

Abstract
Secreted protein acidic and rich in cysteine (SPARC) is a collagen-binding matricellular protein highly expressed during fibrosis. Fibrosis is a prominent component of cardiac aging that reduces myocardial elasticity. Previously, we reported that SPARC deletion attenuated myocardial stiffness and collagen deposition in aged mice. To investigate the mechanisms by which SPARC promotes age-related cardiac fibrosis, we evaluated six groups of mice (n = 5–6/group): young (3–5 mo old), middle-aged (10–12 mo old), and old (18–29 mo old) C57BL/6 wild type (WT) and SPARC-null (Null) mice. Collagen content, determined by picrosirius red staining, increased in an age-dependent manner in WT but not in Null mice. A disintegrin and metalloproteinase with thrombospondin-like motifs 1 (ADAMTS1) increased in middle-aged and old WT compared with young, whereas in Null mice only old animals showed increased ADAMTS1 expression. Versican, a substrate of ADAMTS1, decreased with age only in WT. To assess the mechanisms of SPARC-induced collagen deposition, we stimulated cardiac fibroblasts with SPARC. SPARC treatment increased secretion of collagen I and ADAMTS1 (both the 110-kDa latent and 87-kDa active forms) into the conditioned media as well as the cellular expression of transforming growth factor-β1-induced protein (Tgfbi) and phosphorylated Smad2. An ADAMTS1 blocking antibody suppressed the SPARC-induced collagen I secretion, indicating that SPARC promoted collagen production directly through ADAMTS1 interaction. In conclusion, ADAMTS1 is an important mediator of SPARC-regulated cardiac aging.
Keywords: secreted protein acidic and rich in cysteine, heart, a disintegrin and metalloproteinase with thrombospondin-like motifs 1, fibroblast, matrix metalloproteinase
secreted protein acidic and rich in cysteine (SPARC) is a procollagen-binding protein that participates in procollagen processing to stimulate the formation of insoluble mature cross-linked collagen fibrils. Previous studies in cells of dermal origin suggest that SPARC chaperones procollagen processing by binding to newly secreted procollagen in the extracellular steps (2, 7). One proposed mechanism involves SPARC diminishing procollagen binding to its cell surface receptors, which facilitates procollagen processing and limits improper processing (5, 6, 37, 38). Increased SPARC concentrations are observed in fibrotic areas of the skin, kidney, lung, and left ventricle (LV) (36, 48). Serum SPARC concentrations increase in patients with fibrotic renal injury and in individuals with systemic sclerosis (24). SPARC is an important determinant of procollagen processing in myocardial tissue, and SPARC deletion reduces pressure overload-induced fibrillar collagen deposition and diastolic dysfunction (31). In the aging heart, increases in fibrillar collagen are thought to contribute to increases in myocardial stiffness. SPARC expression in the myocardium increases in old mice and the absence of SPARC suppresses age-related increases in cardiac stiffness and total collagen content (4). In addition, SPARC participates in the deposition of collagen I, III, IV, and VI in aged LV (13).
We have demonstrated that SPARC deletion delays the age-related increases in macrophage infiltration and proinflammatory cytokine expression, indicating that SPARC acts as a mediator of age-related cardiac inflammation (44). In the same study, we found that increases in LV wall thickness and myocyte size in aged WT mice are blunted in the absence of SPARC. The extracellular matrix (ECM) serves as a reservoir for the sequestration of growth factors and cytokines and provides structural support for the myocardium (21). However, our understanding of SPARC functions in the aging myocardium is incomplete. Elucidation of age-dependent changes in the ECM composition is needed to clarify the role of SPARC in cardiac fibrosis, inflammation, and remodeling to prevent or slow age-associated cardiac dysfunction in the elderly. Accordingly, the aims of this study were to investigate the role of SPARC in mediating age-dependent changes in cardiac cell adhesion molecule and ECM expression and explore the mechanisms by which SPARC influences collagen deposition in the LV.
MATERIALS AND METHODS
Animals.
All animal procedures were performed at the Medical University of South Carolina (MUSC) based on the Guide for the Care and Use of Laboratory Animals (National Research Council, National Academies Press, Washington, DC, 2011) and were approved by the Institutional Animal Care and Use Committee at MUSC. C57BL/6 wild type (WT) and SPARC-null (Null) mice were used in this study. Each genotype included three age groups: young (3–5 mo old), middle-aged (10–12 mo old), and old (18–29 mo old). Both male and female mice were included in each group (n = 5–6 per age per genotype). The generation and phenotype of Null mice have been reported previously (34). Hearts were excised under isoflurane anesthesia. The right ventricle was separated from the LV, and the LV was divided into two sections. One section was snap-frozen for RNA extraction, and the second section was fixed in zinc formalin for histological analysis.
RNA extraction and quantitative real-time RT-PCR.
RNA was extracted using TRIzol reagent (15596-026; Invitrogen), and cDNA was synthesized using the RT2 First Strand Kit (330401; Qiagen). RNA levels were quantified using the NanoDrop ND-1000 Spectrophotometer (Thermo Scientific). Real Time RT2-PCR gene array for ECM and adhesion molecules (RT2 Profiler PCR arrays, PAMM-013E; Qiagen) was performed to quantify mRNA expression of 84 genes using RT2 SYBR green Rox quantitative PCR Master Mix (330523; Qiagen). The array performs gene expression analysis with quantitative real-time PCR sensitivity and the multigene profiling capability of microarray. The 84 genes analyzed are listed in Table 1. The relative gene expression of individual target molecules was calculated by normalization of the threshold cycle (CT) values of the target genes to the CT values of the housekeeping gene hypoxanthine-guanine phosphoribosyltransferase 1 (Hprt1).
Table 1.
ECM and adhesion molecules analyzed by gene array
| Gene List | ||||||
|---|---|---|---|---|---|---|
| ADAMTS1 | Contactin 1 | Emilin1 | Integrin-αL | Laminin β3 | MMP-8 | Spp1 |
| ADAMTS2 | Collagen I a1 | Entpd1 | Integrin-αM | Laminin γ1 | MMP-9 | Synaptotagmin-1 |
| ADAMTS5 | Collagen II a1 | Fibulin1 | Integrin-αV | MMP-10 | NCAM1 | Tgfbi |
| ADAMTS8 | Collagen III a1 | Fibronectin 1 | Integrin-αX | MMP-11 | NCAM2 | Thbs1 |
| Catenin-α1 | Collagen IV a1 | Hapln1 | Integrin-β1 | MMP-12 | PECAM1 | Thbs2 |
| Catenin-α2 | Collagen IV a2 | Hc | Integrin-β2 | MMP-13 | Periostin | Thbs3 |
| Catenin-β1 | Collagen IV a3 | ICAM1 | Integrin-β3 | MMP-14 | E-selectin | TIMP1 |
| Cd44 | Collagen V a1 | Integrin-α2 | Integrin-β4 | MMP-15 | L-selectin | TIMP2 |
| Cadherin-1 | Collagen VI a1 | Integrin-α3 | Laminin-α1 | MMP-1a | P-selectin | TIMP3 |
| Cadherin-2 | Versican | Integrin-α4 | Laminin-α2 | MMP-2 | Sgce | Tenascin-C |
| Cadherin-3 | Ctgf | Integrin-α5 | Laminin-α3 | MMP-3 | SPARC | VCAM1 |
| Cadherin-4 | Ecm1 | Integrin-αE | Laminin-β2 | MMP-7 | Spock1 | Vitronectin |
ECM, extracellular matrix; ADAMTS1, a disintegrin and metalloproteinase with thrombospondin-like motifs 1; Ctgf, connective tissue growth factor; Ecm1, extracellular matrix 1; Entpd1, ectonucleoside triphosphate diphosphohydrolase 1; Hapln1, hyaluronan and proteoglycan link protein 1; Hc, hemolytic complement; ICAM1, intercellular adhesion molecule 1; MMP, matrix metalloproteinase; NCAM, neural cell adhesion molecule; Sgce, sarcoglycan epsilon; SPARC, secreted protein acidic and rich in cysteine; Spock1, sparc/osteonectin; Spp1, secreted phosphoprotein 1; Tgfbi, transforming growth factor-β1-induced protein; Thbs, thrombospondin; TIMP, tissue inhibitor of metalloproteinase.
Histology.
Paraffin-embedded LV sections were deparaffinized and rehydrated. Sections were used for quantification of collagen deposition by staining with picrosirius red (PSR). Briefly, dehydrated sections were incubated in 0.2% phosphomolydbic acid and then stained with 0.1% Siris Red in saturated picric acid and washed in 0.01 N hydrochloric acid. Five random regions from each slide were scanned at ×40 magnification, and collagen content was determined by PSR-positive area per total area (Image-Pro Analyzer 7.0).
Cardiac fibroblast cultures.
LV tissue from young (3 mo old) male WT mice was minced into 1- to 2-mm pieces under a sterile laminar flow hood and dissociated into single-cell suspension using collagenase II (600 U/ml; Worthington) and DNase I (60 U/ml; AppliChem) in Hanks' Balanced Salt Solution (Gibco). After 1 h incubation at 37°C, with mechanical dissociation applied every 15 min, and the cell suspension was centrifuged and resuspended in DMEM-F-12 (1:1) media (Gibco) supplemented with 10% FBS and 1% antibiotic solution. The cells were transferred to cell culture flasks (25 cm2) and incubated under standard cell culture conditions (37 °C; 5% CO2). The cells were maintained by passaging using 75-cm2 cell culture flasks when confluence was >80%, and cells from passage 3 were used for the experiments. When cells reached 80% confluence, they were incubated in serum-free media overnight before the initiation of experiments to induce quiescence. Cells were pretreated with anti-a disintegrin and metalloproteinase with thrombospondin-like motifs 1 (ADAMTS1) antibody (15 μg/ml; R & D Systems) or control IgG (15 μg/ml; R & D Systems) for 1 h before incubation with SPARC (20 μg/ml) for 6 h. The media were collected and cells washed with PBS.
Protein extraction and immunoblotting.
Cells were lysed in ice-cold lysis buffer [50 mmol/l Tris, 150 mmol/l NaCl, 1% Triton-X, 0.1% sodium dodecyl sulfate, and 1× Complete Protease Inhibitor Cocktail (Roche), pH 7.5]. Total protein concentrations were determined by the bicinchoninic acid method (Thermo Scientific). Media were concentrated using speed vacuum centrifuges. Immunoblotting was performed according to a standard protocol. Proteins were mixed with sample buffer containing β-mercaptoethanol and heated at 100°C for 5 min. Total protein was run on 4–12% criterion Bis-Tris gels (Bio-Rad Laboratories), electrotransferred to nitrocellulose membranes (Bio-Rad Laboratories), and then stained with Pierce Reversible Protein Stain Kit for Nitrocellulose Membranes (Thermo Scientific). The membranes were incubated with the following primary antibodies overnight and then incubated for 1 h with the appropriate horseradish peroxidase-conjugated secondary antibodies: anti-collagen I antibody (CL50141AP-1; Cedarlane), anti-ADAMTS1 antibody (AF5867; R & D Systemx), anti-transforming growth factor-β1-induced protein (Tgfbi) antibody (ab66957; Abcam), anti-phospho(Ser467)-Smad2 antibody (ab53100; Abcam), and anti-total Smad2 antibody (ab63576; Abcam). The detection of chemiluminescence was carried out with enhanced chemiluminescence, and the bands were quantified by densitometry using ImageJ. For the cellular protein, the amount of each product was normalized to the densitometry of the total protein stain for the entire lane.
Statistics.
Data are expressed as means ± SE. Genotype comparisons of WT and Null mice across ages were performed using two-way ANOVA, followed by the Bonferroni multiple comparisons correction test. One-way ANOVA followed by the Student-Newman-Keuls post hoc test was used for comparisons among young, middle-aged, and old mice and among four in vitro groups. Pearson's correlation was performed to calculate correlation coefficients. Two group comparisons of young WT and old WT were performed using unpaired t-test. P < 0.05 was considered significant.
RESULTS
SPARC deletion suppressed the age-dependent increase in LV cell adhesion molecules.
Because cardiac ECM and associated cell matrix adhesion molecules not only provide structural support but also play important roles in cardiac remodeling, inflammation, and function (29), we measured LV expression of ECM and cell adhesion molecules by gene array. Figures 1A and 2A include adhesion molecules (Fig. 1A) and ECM (Fig. 2A) that changed with age in WT LV. Of the genes listed in Figs. 1A and 2A, SPARC deletion affected the age-dependent changes of four adhesion molecules [cadherin-4, integrin-α2, integrin-αE, and vascular cell adhesion molecule 1 (VCAM1); Fig. 1B] and seven ECM genes (collagens III, IV, and and V as well as ADAMTS1, fibronection1, Tgfbi, and versican; Fig. 2C). Table 2 shows the mRNA levels of adhesion molecules and ECM that displayed similar age-dependent changes in both WT and SPARC-null LV.
Fig. 1.

Age-dependent alterations in cardiac expression of adhesion molecules were suppressed in SPARC-null mice. A: gene array data of adhesion molecules. Table shows adhesion molecules, which changed with age in the left ventricle (LV) of wild type (WT) mice, including the ratio (old WT/young WT) and P value (old WT vs. young WT) of each gene expression. B: among adhesion molecules listed in A, SPARC deletion affected the expression of cadherin-4, integrin-α2, integrin-αE, and vascular cell adhesion molecule 1 (VCAM1). Age-dependent increases in cardiac expression of cadherin-4, integrin αE, and VCAM1 were delayed in secreted protein acidic and rich in cysteine (SPARC)-null (Null) mice compared with WT. Integrin-α2 increased with age only in WT mice. Old Null mice had higher expression of cadherin-4 and integrin-αE than age-matched WT mice. Each gene expression was normalized to hypoxanthine-guanine phosphoribosyltransferase 1 (Hprt1) and shown as 2−ΔCT units. Data are expressed as means ± SE (n = 5–6/group). #P < 0.05 among ages in each genotype; *P < 0.05 vs. age-matched WT.
Fig. 2.

SPARC deletion delayed age-dependent increase in LV expression of a disintegrin and metalloproteinase with thrombospondin-like motifs 1 (ADAMTS1). A: gene array data of extracellular matrix (ECM). Table shows ECM, which changed with age in the LV of WT, including the ratio (old WT/young WT) and P value (old WT vs. young WT) of each gene expression. B: collagen I mRNA levels decreased with age in both WT and Null mice, whereas collagen III and IV showed age-related decrease in WT but not Null mice. The mRNA levels of collagen V were lower in middle-aged and old WT compared with young WT, whereas only the old group showed lower collagen V mRNA expression vs. the young group in SPARC-null mice. C: among ECM listed in A, SPARC deletion abolished cardiac expression of SPARC mRNA and affected the expression of ADAMTS1, fibronectin 1, Tgfbi, and versican. Age-dependent increases in cardiac expression of ADAMTS1 and transforming growth factor β-induced protein (Tgfbi) were delayed in Null mice compared with WT. Middle-aged Null mice had lower expression of ADAMTS1 than age-matched WT mice. Age-dependent decrease in fibronectin 1 and versican was blunted in SPARC-null mice. Each gene expression was normalized to Hprt1 and shown as 2−ΔCT units. Data are expressed as means ± SE (n = 5–6/group). MMP, matrix metalloproteinase; Ctgf, connective tissue growth factor; Ecm1, extracellular matrix 1; Tgfbi, transforming growth factor β-induced protein; Thbs3, thrombospondin-3. #P < 0.05 among ages in each genotype; *P < 0.05 vs. age-matched WT.
Table 2.
The mRNA levels of adhesion molecules and ECM showing age-dependent changes similarly in both WT and SPARC-null mice
| WT |
Null |
|||||
|---|---|---|---|---|---|---|
| Young | Middle-aged | Old | Young | Middle-aged | Old | |
| Adhesion molecules | ||||||
| Catenin-β1 | 1.861 ± 0.127 | 1.636 ± 0.038 | 1.508 ± 0.093# | 2.323 ± 0.296 | 1.710 ± 0.077 | 1.591 ± 0.067# |
| Integrin-α3 | 0.053 ± 0.003 | 0.045 ± 0.002# | 0.039 ± 0.001# | 0.068 ± 0.008 | 0.046 ± 0.003# | 0.041 ± 0.003 |
| Integrin-β1 | 2.429 ± 0.081 | 2.373 ± 0.076 | 1.981 ± 0.142#† | 2.812 ± 0.150 | 2.473 ± 0.075 | 2.346 ± 0.116# |
| ECM | ||||||
| Ctgf | 1.077 ± 0.072 | 1.131 ± 0.108 | 1.803 ± 0.167#† | 0.813 ± 0.129 | 0.901 ± 0.103 | 1.915 ± 0.409#† |
| Ecm1 | 0.558 ± 0.046 | 0.648 ± 0.047 | 0.802 ± 0.068# | 0.623 ± 0.083 | 0.583 ± 0.019 | 1.016 ± 0.107 |
| Laminin-α2 | 0.165 ± 0.012 | 0.158 ± 0.017 | 0.233 ± 0.005#† | 0.143 ± 0.003 | 0.134 ± 0.006 | 0.216 ± 0.020#† |
| Laminin-γ1 | 0.741 ± 0.056 | 0.605 ± 0.017# | 0.570 ± 0.050# | 0.813 ± 0.058 | 0.600 ± 0.046# | 0.609 ± 0.040# |
| MMP-2 | 0.413 ± 0.034 | 0.497 ± 0.018 | 0.589 ± 0.049# | 0.405 ± 0.018 | 0.478 ± 0.038 | 0.7376 ± 0.069# |
| MMP-3 | 0.043 ± 0.006 | 0.061 ± 0.009 | 0.082 ± 0.008# | 0.039 ± 0.004 | 0.050 ± 0.010 | 0.108 ± 0.024# |
| MMP-9 | 0.006 ± 0.001 | 0.008 ± 0.001 | 0.012 ± 0.001# | 0.005 ± 0.001 | 0.006 ± 0.001 | 0.016 ± 0.006# |
| MMP-13 | 0.009 ± 0.001 | 0.005 ± 0.001# | 0.004 ± 0.001# | 0.010 ± 0.001 | 0.004 ± 0.0003# | 0.003 ± 0.001# |
| MMP-15 | 0.712 ± 0.055 | 0.524 ± 0.020# | 0.476 ± 0.025# | 0.827 ± 0.070 | 0.560 ± 0.053# | 0.415 ± 0.034# |
| Thbs3 | 0.035 ± 0.003 | 0.038 ± 0.002 | 0.047 ± 0.004# | 0.036 ± 0.003 | 0.041 ± 0.004 | 0.056 ± 0.005#† |
Data are expressed as means ± SE (n = 5–6/group).
ECM, extracellular matrix; WT; wild type; Ecm1, extracellular matrix 1; MMP, matrix metalloproteinase. Each gene expression was normalized to hypoxanthine-guanine phosphoribosyltransferase 1 and shown as 2−ΔCT units.
P < 0.05 vs. young mice in each genotype;
P < 0.05 vs. middle-aged mice in each genotype.
Figure 1A shows the LV cell adhesion molecule genes that were increased or decreased in an age-dependent manner. Four genes (cadherin-4, integrin-α2, integrin-αE, and VCAM1) increased and three genes (catenin-β1, integrin-α3, and integrin-β1) decreased with age in WT mice (Fig. 1A). As shown in Fig. 1B, the levels of cadherin-4, integrin-αE, and VCAM1 were significantly greater in middle-aged and old WT compared with young WT hearts (P < 0.05 for all), whereas in Null mice, only old hearts showed a greater expression of these molecules vs. young and middle-aged tissue. Levels of integrin-α2 were enhanced with age in WT mice, whereas hearts from mice with deleted expression of SPARC abolished this effect. These results suggest that SPARC expression delays age-dependent cell adhesion molecule alterations. Old Null mice had higher expression of cadherin-4 and integrin-αE than age-matched WT mice, whereas age-associated increases in these genes were delayed in Null mice (Fig. 1B).
SPARC deletion suppressed the age-dependent decreases in fibronectin and versican and increases in ADAMTS1 and Tgfbi.
ECM molecules that demonstrated age-dependent increased or decreased expression are summarized in Fig. 2A. Levels of mRNA encoding collagen I, III, IV, and V decreased with age in WT mice; this is contrary to what is observed at the protein level, where collagen accumulation increases in the aged LV (4). Consistent with previous reports, PSR staining revealed that collagen deposition increased in an age-dependent manner (P < 0.05; Fig. 3A). Cardiac expression of SPARC protein, determined previously by immunohistochemistry (44), and PSR-stained collagen content correlated positively with age. In addition, collagen content correlated significantly with SPARC levels, indicating that age-associated cardiac fibrosis was SPARC dependent (Fig. 3B). As shown in Fig. 2B, the mRNA levels of collagen I and III, the primary fibrillar collagens present in the myocardial ECM, decreased with age in WT heart. Hence, age-dependent cardiac fibrosis did not appear to derive from an increase in transcription. Expression of fibrillar collagen V mRNA was lower in middle-aged and old WT compared with young WT (Fig. 2B). Collagen V has been proposed to serve a seed function in the formation of nascent collagen I-containing fibers (47), suggesting that nascent fibril assembly is reduced with age. These results also suggest that collagen accumulation feeds back to turn off further collagen production. That age-related decreases in collagen III and V mRNA were attenuated in SPARC-null mice support the hypothesis that transcriptional regulation of collagens is not the primary mechanism of SPARC-dependent collagen accumulation in cardiac aging. The mRNA expression of collagen IV showed age-dependent decreases in WT but not SPARC-null mice, indicating that SPARC affects the expression of nonfibrillar collagens as well as fibrillar collagens (Fig. 2B).
Fig. 3.

SPARC deletion inhibited collagen deposition in the aged heart. A: representative images of picosirius red (PSR)-stained sections from the LVs of young, middle-aged, and old WT and Null mice and %PSR-positive area per total area showing age-dependent increase in collagens in WT mice. Scales, 50 μm. #P < 0.05 among ages in each genotype; *P < 0.05 vs. age-matched WT. B: cardiac expression of SPARC protein (%SPARC-positive area/total area) and collagen content increased with age. Pearson correlation analysis revealed significant correlation between SPARC expression and collagen deposition in the aging myocardium.
The mRNA levels of the ECM glycoproteins fibronectin 1 and versican were lower in old WT vs. young and middle-aged WT mice (P < 0.05), whereas there were no age-related changes in Null mice (Fig. 2C). The expression of ADAMTS1 and Tgfbi was significantly greater in middle-aged and old WT compared with young WT mice, whereas in the absence of SPARC expression, only old mice showed a greater expression of these molecules vs. young mice (P < 0.05; Fig. 2C). In addition, middle-aged Null mice had lower expression of ADAMTS1 than age-matched WT mice (P < 0.05; Fig. 2C). These results suggest that SPARC plays a role in decreasing fibronectin 1 and versican expression while increasing the expression of ADAMTS1 and Tgfbi in the aged heart.
SPARC induced collagen secretion through ADAMTS1 production in cardiac fibroblasts.
ADAMTS1, a metalloprotease, regulates ECM turnover (26). SPARC deletion delayed the age-dependent increase in the expression of ADAMTS1 (Fig. 2C), suggesting that SPARC might positively regulate the expression of ADAMTS1 in the LV. To investigate this hypothesis, we treated cardiac fibroblasts with recombinant SPARC protein. In vitro SPARC treatment increased the release of both latent (110 kDa) and active (85 kDa) forms of ADAMTS1 in conditioned media, indicating that SPARC promoted ADAMTS1 production and activation (Fig. 4A). ADAMTS1 has been linked to liver fibrosis, implicating a role for ADAMTS1 in collagen deposition (3).
Fig. 4.

SPARC treatment increased collagen I production by ADAMTS1 expression. Cardiac fibroblasts from young WT mice were stimulated with SPARC (20 μg/ml, 6 h). Media were used to examine the protein expression of ADAMTS1 and collagen I by immunoblotting. A: SPARC increased both latent (110 kDa) and active (87 kDa) forms of ADAMTS1 protein expression in media. B: SPARC increased collagen I protein expression in conditioned media, and function-blocking ADAMTS1 antibodies suppressed SPARC-induced increases in collagen protein expression. The bands were quantified by densitometry. Data are expressed as means ± SE (n = 4–5/group). #P < 0.05 vs. control; †P < 0.05 vs. SPARC-treated group.
To investigate the role of ADAMTS1 in SPARC-induced collagen deposition in the heart, we incubated cardiac fibroblasts with SPARC in the presence or absence of anti-ADAMTS1 antibody. Levels of collagen I increased in the conditioned media of the SPARC-treated group compared with the nontreated group, whereas coincubation with the anti-ADAMTS1 antibody blunted SPARC-induced collagen I secretion, indicating ADAMTS1-mediated collagen deposition by SPARC (Fig. 4B). Because in vivo gene arrays revealed that SPARC deletion suppressed age-related increases in Tgfbi (Fig. 2), we quantified Tgfbi expression in vitro and found that it was also increased in SPARC-treated cells. Interestingly, blocking ADAMTS1 activity had no effect on Tgfbi expression in cardiac fibroblasts (Fig. 5A). Transforming growth factor-β1 (Tgfb1) signaling is initiated by the phosphorylation of Smad2 in one downstream pathway (17). The ratio of phosphorylated Smad2 to total Smad2 was increased in the SPARC-treated cells (Fig. 5B), which would explain the observed increase in Tgfbi expression.
Fig. 5.

SPARC increased Tgfbi and phosphorylated Smad2 expression in cardiac fibroblasts. Cardiac fibroblasts from young WT mice were stimulated with SPARC (20 μg/ml, 6 h), and cellular protein expression of Tgfbi and phosphorylated Smad2 was examined by immunoblotting. A: SPARC increased Tgfbi protein expression. Anti-ADAMTS1 had no effect on the protein expression of Tgfbi. B: SPARC increased phosphorylation of Smad2. Anti-ADAMTS1 blocking antibody had no effect on Smad2 phosphorylation. The bands were quantified by densitometry, and the amount of each product was normalized to the densitometry of the total protein stain in its respective lane. Data are expressed as means ± SE (n = 4–5/group). #P < 0.05 vs. control.
DISCUSSION
Our previous studies suggest that SPARC facilitates cardiac fibrosis and inflammation in the aging heart (4, 44). In the present study, we examined the role of SPARC in age-dependent remodeling of cardiac ECM and expression of adhesion molecules and the mechanisms of SPARC-induced collagen deposition. The present study demonstrated that 1) age-related increases in the expression of two adhesion molecules associated with inflammatory responses in the LV, integrin-αE and VCAM1, were suppressed in SPARC-null mice; 2) age-dependent decreases in fibronectin and versican were blunted by SPARC deletion; 3) SPARC deletion suppressed the age-dependent increase in collagen deposition; 4) the age-associated increase in myocardial expression of ADAMTS1 was delayed in SPARC-null mice; and 5) anti-ADAMTS1 antibody suppressed SPARC-induced collagen secretion in vitro. Combined, our results provide the first evidence linking SPARC and ADAMTS1 production in the myocardium with implications for ECM remodeling in cardiac aging.
Our recent study showed that SPARC promoted macrophage infiltration into LV tissue in aged mice (44). We have also shown that SPARC promoted macrophage polarization into proinflammatory M1 macrophages that was predicted to increase the release of proinflammatory cytokines in the aged heart. Here, we investigated whether SPARC-dependent changes in ECM and adhesion molecules play a role in age-related cardiac inflammation. VCAM1 is a well-known adhesion molecule found in inflammatory lesions (33), and integrin-αE is also reported to be involved in the inflammatory process (15). Our results show that age-dependent increases in VCAM1 and integrin-αE levels were delayed in SPARC-null mice, suggesting that these molecules might contribute to age-related SPARC-induced cardiac inflammation. ADAMTS1, an ECM protease, cleaves proteoglycans such as versican (40). ADAMTS1 expression is upregulated in atherosclerotic leasions in both vascular smooth muscle cells and macrophage/foam cells in human aorta and coronary artery. Overexpression of ADAMTS1 causes increased vascular remodeling in apoE-deficient mice, suggesting an important association between ADAMTS1 and inflammation (23). The present results showing age-dependent increases in myocardial ADAMTS1, which was delayed by the absence of SPARC, suggests that ADAMTS1 might be linked to SPARC-induced cardiac inflammation. Versican, a substrate of ADAMTS1, can bind to growth factors, cytokines, and lipoproteins and interact with a number of matrix proteins, adhesion molecules, and membrane proteins, which suggests wide-ranging potential roles of versican (16, 18, 25, 35). Interestingly, in human atherosclerotic lesions, there is reported to be an increase in macrophages associated with reduced levels of versican compared with control tissue, suggesting a negative correlation between inflammation and versican levels (42). We found an age-dependent decrease in versican in WT LV and no age-dependent changes in expression in SPARC-null LV. Perhaps age-dependent increases in ADAMTS1 expression lead to increased versican degradation, which may mediate SPARC-enhanced cardiac inflammation.
In the present study, we found four integrin genes that showed age-associated changes: integrin-α2 and -αE increased with age, and integrin-α3 and-β1 decreased. Whereas integrin-α3 and -β1 are expressed in both cardiomyocytes and cardiac fibroblasts, integrin-α2 is expressed in cardiac fibroblasts but not in cardiomyocytes. Integrin-αE is expressed in several subsets of inflammatory cells (14, 20, 39). SPARC deletion delayed age-associated increases in integrins-α2 and -αE, indicating that SPARC may exert a greater influence on fibroblasts and inflammatory cells than cardiomyocytes in the aging myocardium.
Cardiac ECM fibrillar collagens, which support myocardial structure and provide mechanical stability, accumulate with age, and excessive accumulation leads to myocardial stiffness and diastolic dysfunction (9, 22, 43, 46). SPARC-null mice show less age-related increases in collagen deposition and cardiac stiffness vs. WT mice, suggesting that SPARC is an important player in age-associated cardiac collagen deposition (4). The present study aimed to investigate the mechanisms by which SPARC affects cardiac fibrosis. Integrin-α2 is reported to contribute to fibrosis in renal fibroblasts (15, 27). In myocardium, SPARC deletion abolished age-related increases in integrin-α2, suggesting a potentially important role of integrin-α2 in SPARC-induced cardiac fibrosis. In addition to integrin-α2, there were five other genes that showed age-associated increases attenuated by SPARC deletion (ADAMTS1, Tgfbi, integrin-αE, cadherin-4, VCAM1). Of note is that versican, a substrate of ADAMTS1, decreased with age in WT but not in SPARC-null, indicating ADAMTS1 activation and concomitant degradation of versican. This finding suggested a potentially important role for ADAMTS1 in the aging myocardium. ADAMTS1 has recently been correlated with collagen I accumulation in human fibrotic and cirrhotic liver, suggesting a fibrogenic role for ADAMTS1 (3). For these reasons, we hypothesized that SPARC may promote collagen deposition by increasing ADAMTS1. To investigate the interaction between SPARC and ADAMTS1, we treated cardiac fibroblasts with SPARC in vitro. SPARC treatment increased ADAMTS1 and collagen I expression in cardiac fibroblasts. Interestingly, pretreatment with blocking anti-ADAMTS1 antibodies suppressed SPARC-induced increases in collagen I in conditioned media. Our data provide evidence that induction of age-dependent collagen deposition by SPARC might depend upon ADAMTS1 production (Fig. 6). SPARC may facilitate cardiac aging by increasing collagen deposition via ADAMTS1 production (Fig. 6). Figure 6 summarizes the role of SPARC in collagen deposition in aging myocardium.
Fig. 6.
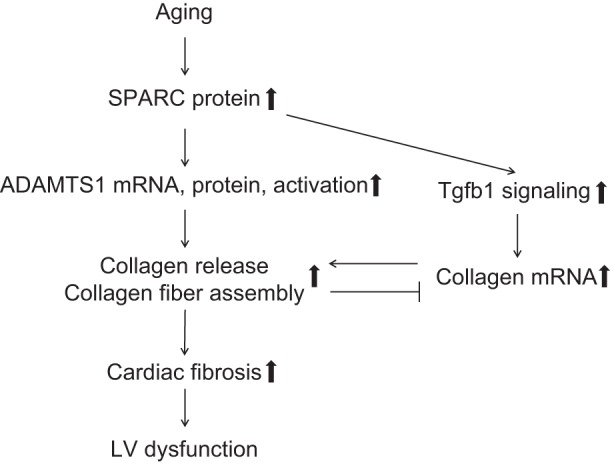
SPARC promotes collagen deposition by increasing ADAMTS1 in aging myocardium. The diagram summarizes the role of SPARC in collagen deposition in aging myocardium. Myocardial expression of SPARC protein increases with advancing age. SPARC stimulates transforming growth factor-β1 (Tgfb1) signaling and the production and activation of ADAMTS1. ADAMTS1 promotes collagen fiber assembly/stabilization and collagen release, which leads to cardiac fibrosis and consequent dysfunction. Collagen deposition may cause a negative feedback to collagen gene transcription in the aged myocardium.
In the present study, we evaluated fibroblasts isolated from young WT mice. In the heart, a major source of SPARC is fibroblasts, and cardiac expression of SPARC increases with age, suggesting that basal levels of SPARC in old WT cardiac fibroblasts are elevated. Increases in endogenous SPARC are predicted to induce ADAMTS1 expression and activation in the steady state, and additional treatment of exogenous SPARC may enhance collagen production in response to SPARC treatment. Since SPARC expression in normal adult animals is limited largely to remodeling tissue, basal expression of SPARC is anticipated to be low (8). Hence, SPARC treatment will likely cause similar effects in cardiac fibroblasts isolated from young SPARC-null mice compared with those from young WT mice.
SPARC stimulates Tgfb-mediated Smad2 activation in epithelial cells, and the aged myocardium contains elevated Tgfbi expression and Smad2 phosphorylation (10, 41). The present study demonstrated that age-related increases in Tgfbi expression were delayed in SPARC-null mice, indicating that activation of Tgfb signaling might be suppressed by SPARC deletion in the aging heart. Since the phosphorylation of Smad2 to phospho-Smad2 is mediated by Tgfb signaling (17), SPARC deletion may suppress Smad2 phosphorylation in the aging heart. Our in vitro study showed that SPARC-stimulated Tgfb signaling was independent of ADAMTS1 (Fig. 6). Future investigation into the up- and downstream of ADAMTS1 production and activation will provide an important insight for potential treatments of age-related cardiac fibrosis.
The present study presented the fact that collagen gene expression decreased with age but did not completely fill the gaps with regard to SPARC roles in posttranscriptional events. SPARC promotes the formation of cross-linked mature collagen fibrils by binding to newly secreted procollagens in the extracellular space and thereby facilitates collagen deposition into the ECM (5, 6, 37, 38). Increases in soluble (non-cross-linked) collagen, which accounts for a minor fraction of the total collagen, are detected in old SPARC null vs. WT hearts (4). Increases in soluble collagen in the absence of SPARC suggested a deficit in collagen incorporation into insoluble ECM. The accumulation of collagen in aging hearts (Fig. 3) might result in a negative feedback to decreases in mRNA expression, as was observed in WT hearts (Fig. 6).
Increased cardiac activity of matrix metalloproteinase (MMP)-2 and MMP-9 is associated with cardiac remodeling and heart failure (11, 28, 32, 45). It has been reported that SPARC increases the expression and activity of MMP-2 and MMP-9 (1, 30). Age-related increases in mRNA levels of MMP-2 and MMP-9 might be downstream of increases in myocardial SPARC. Previous studies have shown that MMP-9 deletion reduces Tgfb activation and results in suppressed cardiac fibrosis in the aged heart (10), raising the possibility that SPARC-induced cardiac fibrosis might be mediated at least in part by increasing MMP-9 activity. Since SPARC deletion had no effects on MMP-9 mRNA levels, investigation into MMP-9 activity might provide further insights into the mechanisms of SPARC-dependent cardiac fibrosis. MMP-13, also called collagenase-3, degrades intact fibrillar collagens such as collagen I and II, and is the major collagenase expressed in mice (12, 19). Age-dependent decreases in MMP-13 might result in insufficient collagen degradation and play a role in collagen deposition in the aging heart.
In conclusion, we demonstrated that SPARC augmented age-dependent collagen deposition by enhancing ADAMTS1 levels in the heart. Increased ADAMTS1 stimulates versican cleavage, with subsequent effects on cardiac inflammation. Suppressing the effects of SPARC may provide novel therapeutic strategies for the prevention and treatment of cardiac dysfunction in elderly patients.
GRANTS
We acknowledge support from American Heart Association for Grant no. 14SDG18860050, from the Biomedical Laboratory Research, Development Service of the Veterans Affairs Office of Research and Development Awards 1I01BX001385 and 5I01BX000505, and from the National Institutes of Health for Grants HL-075360, HL-051971, and GM-104357.
DISCLOSURES
The authors declare that there are no conflicts of interests, financial or otherwise.
AUTHOR CONTRIBUTIONS
H.T., L.E.d.C.B., M.L.L., and A.D.B. conception and design of research; H.T. and C.F.B. performed experiments; H.T. and L.E.d.C.B. analyzed data; H.T., L.E.d.C.B., M.R.Z., M.L.L., and A.D.B. interpreted results of experiments; H.T. prepared figures; H.T., L.E.d.C.B., M.R.Z., M.L.L., and A.D.B. drafted manuscript; H.T., L.E.d.C.B., C.F.B., M.R.Z., M.L.L., and A.D.B. edited and revised manuscript; H.T., L.E.d.C.B., C.F.B., M.R.Z., M.L.L., and A.D.B. approved final version of manuscript.
REFERENCES
- 1.Arnold S, Mira E, Muneer S, Korpanty G, Beck AW, Holloway SE, Manes S, Brekken RA. Forced expression of MMP9 rescues the loss of angiogenesis and abrogates metastasis of pancreatic tumors triggered by the absence of host SPARC. Exp Biol Med (Maywood) 233: 860–873, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bornstein P, Sage EH. Matricellular proteins: extracellular modulators of cell function. Curr Opin Cell Biol 14: 608–616, 2002. [DOI] [PubMed] [Google Scholar]
- 3.Bourd-Boittin K, Bonnier D, Leyme A, Mari B, Tuffery P, Samson M, Ezan F, Baffet G, Theret N. Protease profiling of liver fibrosis reveals the ADAM metallopeptidase with thrombospondin type 1 motif, 1 as a central activator of transforming growth factor beta. Hepatology 54: 2173–2184, 2011. [DOI] [PubMed] [Google Scholar]
- 4.Bradshaw AD, Baicu CF, Rentz TJ, Van Laer AO, Bonnema DD, Zile MR. Age-dependent alterations in fibrillar collagen content and myocardial diastolic function: role of SPARC in post-synthetic procollagen processing. Am J Physiol Heart Circ Physiol 298: H614–H622, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bradshaw AD, Puolakkainen P, Dasgupta J, Davidson JM, Wight TN, Helene Sage E. SPARC-null mice display abnormalities in the dermis characterized by decreased collagen fibril diameter and reduced tensile strength. J Invest Dermatol 120: 949–955, 2003. [DOI] [PubMed] [Google Scholar]
- 6.Bradshaw AD, Reed MJ, Sage EH. SPARC-null mice exhibit accelerated cutaneous wound closure. J Histochem Cytochem 50: 1–10, 2002. [DOI] [PubMed] [Google Scholar]
- 7.Bradshaw AD, Sage EH. SPARC, a matricellular protein that functions in cellular differentiation and tissue response to injury. J Clin Invest 107: 1049–1054, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brekken RA, Sage EH. SPARC, a matricellular protein: at the crossroads of cell-matrix. Matrix Biol 19: 569–580, 2000. [DOI] [PubMed] [Google Scholar]
- 9.Burgess ML, McCrea JC, Hedrick HL. Age-associated changes in cardiac matrix and integrins. Mech Ageing Dev 122: 1739–1756, 2001. [DOI] [PubMed] [Google Scholar]
- 10.Chiao YA, Ramirez TA, Zamilpa R, Okoronkwo SM, Dai Q, Zhang J, Jin YF, Lindsey ML. Matrix metalloproteinase-9 deletion attenuates myocardial fibrosis and diastolic dysfunction in ageing mice. Cardiovasc Res 96: 444–455, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cochain C, Auvynet C, Poupel L, Vilar J, Dumeau E, Richart A, Recalde A, Zouggari Y, Yin KY, Bruneval P, Renault G, Marchiol C, Bonnin P, Levy B, Bonecchi R, Locati M, Combadiere C, Silvestre JS. The chemokine decoy receptor D6 prevents excessive inflammation and adverse ventricular remodeling after myocardial infarction. Arterioscler Thromb Vasc Biol 32: 2206–2213, 2012. [DOI] [PubMed] [Google Scholar]
- 12.Creemers EE, Cleutjens JP, Smits JF, Daemen MJ. Matrix metalloproteinase inhibition after myocardial infarction: a new approach to prevent heart failure? Circ Res 89: 201–210, 2001. [DOI] [PubMed] [Google Scholar]
- 13.de Castro Bras LE, Toba H, Baicu CF, Zile MR, Weintraub ST, Lindsey ML, Bradshaw AD. Age and SPARC change the extracellular matrix composition of the left ventricle. Biomed Res Int 2014: 810562, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Goldsmith EC, Bradshaw AD, Zile MR, Spinale FG. Myocardial fibroblast-matrix interactions and potential therapeutic targets. J Mol Cell Cardiol 70: 92–99, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Goodman SL, Picard M. Integrins as therapeutic targets. Trends Pharmacol Sci 33: 405–412, 2012. [DOI] [PubMed] [Google Scholar]
- 16.Hakala JK, Oorni K, Pentikainen MO, Hurt-Camejo E, Kovanen PT. Lipolysis of LDL by human secretory phospholipase A(2) induces particle fusion and enhances the retention of LDL to human aortic proteoglycans. Arterioscler Thromb Vasc Biol 21: 1053–1058, 2001. [DOI] [PubMed] [Google Scholar]
- 17.Heldin CH, Miyazono K, ten Dijke P. TGF-beta signalling from cell membrane to nucleus through SMAD proteins. Nature 390: 465–471, 1997. [DOI] [PubMed] [Google Scholar]
- 18.Hurt-Camejo E, Olsson U, Wiklund O, Bondjers G, Camejo G. Cellular consequences of the association of apoB lipoproteins with proteoglycans. Potential contribution to atherogenesis. Arterioscler Thromb Vasc Biol 17: 1011–1017, 1997. [DOI] [PubMed] [Google Scholar]
- 19.Hutchinson KR, Stewart JA Jr, Lucchesi PA. Extracellular matrix remodeling during the progression of volume overload-induced heart failure. J Mol Cell Cardiol 48: 564–569, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Israeli-Rosenberg S, Manso AM, Okada H, Ross RS. Integrins and integrin-associated proteins in the cardiac myocyte. Circ Res 114: 572–586, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jacob MP. Extracellular matrix remodeling and matrix metalloproteinases in the vascular wall during aging and in pathological conditions. Biomed Pharmacother 57: 195–202, 2003. [DOI] [PubMed] [Google Scholar]
- 22.Jalil JE, Doering CW, Janicki JS, Pick R, Shroff SG, Weber KT. Fibrillar collagen and myocardial stiffness in the intact hypertrophied rat left ventricle. Circ Res 64: 1041–1050, 1989. [DOI] [PubMed] [Google Scholar]
- 23.Jonsson-Rylander AC, Nilsson T, Fritsche-Danielson R, Hammarstrom A, Behrendt M, Andersson JO, Lindgren K, Andersson AK, Wallbrandt P, Rosengren B, Brodin P, Thelin A, Westin A, Hurt-Camejo E, Lee-Sogaard CH. Role of ADAMTS-1 in atherosclerosis: remodeling of carotid artery, immunohistochemistry, and proteolysis of versican. Arterioscler Thromb Vasc Biol 25: 180–185, 2005. [DOI] [PubMed] [Google Scholar]
- 24.Kanauchi M, Nishioka M, Dohi K. Secreted protein acidic and rich in cysteine (SPARC) in patients with diabetic nephropathy and tubulointerstitial injury. Diabetologia 43: 1076–1077, 2000. [DOI] [PubMed] [Google Scholar]
- 25.Kawashima H, Hirose M, Hirose J, Nagakubo D, Plaas AH, Miyasaka M. Binding of a large chondroitin sulfate/dermatan sulfate proteoglycan, versican, to L-selectin, P-selectin, and CD44. J Biol Chem 275: 35448–35456, 2000. [DOI] [PubMed] [Google Scholar]
- 26.Kelwick R, Desanlis I, Wheeler GN, Edwards DR. The ADAMTS (A Disintegrin and Metalloproteinase with Thrombospondin motifs) family. Genome Biol 16: 113, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kondo S, Kagami S, Urushihara M, Kitamura A, Shimizu M, Strutz F, Muller GA, Kuroda Y. Transforming growth factor-beta1 stimulates collagen matrix remodeling through increased adhesive and contractive potential by human renal fibroblasts. Biochim Biophys Acta 1693: 91–100, 2004. [DOI] [PubMed] [Google Scholar]
- 28.Lindsey ML, Zamilpa R. Temporal and spatial expression of matrix metalloproteinases and tissue inhibitors of metalloproteinases following myocardial infarction. Cardiovasc Ther 30: 31–41, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Martos R, Baugh J, Ledwidge M, O'Loughlin C, Conlon C, Patle A, Donnelly SC, McDonald K. Diastolic heart failure: evidence of increased myocardial collagen turnover linked to diastolic dysfunction. Circulation 115: 888–895, 2007. [DOI] [PubMed] [Google Scholar]
- 30.McClung HM, Thomas SL, Osenkowski P, Toth M, Menon P, Raz A, Fridman R, Rempel SA. SPARC upregulates MT1-MMP expression, MMP-2 activation, and the secretion and cleavage of galectin-3 in U87MG glioma cells. Neurosci Lett 419: 172–177, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.McCurdy SM, Dai Q, Zhang J, Zamilpa R, Ramirez TA, Dayah T, Nguyen N, Jin YF, Bradshaw AD, Lindsey ML. SPARC mediates early extracellular matrix remodeling following myocardial infarction. Am J Physiol Heart Circ Physiol 301: H497–H505, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Moshal KS, Rodriguez WE, Sen U, Tyagi SC. Targeted deletion of MMP-9 attenuates myocardial contractile dysfunction in heart failure. Physiol Res 57: 379–384, 2008. [DOI] [PubMed] [Google Scholar]
- 33.Nakashima Y, Raines EW, Plump AS, Breslow JL, Ross R. Upregulation of VCAM-1 and ICAM-1 at atherosclerosis-prone sites on the endothelium in the ApoE-deficient mouse. Arterioscler Thromb Vasc Biol 18: 842–851, 1998. [DOI] [PubMed] [Google Scholar]
- 34.Norose K, Clark JI, Syed NA, Basu A, Heber-Katz E, Sage EH, Howe CC. SPARC deficiency leads to early-onset cataractogenesis. Invest Ophthalmol Vis Sci 39: 2674–2680, 1998. [PubMed] [Google Scholar]
- 35.Olin AI, Morgelin M, Sasaki T, Timpl R, Heinegard D, Aspberg A. The proteoglycans aggrecan and Versican form networks with fibulin-2 through their lectin domain binding. J Biol Chem 276: 1253–1261, 2001. [DOI] [PubMed] [Google Scholar]
- 36.Pichler RH, Hugo C, Shankland SJ, Reed MJ, Bassuk JA, Andoh TF, Lombardi DM, Schwartz SM, Bennett WM, Alpers CE, Sage EH, Johnson RJ, Couser WG. SPARC is expressed in renal interstitial fibrosis and in renal vascular injury. Kidney Int 50: 1978–1989, 1996. [DOI] [PubMed] [Google Scholar]
- 37.Puolakkainen P, Bradshaw AD, Kyriakides TR, Reed M, Brekken R, Wight T, Bornstein P, Ratner B, Sage EH. Compromised production of extracellular matrix in mice lacking secreted protein, acidic and rich in cysteine (SPARC) leads to a reduced foreign body reaction to implanted biomaterials. Am J Pathol 162: 627–635, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rentz TJ, Poobalarahi F, Bornstein P, Sage EH, Bradshaw AD. SPARC regulates processing of procollagen I and collagen fibrillogenesis in dermal fibroblasts. J Biol Chem 282: 22062–22071, 2007. [DOI] [PubMed] [Google Scholar]
- 39.Ross RS, Borg TK. Integrins and the myocardium. Circ Res 88: 1112–1119, 2001. [DOI] [PubMed] [Google Scholar]
- 40.Sandy JD, Westling J, Kenagy RD, Iruela-Arispe ML, Verscharen C, Rodriguez-Mazaneque JC, Zimmermann DR, Lemire JM, Fischer JW, Wight TN, Clowes AW. Versican V1 proteolysis in human aorta in vivo occurs at the Glu441-Ala442 bond, a site that is cleaved by recombinant ADAMTS-1 and ADAMTS-4. J Biol Chem 276: 13372–13378, 2001. [DOI] [PubMed] [Google Scholar]
- 41.Schiemann BJ, Neil JR, Schiemann WP. SPARC inhibits epithelial cell proliferation in part through stimulation of the transforming growth factor-beta-signaling system. Mol Biol Cell 14: 3977–3988, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Theocharis AD, Tsolakis I, Hjerpe A, Karamanos NK. Human abdominal aortic aneurysm is characterized by decreased versican concentration and specific downregulation of versican isoform V(0). Atherosclerosis 154: 367–376, 2001. [DOI] [PubMed] [Google Scholar]
- 43.Thomas DP, McCormick RJ, Zimmerman SD, Vadlamudi RK, Gosselin LE. Aging- and training-induced alterations in collagen characteristics of rat left ventricle and papillary muscle. Am J Physiol Heart Circ Physiol 263: H778–H783, 1992. [DOI] [PubMed] [Google Scholar]
- 44.Toba H, de Castro Bras LE, Baicu CF, Zile MR, Lindsey ML, Bradshaw AD. Secreted protein acidic and rich in cysteine facilitates age-related cardiac inflammation and macrophage M1 polarization. Am J Physiol Cell Physiol 308: C972–C982, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tyagi SC, Kumar SG, Haas SJ, Reddy HK, Voelker DJ, Hayden MR, Demmy TL, Schmaltz RA, Curtis JJ. Post-transcriptional regulation of extracellular matrix metalloproteinase in human heart end-stage failure secondary to ischemic cardiomyopathy. J Mol Cell Cardiol 28: 1415–1428, 1996. [DOI] [PubMed] [Google Scholar]
- 46.Weber KT, Sun Y, Tyagi SC, Cleutjens JP. Collagen network of the myocardium: function, structural remodeling and regulatory mechanisms. J Mol Cell Cardiol 26: 279–292, 1994. [DOI] [PubMed] [Google Scholar]
- 47.Wenstrup RJ, Florer JB, Brunskill EW, Bell SM, Chervoneva I, Birk DE. Type V collagen controls the initiation of collagen fibril assembly. J Biol Chem 279: 53331–53337, 2004. [DOI] [PubMed] [Google Scholar]
- 48.Wu LL, Cox A, Roe CJ, Dziadek M, Cooper ME, Gilbert RE. Secreted protein acidic and rich in cysteine expression after subtotal nephrectomy and blockade of the renin-angiotensin system. J Am Soc Nephrol 8: 1373–1382, 1997. [DOI] [PubMed] [Google Scholar]