.1038/sj.onc.1207230. [DOI] [PubMed] [Google Scholar]
- 12.Wajant H, Pfizenmaier K, Scheurich P. Tumor necrosis factor signaling. Cell Death Differ. 2003;10:45–65. doi: 10.1038/sj.cdd.4401189. [DOI] [PubMed] [Google Scholar]
- 13.Ashkenazi A, Salvesen G. Regulated cell death: signaling and mechanisms. Annu Rev Cell Dev Biol. 2014;30:337–356. doi: 10.1146/annurev-cellbio-100913-013226. [DOI] [PubMed] [Google Scholar]
- 14.Dutta J, Fan Y, Gupta N, Fan G, Gélinas C. Current insights into the regulation of programmed cell death by NF-kappaB. Oncogene. 2006;25:6800–6816. doi: 10.1038/sj.onc.1209938. [DOI] [PubMed] [Google Scholar]
- 15.Huxford T, Huang DB, Malek S, Ghosh G. The crystal structure of the IkappaBalpha/NF-kappaB complex reveals mechanisms of NF-kappaB inactivation. Cell. 1998;95:759–770. doi: 10.1016/S0092-8674(00)81699-2. [DOI] [PubMed] [Google Scholar]
- 16.Karin M, Ben-Neriah Y. Phosphorylation meets ubiquitination: the control of NF-[kappa]B activity. Annu Rev Immunol. 2000;18:621–663. doi: 10.1146/annurev.immunol.18.1.621. [DOI] [PubMed] [Google Scholar]
- 17.Irmler M, Thome M, Hahne M, Schneider P, Hofmann K, Steiner V, Bodmer JL, Schröter M, Burns K, Mattmann C, et al. Inhibition of death receptor signals by cellular FLIP. Nature. 1997;388:190–195. doi: 10.1038/40657. [DOI] [PubMed] [Google Scholar]
- 18.Chu ZL, McKinsey TA, Liu L, Gentry JJ, Malim MH, Ballard DW. Suppression of tumor necrosis factor-induced cell death by inhibitor of apoptosis c-IAP2 is under NF-kappaB control. Proc Natl Acad Sci USA. 1997;94:10057–10062. doi: 10.1073/pnas.94.19.10057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tang G, Minemoto Y, Dibling B, Purcell NH, Li Z, Karin M, Lin A. Inhibition of JNK activation through NF-kappaB target genes. Nature. 2001;414:313–317. doi: 10.1038/35104568. [DOI] [PubMed] [Google Scholar]
- 20.Chen YR, Tan TH. The c-Jun N-terminal kinase pathway and apoptotic signaling (Review) Int J Oncol. 2000;16:651–662. doi: 10.3892/ijo.16.4.651. [DOI] [PubMed] [Google Scholar]
- 21.Davis RJ. Signal transduction by the JNK group of MAP kinases. Cell. 2000;103:239–252. doi: 10.1016/S0092-8674(00)00116-1. [DOI] [PubMed] [Google Scholar]
- 22.Dhanasekaran DN, Johnson GL. MAPKs: function, regulation, role in cancer and therapeutic targeting. Oncogene. 2007;26:3097–3099. doi: 10.1038/sj.onc.1210395. [DOI] [PubMed] [Google Scholar]
- 23.Fan M, Chambers TC. Role of mitogen-activated protein kinases in the response of tumor cells to chemotherapy. Drug Resist Updat. 2001;4:253–267. doi: 10.1054/drup.2001.0214. [DOI] [PubMed] [Google Scholar]
- 24.Tournier C, Hess P, Yang DD, Xu J, Turner TK, Nimnual A, Bar-Sagi D, Jones SN, Flavell RA, Davis RJ. Requirement of JNK for stress-induced activation of the cytochrome c-mediated death pathway. Science. 2000;288:870–874. doi: 10.1126/science.288.5467.870. [DOI] [PubMed] [Google Scholar]
- 25.Lei K, Davis RJ. JNK phosphorylation of Bim-related members of the Bcl2 family induces Bax-dependent apoptosis. Proc Natl Acad Sci USA. 2003;100:2432–2437. doi: 10.1073/pnas.0438011100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chen LF, Greene WC. Shaping the nuclear action of NF-kappaB. Nat Rev Mol Cell Biol. 2004;5:392–401. doi: 10.1038/nrm1368. [DOI] [PubMed] [Google Scholar]
- 27.Jacobs MD, Harrison SC. Structure of an IkappaBalpha/NF-kappaB complex. Cell. 1998;95:749–758. doi: 10.1016/S0092-8674(00)81698-0. [DOI] [PubMed] [Google Scholar]
- 28.Yokouchi M, Hiramatsu N, Hayakawa K, Okamura M, Du S, Kasai A, Takano Y, Shitamura A, Shimada T, Yao J, Kitamura M. Involvement of selective reactive oxygen species upstream of proapoptotic branches of unfolded protein response. J Biol Chem. 2008;283:4252–4260. doi: 10.1074/jbc.M705951200. [DOI] [PubMed] [Google Scholar]
- 29.Liu F, Wang B, Li L, Dong F, Chen X, Li Y, Dong X, Wada Y, Kapron CM, Liu J. Low-dose cadmium upregulates VEGF expression in lung adenocarcinoma cells. Int J Environ Res Public Health. 2015;12:10508–10521. doi: 10.3390/ijerph120910508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dong F, Zhou X, Li C, Yan S, Deng X, Cao Z, Li L, Tang B, Allen TD, Liu J. Dihydroartemisinin targets VEGFR2 via the NF-κB pathway in endothelial cells to inhibit angiogenesis. Cancer Biol Ther. 2014;15:1479–1488. doi: 10.4161/15384047.2014.955728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Javelaud D, Besançon F. NF-kappa B activation results in rapid inactivation of JNK in TNF alpha-treated Ewing sarcoma cells: a mechanism for the anti-apoptotic effect of NF-kappa B. Oncogene. 2001;20:4365–4372. doi: 10.1038/sj.onc.1204570. [DOI] [PubMed] [Google Scholar]
- 32.Du L, Dong F, Guo L, Hou Y, Yi F, Liu J, Xu D. Interleukin-1β increases permeability and upregulates the expression of vascular endothelial-cadherin in human renal glomerular endothelial cells. Mol Med Rep. 2015;11:3708–3714. doi: 10.3892/mmr.2015.3172. [DOI] [PubMed] [Google Scholar]
- 33.Li L, Dong F, Xu D, Du L, Yan S, Hu H, Lobe CG, Yi F, Kapron CM, Liu J. Short-term, low-dose cadmium exposure induces hyperpermeability in human renal glomerular endothelial cells. J Appl Toxicol. 2016;36:257–265. doi: 10.1002/jat.3168. [DOI] [PubMed] [Google Scholar]
- 34.Morales AI, Vicente-Sánchez C, Jerkic M, Santiago JM, Sánchez-González PD, Pérez-Barriocanal F, López-Novoa JM. Effect of quercetin on metallothionein, nitric oxide synthases and cyclooxygenase-2 expression on experimental chronic cadmium nephrotoxicity in rats. Toxicol Appl Pharmacol. 2006;210:128–135. doi: 10.1016/j.taap.2005.09.006. [DOI] [PubMed] [Google Scholar]
- 35.Bubici C, Papa S, Pham CG, Zazzeroni F, Franzoso G. NF-kappaB and JNK: an intricate affair. Cell Cycle. 2004;3:1524–1529. doi: 10.4161/cc.3.12.1321. [DOI] [PubMed] [Google Scholar]
- 36.Beg AA, Baltimore D. An essential role for NF-kappaB in preventing TNF-alpha-induced cell death. Science. 1996;274:782–784. doi: 10.1126/science.274.5288.782. [DOI] [PubMed] [Google Scholar]
- 37.Jeong EM, Moon CH, Kim CS, Lee SH, Baik EJ, Moon CK, Jung YS. Cadmium stimulates the expression of ICAM-1 via NF-kappaB activation in cerebrovascular endothelial cells. Biochem Biophys Res Commun. 2004;320:887–892. doi: 10.1016/j.bbrc.2004.05.218. [DOI] [PubMed] [Google Scholar]
- 38.Chen DJ, Xu YM, Du JY, Huang DY, Lau AT. Cadmium induces cytotoxicity in human bronchial epithelial cells through upregulation of eIF5A1 and NF-kappaB. Biochem Biophys Res Commun. 2014;445:95–99. doi: 10.1016/j.bbrc.2014.01.146. [DOI] [PubMed] [Google Scholar]
- 39.Huang TT, Wuerzberger-Davis SM, Wu ZH, Miyamoto S. Sequential modification of NEMO/IKKgamma by SUMO-1 and ubiquitin mediates NF-kappaB activation by genotoxic stress. Cell. 2003;115:565–576. doi: 10.1016/S0092-8674(03)00895-X. [DOI] [PubMed] [Google Scholar]
- 40.Wang CY, Mayo MW, Korneluk RG, Goeddel DV, Baldwin AS., Jr NF-kappaB antiapoptosis: induction of TRAF1 and TRAF2 and c-IAP1 and c-IAP2 to suppress caspase-8 activation. Science. 1998;281:1680–1683. doi: 10.1126/science.281.5383.1680. [DOI] [PubMed] [Google Scholar]
- 41.De Smaele E, Zazzeroni F, Papa S, Nguyen DU, Jin R, Jones J, Cong R, Franzoso G. Induction of gadd45beta by NF-kappaB downregulates pro-apoptotic JNK signalling. Nature. 2001;414:308–313. doi: 10.1038/35104560. [DOI] [PubMed] [Google Scholar]
- 42.Zong WX, Edelstein LC, Chen C, Bash J, Gélinas C. The prosurvival Bcl-2 homolog Bfl-1/A1 is a direct transcriptional target of NF-kappaB that blocks TNFalpha-induced apoptosis. Genes Dev. 1999;13:382–387. doi: 10.1101/gad.13.4.382. [DOI] [PMC free article] [PubMed] [Google Scholar]