

Abstract
The battle for survival between bacteria and the viruses that infect them (phages) has led to the evolution of many bacterial defence systems and phage-encoded antagonists of these systems. Clustered regularly interspaced short palindromic repeats (CRISPR) and the CRISPR-associated (cas) genes comprise an adaptive immune system that is one of the most widespread means by which bacteria defend themselves against phages1–3. We identified the first examples of proteins produced by phages that inhibit a CRISPR–Cas system4. Here we performed biochemical and in vivo investigations of three of these anti-CRISPR proteins, and show that each inhibits CRISPR–Cas activity through a distinct mechanism. Two block the DNA-binding activity of the CRISPR–Cas complex, yet do this by interacting with different protein subunits, and using steric or non-steric modes of inhibition. The third anti-CRISPR protein operates by binding to the Cas3 helicase–nuclease and preventing its recruitment to the DNA-bound CRISPR–Cas complex. In vivo, this anti-CRISPR can convert the CRISPR–Cas system into a transcriptional repressor, providing the first example—to our knowledge—of modulation of CRISPR–Cas activity by a protein interactor. The diverse sequences and mechanisms of action of these anti-CRISPR proteins imply an independent evolution, and foreshadow the existence of other means by which proteins may alter CRISPR–Cas function.
CRISPR–Cas RNA-guided immune systems are widespread in prokaryotes, and play a major part in microbial evolution2,3. In these systems, CRISPR arrays are transcribed and processed to generate small CRISPR RNAs (crRNAs), which combine with Cas proteins to form crRNA-guided surveillance complexes2,5. In the type I-F CRISPR–Cas system, the Csy4 protein is a CRISPR-specific endoribonuclease that binds to and cleaves each repeat sequence in the pre-crRNA6. Csy4 remains associated with the 3′ end of the mature 60-nucleotide crRNA and then assembles with Csy1, Csy2 and Csy3 proteins to form a 350 kilodalton (kDa) surveillance complex7,8. This complex relies on a 32-nucleotide segment of the crRNA for complementary base pairing to invading DNA sequences, known as protospacers. Binding of target DNA by the Csy complex leads to the recruitment of the nuclease–helicase protein Cas3 and subsequent phage genome degradation9,10. We previously identified five unique type I-F anti-CRISPR proteins4. Here we determine the mechanisms by which three of these proteins function.
Three type I-F anti-CRISPRs, AcrF1 (11 kDa, encoded by gene 35 from phage JBD30), AcrF2 (13 kDa, encoded by gene 30 from phage D3112), and AcrF3 (16 kDa, encoded by gene 35 from phage JBD5), could be expressed in Escherichia coli and purified to homogeneity. Using a previously described E. coli expression system7, we also purified the 350 kDa Pseudomonas aeruginosa Csy complex, including a crRNA and the four Csy proteins. This complex was mixed in vitro with each purified anti-CRISPR protein, and fractionated by size-exclusion chromatography (SEC). AcrF1 and AcrF2 co-eluted with the Csy complex (Fig. 1a and Extended Data Fig. 1), indicating a direct interaction. AcrF3 did not co-elute with the Csy complex (Fig. 1b). The lack of AcrF3 binding to the Csy complex suggested that it might inhibit the CRISPR–Cas system by interactingwith Cas3, the helicase–nuclease that is responsible for target DNA destruction after recognition by the Csy complex. Supporting this hypothesis, AcrF3 co-eluted with purified Cas3, while AcrF1 did not (Fig. 1c and Extended Data Fig. 2). These experiments demonstrate that each of the three tested anti-CRISPR proteins can bind to either the Csy complex or Cas3.
Figure 1. Anti-CRISPR proteins inhibit CRISPR–Cas function by directly interacting with the Csy complex or Cas3.

a, b, Purified Csy complex was incubated with purified AcrF1 (a) or AcrF3 (b) and the mixture was fractionated by SEC. Fractions were analysed by SDS–polyacrylamide gel electrophoresis (SDS–PAGE) and are numbered according to their elution position (see Extended Data Fig. 1 for SEC of the Csy complex alone or with AcrF2). The purified Csy complex or anti-CRISPR (ACR) are shown in the second (Csy) and last (ACR) lanes, respectively. c, Purified Cas3 was incubated with (right) or without (left) AcrF3 and fractionated by SEC. The eluting fractions were analysed by SDS–PAGE as described earlier. The input (In) lanes show the protein mixture that was loaded onto the SEC column. MBP, maltose-binding protein. d, dsDNA binding by the Csy complex was assayed using an EMSA. Csy complex was present in all reactions except for lanes 1 and 6. Other components added to each reaction are designated above the lanes. In the lanes coloured red and blue, the designated components were premixed before the addition of DNA. ATP was added to the Csy–DNA–Cas3 reaction either before the addition of Cas3 (lanes 11, 12) or after (lane 13). The supershifted species resulting from Cas3 addition did not migrate into the gel upon prolonged electrophoresis, but it is dissociated by the addition of ATP (lane 13), demonstrating that the supershift is not caused by aggregated inactive protein.
The Csy complex recognizes foreign DNA targets through sequential recognition of a protospacer adjacent motif (PAM) and crRNA-guided base pairing to a target11. We performed electrophoretic mobility shift assays (EMSAs) to demonstrate that the interaction of AcrF1 and AcrF2 with the Csy complex blocked its ability to bind a 50 base pair (bp) double-stranded DNA (dsDNA) target containing a PAM and a sequence identical to the crRNA spacer (Fig. 1d). We used isothermal titration calorimetry to show that these anti-CRISPRs also blocked binding of the Csy complex to an 8-nucleotide single-stranded DNA (ssDNA) target complementary to the functionally crucial ‘seed’ region12 of the crRNA (Extended Data Fig. 3). AcrF3, which does not interact with the Csy complex, did not inhibit the DNA-binding activity of the Csy complex (Fig. 1d, lane 5, and Extended Data Fig. 3).
To probe the potential role of AcrF3 in blocking Cas3 activity, we mixed purified Cas3 with the Csy complex and target DNA. In this instance, a supershifted species appeared in the EMSA gel that we presumed comprised the Csy complex, DNA and Cas3 (Fig. 1d, lane 7; a reaction containing only Cas3 and DNA did not display this species, lane 6). Importantly, pre-incubation of Cas3 with AcrF3 prevented formation of the supershifted complex (Fig. 1d, lane 10), indicating that this anti-CRISPR blocks recruitment of Cas3 to the Csy–DNA complex. Pre-incubation of Cas3 with AcrF1 or AcrF2 did not have this effect (Fig. 1d, lanes 8, 9). Further corroborating the presence of Cas3 in the supershifted complex, the addition of ATP prevented formation of this species (Fig. 1d, lane 11) and destabilized a preformed complex (lane 13), probably owing to the activation of the ATP-dependent helicase activity of Cas3, as described for the type I-E CRISPR–Cas system10.
To demonstrate that the described anti-CRISPR mechanisms operate in vivo, we targeted the Csy complex to the promoter of the phzM gene, which is required in P. aeruginosa for production of the blue–green pigment pyocyanin13. Binding of the phzM promoter by a Csy complex in the absence of Cas3 activity was expected to repress transcription, as was previously observed for a type I-E CRISPR–Cas system14,15. Consistent with this expectation, targeting of the phzM promoter in cells containing a prophage expressing acrF3 resulted in cultures with a complete lack of pigment production, similar to a strain lacking Cas3 (Fig. 2a; the somewhat higher pigment production in the Δcas3 strain is probably due to reduced Csy function16). By contrast, the expression of acrF1 and acrF2, which inhibit DNA binding by the Csy complex, resulted in blue–green cultures, as did expression of the phzM promoter targeting crRNA in cells lacking Csy3. Quantitative polymerase chain reaction with reverse transcription (RT–qPCR) experiments showed that these changes in pyocyanin production correlated with reduced transcription of the phzM gene (Extended Data Fig. 4). These results demonstrate that the expression of acrF3 blocks Cas3 activity in vivo, causing the Csy complex to function as a transcriptional repressor. Further in vivo experiments showed that phages dependent on acrF1 and acrF2 for viability4 were markedly inhibited by overexpression of the Csy complex subunits (Fig. 2b). The elevated level of Csy proteins probably increases the number of active Csy complexes and/or binds and titrates out anti-CRISPR molecules, resulting in insufficient levels of anti-CRISPR proteins to support robust phage replication. Phages dependent on acrF3 were not affected under these conditions because this anti-CRISPR protein binds to Cas3, the level of which is unchanged (overexpression of Cas3 inhibited cell growth). Interestingly, Csy subunit overexpression also inhibited a phage expressing acrF4 (gene 37 from phage JBD26), an anti-CRISPR protein that could not be purified. In addition, expression of this anti-CRISPR in the transcriptional repression assay resulted in a blue–green culture (Fig. 2a). These complementary results imply that AcrF4 binds the Csy complex, which we have experimentally confirmed (Extended Data Fig. 5). We conclude that our in vivo experiments are able to distinguish the effects of anti-CRISPR proteins that inactivate the Csy complex from those that inhibit Cas3.
Figure 2. Anti-CRISPR proteins interact with Cas proteins in vivo.
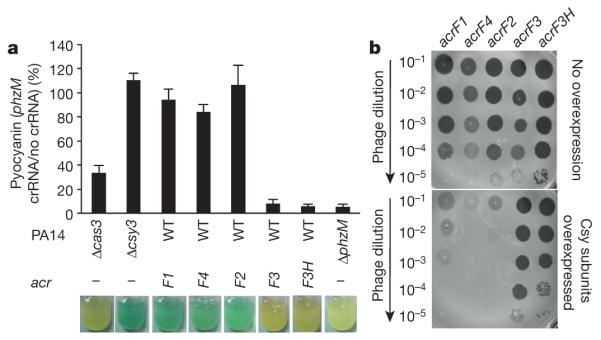
a, The phzM promoter was targeted by a plasmid-encoded crRNA in P. aeruginosa. The production of pyocyanin was quantified in different PA14 mutant backgrounds (Δcas3, Δcsy3 or ΔphzM) or during the expression of the indicated anti-CRISPR from a prophage. The amount of pyocyanin produced in the presence of a plasmid producing the crRNA is shown as a percentage of the same strain with the empty plasmid vector. An average of three independent experiments is shown with error bars representing the standard deviation (s.d.). Representative pictures of cultures are shown. The pyocyanin ratio for the ΔphzM mutant was derived by comparing it to the value for the Δcsy3 mutant. The prophage expressing acrF3 also encoded another anti-CRISPR, the functional mechanism of which is not known. To bolster our conclusions pertaining to acrF3, we also tested a prophage that expresses an 86% identical homologue of acrF3, designated acrF3H, and no other anti-CRISPR. WT, wild type. b, Lysates of phages expressing the indicated anti-CRISPR proteins were spotted in tenfold serial dilutions on bacterial lawns of wild-type P. aeruginosa PA14 (top) or the same strain bearing a plasmid that overexpresses the Csy subunits (bottom). These phages would be targeted by the CRISPR–Cas system in the absence of anti-CRISPR activity.
AcrF1 and AcrF2 both prevent DNA binding by the Csy complex, but might achieve this outcome through different mechanisms. The Csy complex assembles with a Csy1–Csy2 heterodimer bound at the 5′ end of the crRNA and a Csy4 monomer bound to the 3′ end, with six Csy3 subunits arrayed along the backbone of the spacer region in between (Fig. 3a)6,8. By purifying the Csy1–Csy2 heterodimer on its own and mixing it with purified anti-CRISPR proteins, we found that it co-eluted with AcrF2 in SEC experiments, but not with AcrF1 (Fig. 3b and Extended Data Fig. 6a). By contrast, AcrF1, but not AcrF2, bound Csy3 (Fig. 3c and Extended Data Fig. 6b). Csy3 eluted in monomeric and multimeric forms in SEC experiments, with AcrF1 binding predominantly to the multimeric fraction (Fig. 3c). The presence of distinct binding sites for AcrF1 and AcrF2 on the intact Csy complex was corroborated through competition experiments showing that both anti-CRISPR proteins could simultaneously bind the Csy complex and that the presence of one had no effect on the binding ability of the other (Extended Data Fig. 6c). RNase A treatment of the Csy complex, which resulted in Csy4 dissociation, had no effect on the binding of either anti-CRISPR (Extended Data Fig. 7). Quantification of the co-eluted fractions of AcrF1 or AcrF2 with the Csy complex by protein gel electrophoresis revealed the stoichiometry of AcrF1 to be 2.6 ± 0.3 proteins per Csy complex, while AcrF2 was 0.8 ± 0.1 (Extended Data Fig. 7c). To verify these stoichiometries, we created Csy complexes with shorter (16 nucleotides; Csy16 complex) and longer spacer regions (48 nucleotides; Csy48 complex). The purified Csy16 complex contained fewer molecules of Csy3 (4 ± 0.7) than wild type, and the Csy48 complex contained a proportionally greater number (9 ± 0.8) (Fig. 3d and Extended Data Fig. 8). Concomitant with the altered number of Csy3 molecules in the Csy16 and Csy48 complexes, we observed corresponding changes in the number of AcrF1 molecules bound, with the ratio of Csy3 to AcrF1 remaining constant. These results imply that AcrF1 binds along the full length of the Csy3 ‘spine’ of the complex. Its binding sites are probably at the interaction interfaces of the Csy3 subunits, which would account for the 2:1 Csy3/AcrF1 stoichiometry and for AcrF1 binding to only the multimeric Csy3 fraction (Fig. 3c). In contrast to AcrF1, the number of AcrF2 molecules bound to the altered Csy complexes did not change as the number of Csy3 molecules increased or decreased, consistent with AcrF2 binding to the Csy1–Csy2 heterodimer.
Figure 3. AcrF1 and AcrF2 bind distinct Csy complex subunits.

a, A schematic of the crRNA showing the repeat-derived regions of the crRNA (black) and the 32-nucleotide (nt) spacer region (red). The coloured circles represent the Csy1–4 subunits. b, c, Purified 6×His/MBP-tagged Csy1–Csy2 heterodimer (b) or Csy3 (c) was fractionated by SEC in the presence (right) or absence (left) of the indicated anti-CRISPR proteins. The SEC fractions were analysed by SDS–PAGE. The ‘In’ lanes show the protein mixture that was loaded onto the SEC column and fractions are numbered. d, Purified Csy complexes with 16-, 32-, or 48-nucleotide crRNA spacer regions were bound to AcrF1 or AcrF2 and fractionated by SEC. The stoichiometry of the bound anti-CRISPR proteins was quantified through densitometry of the Coomassie blue stained gels. An average of three independent experiments is shown with error bars representing s.d.
To define further the sites of action of the anti-CRISPR proteins on the Csy complex, we performed DNA-binding assays using ssDNA molecules complementary to subregions of the crRNA spacer. As shown in Fig. 4a, AcrF1 inhibited binding to all the ssDNA molecules tested. By contrast, AcrF2 prevented binding to a 24-nucleotide ssDNA molecule complementary to the 5′ end of the crRNA, including the seed region, but did not inhibit binding to a 16-nucleotide ssDNA complementary to the 3′ end of the spacer. Binding to a 26-nucleotide ssDNA binding the 3′ end was only partially inhibited. These data suggest that AcrF2 inhibits DNA binding by sterically blocking the 5′ end of the crRNA spacer through its interaction with Csy1–Csy2, which is expected to be bound to this region of the crRNA17,18. Addition of AcrF2 to a Csy complex that had been pre-saturated with target DNA resulted in an approximately 60% decrease in the binding level of this anti-CRISPR, suggesting that AcrF2 and DNA compete for an overlapping binding interface (Fig. 4b and Extended Data Fig. 9a). Consistent with this result, addition of AcrF2 to a DNA-bound Csy complex resulted in appreciably decreased DNA binding as detected by EMSA (Fig. 4c). Parallel experiments performed with AcrF1 showed that the binding of AcrF1 to the Csy complex was not affected by prior binding to DNA (Fig. 4b). We conclude that the interaction of AcrF1 with the full length of the spine of the complex formed by multiple Csy3 molecules and the crRNA accounts for its ability to block binding to all dsDNA and ssDNA molecules tested. Furthermore, the ability of AcrF1 and DNA to bind the Csy complex simultaneously suggests an allosteric mechanism for the activity of this anti-CRISPR. Thus, the mechanisms of AcrF1 and AcrF2 are distinct, using different Csy protein-binding partners, stoichiometry and DNA occlusion mechanisms (that is, steric versus allosteric).
Figure 4. Two anti-CRISPR proteins inhibit target recognition via unique mechanisms.
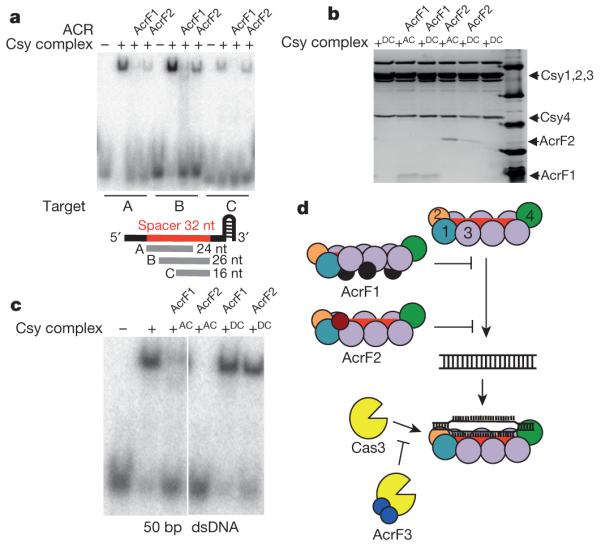
a, EMSA experiments were used to assay binding of the Csy complex to three different ssDNA oligonucleotides (labelled A, B and C) that are complementary to different regions of the crRNA spacer as shown in the schematic (see Extended Data Fig. 9b). Where noted, the Csy complex was pre-incubated with the indicated anti-CRISPR. b, c, Apo–Csy complex (AC) or DNA-bound Csy complex (DC) was incubated with AcrF1 or AcrF2. b, This mixture was fractionated by SEC and fractions were visualized by SDS–PAGE. c, An EMSA experiment is shown with binding to dsDNA in the same experimental setup as in b. d, A model summarizing anti-CRISPR mechanisms. Arrows indicate the steps of the uninhibited CRISPR–Cas interference pathway. Numbers in the Csy complex indicate the Csy subunits. The lines with flat ends indicate the step in the CRISPR–Cas pathway blocked by each anti-CRISPR. The manner in which each anti-CRISPR binds to CRISPR–Cas components is also shown. AcrF1 makes the whole crRNA inaccessible while AcrF2 occludes the 5′ end.
We provide the first insight into the mechanisms by which proteins can inhibit a CRISPR–Cas system. The diverse and distinct mechanisms discovered here (Fig. 4d) reflect the deep evolutionary roots of the virus–host arms race. Anti-CRISPR proteins, both known4,19 and yet to be discovered, will provide an extensive set of valuable tools both better to understand and to manipulate CRISPR–Cas systems. One example is our finding that AcrF3 converts the CRISPR–Cas system into a gene regulator by blocking Cas3 recruitment. Since CRISPR–Cas systems perform a variety of roles beyond destroying foreign DNA20, many important functions may be fulfilled by proteins that interact with CRISPR–Cas components and thus alter the activity of the system.
METHODS
Protein purification
All proteins were affinity purified using Ni-NTA beads (Qiagen) to isolate recombinant proteins bearing a terminal 6×His tag. Anti-CRISPR proteins were expressed from the p15TV-L vector (NCBI accession number EF456736), which possesses a T7 promoter and an amino-terminal 6×His tag. Constructs expressing Csy1–4 containing a 6×His tag on either Csy3 or Csy4 were co-expressed with a construct producing a crRNA as previously described7. Individual Cas proteins (Csy1–Csy2, Csy3, and Cas3) were expressed from pHMGWA (NCBI accession number EU680841), which also has a T7 promoter. The proteins in this vector were tagged with a maltose-binding protein and 6×His.
Cultures of E. coli BL21 containing a plasmid expressing a protein of interest were grown to an optical density (OD600 nm) of 0.5 and then induced with 1 mM isopropyl-β-D-thiogalactoside (IPTG) for 3 h at 37 °C (anti-CRISPRs, Csy3) or for 16 h at room temperature (Csy complex, Csy1–Csy2, Cas3). Cells were collected by centrifugation at 5,000g for 10 min and resuspended in a binding buffer (20 mM Tris, pH 7.5, 250 mM NaCl, 5 mM imidazole, 1 mM dithiothreitol (DTT) and 1 mM PMSF). The cells were lysed by sonication and the resulting lysate was centrifuged at 15,000g for 15 min to remove cell debris. The supernatant was mixed with Ni-NTA beads that had been washed in binding buffer (without DTT) five times. Binding to the beads proceeded for 1 h at 4 °C under gentle rotation, at which point the lysate and beads were passed through a column, washed 3–5 times with binding buffer containing 30 mM imidazole and ultimately eluted in buffer containing 250 mM imidazole. Colourimetric Bradford assays were conducted during the procedure to determine the number of washes to perform and elution fractions to collect. Purified protein was dialysed into the binding buffer containing 5 mM imidazole to remove excess imidazole and visualized on Coomassie blue R250 stained SDS–PAGE gels. Cas3 was purified following the same general protocol but in a buffer optimized for this protein (50 mM HEPES, pH 7.5, 500 mM NaCl, 5% glycerol, 1 mM DTT, supplemented with 1 mM PMSF and 150 μM NiSO4 in the lysis buffer). Purified Cas3 was concentrated and buffer exchanged in an Amicon Ultra centrifugal filter (Millipore) into a different buffer (20 mM HEPES, pH 7.5, 300 mM KCl, 5% glycerol, 1 mM DTT) for protein interaction assays. Csy1–Csy2 also purified in the same buffer as Cas3 (with NiSO4 omitted). Purified Csy1–Csy2 was then dialysed into a different buffer (20 mM HEPES, pH 7.5, 250 mM NaCl, 5% glycerol, 1 mM DTT) for protein interaction experiments.
Size-exclusion chromatography
Affinity-purified proteins were fractionated by SEC using a GE Life Sciences Superdex 200 10/300 column. Fractions were collected in 0.5 ml volumes and monitored by optical density at 280 nm. SDS–PAGE gels were stained with silver nitrate or Coomassie blue R250 to identify proteins. In interaction experiments, purified proteins were mixed together before fractionation by SEC and co-eluting proteins were identified by SDS–PAGE. The Csy complex or Csy proteins and an anti-CRISPR protein of interest were generally incubated together for 1 h at 4 °C. This mixture was then applied to the SEC column at room temperature. A fraction of the input (~0.5%) was also kept for SDS–PAGE analysis.
Anti-CRISPR stoichiometry
The purified Csy complex was incubated with ~10-fold molar excess of purified anti-CRISPR proteins. This mixture was fractionated by SEC as described earlier. The Csy complex peak fraction was run on SDS–PAGE gels in twofold serial dilutions. The protein bands were identified with Coomassie blue R250. Image Lab Software (Bio-Rad) was used to quantify band intensities and calculate the relative stoichiometries of the various subunits and anti-CRISPRs, after adjusting for molecular weight and comparing dilutions. Our estimates of the absolute stoichiometries of the Csy subunits is based on the stoichiometry of the Csy complex established in previous publications7,8.
RNase A treatment of the Csy complex
Pancreatic RNase A (73 μM) was used to treat the Csy complex (4 μM) for 30 min at 37 °C. After digestion, the treated Csy complex was fractionated by SEC in the absence or presence of an anti-CRISPR protein. Fractions from SEC were analysed on Coomassie stained SDS–PAGE gels to visualize proteins and SYBR Gold stained TBE-Urea gels to visualize nucleic acid.
Isothermal titration calorimetry
Purified Csy complex was added to the isothermal titration calorimetry (ITC) chamber at a concentration of 7.5 μM. The DNA ligand (8-nucleotide ssDNA) was placed in the injection syringe at a concentration of 75 μM. After a null injection of 0.3 μl of titrant, 3 μl of titrant were injected 13 times, with 120 s intervals between the injections to establish a baseline. The DNA titrant and Csy complex were in the same buffer (20 mM Tris, pH 7.5, 250 mM NaCl, 5 mM imidazole) and the experiment was temperature controlled at 25 °C. To assess the role of AcrF1 in interfering with the interaction between the Csy complex and a DNA target, the Csy complex was first incubated with a ~10-fold molar excess of anti-CRISPR proteins for 1 h at 4 °C. This mixture was then applied to the chamber, the temperature equilibrated to 25 °C and the DNA titration performed.
Electrophoretic mobility shift assay
A 50-nucleotide ssDNA molecule was synthesized (Eurofins Genomics) that contains 32 nucleotides of complementarity to the crRNA in the purified Csy complex. The DNA (200 nM) was phosphorylated in a T4 polynucleotide kinase reaction with [γ-32P]ATP. The reaction was stopped with 12 mM EDTA and GE MicroSpin G-25 columns were used to remove remaining radiolabelled nucleotides. To generate dsDNA, the labelled strand was heated to 98 °C in the presence of a twofold excess of an unlabelled complementary strand and allowed to return slowly to room temperature. Csy complex–DNA-binding reactions were conducted in a binding buffer (10 mM HEPES, pH 7.5, 1 mM MgCl2, 20 mM KCl, 1 mM TCEP, bromophenol blue and 6% glycerol) at 37 °C for 15 min. The concentration of the Csy complex used in EMSA experiments varied, depending on the oligonucleotide target being used. For 50 bp dsDNA EMSA reactions, 100 nM of the Csy complex was routinely used in reactions, with <1 nM labelled DNA. Anti-CRISPR proteins were used at a tenfold molar excess compared to the Csy complex and allowed to incubate with Apo–Csy complex or DNA-bound Csy complex for 1 h. After the appropriate incubation, the reactions were resolved on native 6% polyacrylamide TBE gels. Gels were wrapped in Saran wrap and visualized with a phosphoscreen and Typhoon imager. Optimal exposures were ~2–3 h.
For EMSA experiments involving Cas3, the Csy complex and target DNA were prebound as described above. 6×His-tagged Cas3 was purified by Ni-NTA chromatography (6×His) followed by SEC, concentrated, transferred into the EMSA reaction buffer, flash frozen in small volumes (50 μl) and stored at −70 °C. Cas3 was added to the EMSA reaction at a final concentration of 400 nM and incubated for 30 min at 37 °C. ATP was added at a final concentration of 2 mM and all reactions with Cas3 also contained 100 μM CoCl2.
Pyocyanin repression
A crRNA was designed to target the promoter region of phzM, a gene required for the biosynthesis of the blue–green pigment pyocyanin. Two complementary oligonucleotides were synthesized containing two 28 bp PA14 CRISPR repeat sequences, flanking a 32 bp sequence with perfect complementarity to the −35/−10 region of the phzM promoter (position 813576–813607 in the PA14 genome). The spacer was designed to produce a crRNA that would bind to the non-template strand, in a position where the protospacer adjacent motif (GG) is present. The oligonucleotides were annealed and cloned into an arabinose inducible P. aeruginosa expression vector, pHERD30T. This construct was then used to transform PA14 strains possessing single cas gene knockouts or wild-type PA14 possessing prophages expressing various anti-CRISPRs. Individual transformants were grown overnight (~20 h) in 2 ml of King’s A media in 50 μg ml−1 gentamicin and 0.025% arabinose, to induce expression of the crRNA. Pyocyanin was extracted with an equal volume of chloroform, and then mixed with 1 ml of 0.2 M HCl, producing a pink–red colour proportional to the amount of pyocyanin, which was quantitated by measuring absorbance at 520 nm. Anti-CRISPR proteins were expressed from the following prophages: JBD30 (AcrF1), D3112 (AcrF2), JBD26 (AcrF4), JBD5 (AcrF3 and AcrF5), and JBD88a (AcrF3H). Since phage JBD5 contains two type I-F anti-CRISPR proteins, phage JBD88a (possessing a homologue of AcrF3 with 86% protein sequence identity) was also used.
Competition experiments
To determine whether the two anti-CRISPR proteins that bind to the Csy complex compete with each other for the same binding site, the first anti-CRISPR was added for 1 h at 4 °C and then the second for the same amount of time. This entire mixture was then fractionated by SEC.
To determine whether DNA and anti-CRISPR proteins compete for the same binding site, the purified Csy complex (4.5 μM) was mixed with a 50 bp dsDNA target (10 μM) and incubated for 15 min at 37 °C in the same buffer in which the proteins were purified (20 mM Tris, pH 7.5, 250 mM NaCl, 5 mM imidazole). This DNA-bound Csy complex was then mixed with a tenfold molar excess of AcrF1, AcrF2, or an equivalent volume of buffer and incubated for 1 h at 4 °C. This mixture was fractionated by SEC. The fraction containing the Csy complex was analysed on Coomassie blue stained SDS–PAGE gels or SYBR Gold stained TBE-Urea gels.
Plaque assays with Csy subunit overexpression
To assess the consequence of Csy protein overexpression on phages possessing distinct anti-CRISPR proteins in vivo, a pHERD30T derived plasmid expressing the csy1, csy2, csy3 and csy4 genes was used to transform P. aeruginosa strain PA14. Phage lysates were spotted in tenfold serial dilutions onto a lawn of PA14 containing empty vector, or the plasmid expressing the csy genes. Phages JBD30, JBD26, D3112 and JBD88a all have protospacers that display 100% identity to spacers 17 and 20 in the PA14 CRISPR2 locus4. JBD5 has a protospacer matching CRISPR2 spacer 1 that has been shown to be targeted4,21.
RT–qPCR
RT–qPCR reactions were conducted as described previously4. Briefly, total RNA was extracted and DNase treated. One nanogram of total RNA was subjected to a reverse transcription reaction and qPCR, using primers specific to phzM or a control, rpsL. The efficient removal of DNA from the RNA preparation was confirmed by including controls for each sample without reverse transcriptase added.
AcrF2 misannotation
The D3112 phage genome has an annotated open reading frame identified as gene 30, which is a predicted 90 amino acid protein (NCBI accession number NC_005178). This version of the gene was previously identified as an anti-CRISPR, although overexpression from a plasmid was required for activity4. A nucleotide alignment of the anti-CRISPR region of many phages revealed that all phage anti-CRISPR operons possess a start codon (ATG) at the same position for the first anti-CRISPR gene, except phage D3112. Phages D3112 and MP29 (which has a D3112 gene 30 homologue), had the start position annotated downstream of this commonly used ATG, at a second ATG, in frame with the first, resulting in a putative truncation of six amino acid residues. Re-cloning of the gene to include these six residues resulted in a construct that had full anti-CRISPR activity in the absence of overexpression. Thus, this 96-residue protein (sequence shown later, with new residues in bold) is the version that was used in all downstream experiments presented here and in affinity purification, after addition of the appropriate tag. All other anti-CRISPR protein sequences are as reported in ref. 4. AcrF2: MTKTAQMIAQQHKDTVAACEAAEAIAIAKDQVWDGEGYT KYTFDDNSVLIQSGTTQYAMDADDADSIKGYADWLDDEARSAEASEIER LLESVEEE.
Statistics, reagents and data deposition
To assess interactions between anti-CRISPR proteins and the Csy complex or purified Cas proteins, mixed components were fractionated by SEC. Each result shown in the manuscript was obtained on at least two independent occasions. ITC, EMSA and plaque assays were all replicated at least three times. No statistical methods were used to predetermine sample size. The experiments were not randomized. The investigators were not blinded to allocation during experiments and outcome assessment.
The sequences of the anti-CRISPR proteins are present in ref. 4, with full genomes for phages JBD30, D3112, JBD5, JBD26 and JBD88a available on NCBI (accession numbers: NC_020198, NC_005178, NC_020202, JN811560 and NC_020200, respectively).
Extended Data
Extended Data Figure 1. AcrF2 interacts with the Csy complex.

a, b, Purified Csy complex was fractionated by SEC alone (a) or in the presence of AcrF2 (b). Fractions were analysed on a silver nitrate stained SDS–PAGE gel. The input (IN) and fractions are shown.
Extended Data Figure 2. AcrF3, not AcrF1, interacts with Cas3.

a, Cas3 was fractionated by SEC alone or in the presence of AcrF3 or AcrF1. Overlays of plots of elution volume versus optical density at 280 nm of the column eluates are shown. The numbers represent the fractions that were selected for analysis. b–e, Silver nitrate stained SDS–PAGE gels are shown from SEC experiments with Cas3 (b), AcrF3 (c), Cas3 with AcrF3 (d) or Cas3 with AcrF1 (e). The sample that was loaded onto the SEC column is shown as input (In) and fractions from the same elution positions are indicated numerically. AcrF3 is seen eluting in fractions 4–8 only in the presence of Cas3. There is also a visible shift in the Cas3 elution profile in the presence of AcrF3 but not AcrF1 (fractions 3–5).
Extended Data Figure 3. AcrF1 and AcrF2 prevent target recognition by the Csy complex.

Isothermal titration calorimetry (ITC) assays showing the Csy complex binding to an 8-nucleotide ssDNA target that comprises the seed region. No binding is observed in the presence of AcrF1, AcrF2 or with a non-target (the reverse complement sequence of the target) ssDNA substrate. A representative run is shown for each condition with the dissociation constant (Kd) value and error of fit from that particular run. Over multiple runs (n = 6) with the Csy complex binding to the ssDNA ligand, the average Kd value was 90 nM ± 37.
Extended Data Figure 4. Expression of phzM is repressed by the Csy complex.
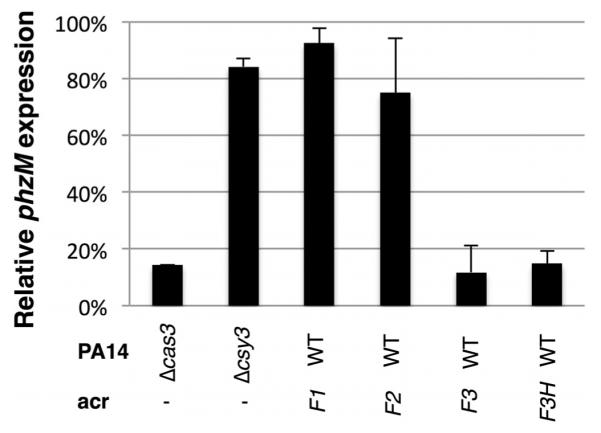
The Csy complex was targeted to the promoter of the gene phzM, and repression efficiency was assayed by RT–qPCR (see Methods). The per cent repression of phzM in the indicated strains expressing a phzM-targeting crRNA relative to wild-type (WT) PA14 with an empty plasmid is shown. All values were normalized to rpsL, a gene encoding a ribosomal protein. Means ± s.d. are shown.
Extended Data Figure 5. AcrF4 interacts with the Csy complex.

Untagged AcrF4 was expressed in E. coli BL21 cells and a crude lysate of these cells was mixed with the Csy complex bound to Ni-NTA beads via a 6×His tag on Csy3. a, The flow through (FT), wash 1 (W1), and two elution fractions (E1, E2) from the Ni-NTA column are shown, as well as a comparison to pure Csy complex. b, The Ni-NTA elution fractions were fractionated by SEC, demonstrating a stable interaction between the Csy complex and AcrF4. The input (In) lane shows the sample that was loaded on the SEC column and numbered fractions are analysed on SDS–PAGE gels.
Extended Data Figure 6. AcrF1 and AcrF2 bind the Csy complex at distinct locations.

a, Purified Csy1–Csy2 heterodimer with an MBP and 6×His tag fused to Csy1 was fractionated by SEC in the presence or absence of AcrF1 (boxes indicate the Csy1–Csy2 peak). b, Purified MBP/6×His-tagged Csy3 was fractionated in the presence or absence of AcrF2. These are complementary experiments to those seen in Fig. 3b and c, respectively. Input (In) and selected fractions are shown on SDS–PAGE gels. c, AcrF1 and AcrF2 were incubated with the Csy complex singly or in combination. Asterisks designate which anti-CRISPR was added first to the reactions containing both anti-CRISPR proteins. The addition order did not affect the result since there is no competition for binding sites between these two anti-CRISPR proteins. After incubation, each mixture was fractionated by SEC and the peak Csy complex fraction is shown on an SDS–PAGE gel. In each experiment the anti-CRISPR proteins are in excess relative to the Csy complex.
Extended Data Figure 7. AcrF1 and AcrF2 interact with an RNase-A-treated Csy complex.

a, The Csy complex was treated with a low concentration (600 nM, +) of RNase A or a high concentration of RNase A (70 μM, ++). This mixture was fractionated by SEC, revealing Csy4 dissociation at the higher RNase A concentration. Pre-treatment of the Csy complex with RNase A, with the subsequent addition of AcrF1 or AcrF2 followed by SEC fractionation was then conducted. Peak Csy complex fractions are shown on an SDS–PAGE gel. b, A TBE-urea denaturing gel is shown, stained with SYBR gold, showing the native crRNA in the Csy complex and the protected fragments remaining after 70 μM RNase A treatment. c, Quantification of Coomassie blue stained gels from three independent preparations of the respective proteins is shown. Anti-CRISPR proteins bound with unaltered stoichiometry to RNase-A-pre-treated Csy complexes. Error bars represent s.d.
Extended Data Figure 8. Twofold dilutions used to quantify anti-CRISPR binding stoichiometry.

Csy complexes with crRNA molecules possessing spacers of differing lengths (16, 32, or 48 nucleotides) were purified and fractionated by SEC in the presence of AcrF1. A representative Coomassie blue stained SDS–PAGE gel is shown, with twofold dilutions of the peak fraction containing the Csy complex and co-eluting AcrF1. Arrows on the bottom of the gel indicate comparable dilutions based on the levels of Csy1. Note the increasing abundance of Csy3 and AcrF1. b, Lanes with arrows from the gel in a are shown next to each other for comparison.
Extended Data Figure 9. dsDNA binds to the Csy complex after SEC fractionation.

a, The same samples from Fig. 4a were run on a denaturing TBE-urea gel, stained with SYBR gold, to reveal the crRNA (two species are apparent), and the Csy-complex-bound 50 bp dsDNA. In these experiments, DNA was prebound to the Csy complex, and AcrF1 or AcrF2 were subsequently added to the DNA-saturated Csy complex. This mixture was then fractionated by SEC and the Csy-complex-containing peak fractions were analysed. b, A schematic showing the crRNA sequence with repeat-derived regions shown in black and the variable 32-nucleotide spacer region in red. The seed-interacting region that is critical for target recognition (nucleotides 1–5, 7, 8) is in bold. DNA oligonucleotides used in this study are shown, with labels ‘A’, ‘B’ and ‘C’ corresponding to the targets shown in Fig. 4c. The 8-nucleotide ssDNA substrate was used in ITC experiments (Extended Data Fig. 3), and the 50 bp dsDNA in EMSAs (Figs 1d and 4b).
Acknowledgements
We thank W. Navarre and E. Westra for reading the manuscript. This work was supported by an Operating Grant to A.R.D. (MOP-130482) and to K.L.M. (MOP-136845), both of which were from the Canadian Institutes of Health Research (CIHR). J.B.-D. was supported by a CIHR Canada Graduate Scholarship Doctoral Award and an Ontario Graduate Scholarship award. Research in the Wiedenheft laboratory is supported by the National Institutes of Health (P20GM103500 and R01GM108888), the National Science Foundation EPSCoR (EPS-110134), the M.J. Murdock Charitable Trust, and the Montana State University Agricultural Experimental Station.
Footnotes
Online Content Methods, along with any additional Extended Data display items and Source Data, are available in the online version of the paper; references unique to these sections appear only in the online paper.
Author Contributions J.B.-D. designed, performed and supervised experiments and wrote the manuscript. B.G., S.S., M.D., and Y.H.-R. performed experiments. M.F.R. and B.W. provided essential reagents and experimental assistance. K.L.M. supervised experiments. A.R.D. designed and supervised experiments and wrote the manuscript.
The authors declare no competing financial interests.
Readers are welcome to comment on the online version of the paper.
References
- 1.Barrangou R, et al. CRISPR provides acquired resistance against viruses in prokaryotes. Science. 2007;315:1709–1712. doi: 10.1126/science.1138140. [DOI] [PubMed] [Google Scholar]
- 2.Makarova KS, et al. Evolution and classification of the CRISPR-Cas systems. Nature Rev. Microbiol. 2011;9:467–477. doi: 10.1038/nrmicro2577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jore MM, Brouns SJJ, van der Oost J. RNA in defense: CRISPRs protect prokaryotes against mobile genetic elements. Cold Spring Harb. Perspect. Biol. 2012;4:a003657. doi: 10.1101/cshperspect.a003657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bondy-Denomy J, Pawluk A, Maxwell KL, Davidson AR. Bacteriophage genes that inactivate the CRISPR/Cas bacterial immune system. Nature. 2013;493:429–432. doi: 10.1038/nature11723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.van der Oost J, Westra ER, Jackson RN, Wiedenheft B. Unravelling the structural and mechanistic basis of CRISPR–Cas systems. Nature Rev. Microbiol. 2014;12:479–492. doi: 10.1038/nrmicro3279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Haurwitz RE, Jinek M, Wiedenheft B, Zhou K, Doudna JA. Sequence- and structure-specific RNA processing by a CRISPR endonuclease. Science. 2010;329:1355–1358. doi: 10.1126/science.1192272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wiedenheft B, et al. RNA-guided complex from a bacterial immune system enhances target recognition through seed sequence interactions. Proc. Natl Acad. Sci. USA. 2011;108:10092–10097. doi: 10.1073/pnas.1102716108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.van Duijn E, et al. Native tandem and ion mobility mass spectrometry highlight structural and modular similarities in clustered-regularly-interspaced shot-palindromic-repeats (CRISPR)-associated protein complexes from Escherichia coli and Pseudomonas aeruginosa. Mol. Cell. Proteomics. 2012;11:1430–1441. doi: 10.1074/mcp.M112.020263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Westra ER, et al. CRISPR immunity relies on the consecutive binding and degradation of negatively supercoiled invader DNA by Cascade and Cas3. Mol. Cell. 2012;46:595–605. doi: 10.1016/j.molcel.2012.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Huo Y, et al. Structures of CRISPR Cas3 offer mechanistic insights into Cascade-activated DNA unwinding and degradation. Nature Struct. Mol. Biol. 2014;21:771–777. doi: 10.1038/nsmb.2875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rollins MF, Schuman JT, Paulus K, Bukhari HST, Wiedenheft B. Mechanism of foreign DNA recognition by a CRISPR RNA-guided surveillance complex from Pseudomonas aeruginosa. Nucleic Acids Res. 2015;43:2216–2222. doi: 10.1093/nar/gkv094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Semenova E, et al. Interference by clustered regularly interspaced short palindromic repeat (CRISPR) RNA is governed by a seed sequence. Proc. Natl Acad. Sci. USA. 2011;108:10098–10103. doi: 10.1073/pnas.1104144108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wurtzel O, et al. The single-nucleotide resolution transcriptome of Pseudomonas aeruginosa grown in body temperature. PLoS Pathog. 2012;8:e1002945. doi: 10.1371/journal.ppat.1002945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Luo ML, Mullis AS, Leenay RT, Beisel CL. Repurposing endogenous type I CRISPR-Cas systems for programmable gene repression. Nucleic Acids Res. 2015;43:674–681. doi: 10.1093/nar/gku971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rath D, Amlinger L, Hoekzema M, Devulapally PR, Lundgren M. Efficient programmable gene silencing by Cascade. Nucleic Acids Res. 2015;43:237–246. doi: 10.1093/nar/gku1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cady KC, O’Toole GA. Non-identity-mediated CRISPR-bacteriophage interaction mediated via the Csy and Cas3 proteins. J. Bacteriol. 2011;193:3433–3445. doi: 10.1128/JB.01411-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jackson RN, et al. Crystal structure of the CRISPR RNA-guided surveillance complex from Escherichia coli. Science. 2014;345:1473–1479. doi: 10.1126/science.1256328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mulepati S, Héroux A, Bailey S. Crystal structure of a CRISPR RNA-guided surveillance complex bound to a ssDNA target. Science. 2014;345:1479–1484. doi: 10.1126/science.1256996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pawluk A, Bondy-Denomy J, Cheung VHW, Maxwell KL, Davidson AR. A new group of phage anti-CRISPR genes inhibits the type I-E CRISPR-Cas system of Pseudomonas aeruginosa. MBio. 2014;5:e00896. doi: 10.1128/mBio.00896-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Westra ER, Buckling A, Fineran PC. CRISPR–Cas systems: beyond adaptive immunity. Nature Rev. Microbiol. 2014;12:317–326. doi: 10.1038/nrmicro3241. [DOI] [PubMed] [Google Scholar]
- 21.Cady KC, Bondy-Denomy J, Heussler GE, Davidson AR, O’Toole GA. The CRISPR/Cas adaptive immune system of Pseudomonas aeruginosa mediates resistance to naturally occurring and engineered phages. J. Bacteriol. 2012;194:5728–5738. doi: 10.1128/JB.01184-12. [DOI] [PMC free article] [PubMed] [Google Scholar]