

Abstract
BMAL1 is a core component of the transcription/translation machinery that regulates central and peripheral circadian rhythms that coordinate behavior and metabolism, respectively. Our objective was to determine the impact of BMAL1 in adipose alone or in combination with liver on metabolic phenotypes. Control, adipose-Bmal1 knockout (ABKO), and liver- and adipose-Bmal1 knockout (LABKO) female mice were placed in TSE System metabolic chambers for metabolic phenotyping. A second cohort of male mice was fed a control or diabetogenic diet, and body weight and composition, glucose tolerance, insulin sensitivity, and serum and hepatic lipids were measured. Both female ABKO and LABKO mice exhibited increased food consumption compared with control mice. ABKO mice also exhibited increased overall activity predominantly during the light phase compared with both control and LABKO mice and were protected from increased weight gain. When the male cohort was challenged with a diabetogenic diet, LABKO mice had increased body weight due to increased fat mass compared with control and ABKO mice. However, these mice did not present further impairments in glycemic control, adipose inflammation, or liver injury. LABKO mice had increased hepatic cholesterol and elevated expression of cholesterol synthesis and uptake genes. Our data indicate that deletion of this allele in adipose or in combination with liver alters feeding behavior and locomotor activity. However, obesity is exacerbated only with the combination of liver and adipose deletion.
Keywords: brain and muscle arnt-like protein-1, circadian, obesity, insulin resistance, cholesterol
coordinated signaling between tissues in the periphery and the central nervous system is required to properly regulate metabolic processes and thus maintain energy homeostasis (26). The central clock, housed in the suprachiasmatic nucleus, is entrained by the light-dark cycle, is responsible for controlling rhythmic behavior, and facilitates the synchronization of peripheral clocks (23). Epidemiological studies suggest a strong association between altered circadian behavior and the development of metabolic diseases (12, 28, 29). Global clock disruptions in mice have resulted in a variety of behavioral and metabolic disturbances (22, 27, 31, 35, 37). Furthermore, high-fat diet has been shown to disrupt circadian gene expression in mice (13, 21). Restricting high-fat feeding to certain times of the day was protective against obesity-related phenotypes, suggesting that timing of caloric intake impacts metabolism more so than caloric density or macronutrient composition of the diet (10). Indeed, a number of nuclear receptor subfamilies in metabolic tissues, including white adipose tissue, brown adipose tissue, liver, and skeletal muscle, fluctuate rhythmically at the mRNA level (36). Similarly, factors involved in energy regulation such as glucose, insulin, and leptin oscillate in rhythmic patterns (14, 22, 34).
Coordinated functions of adipose tissue and the liver are essential for the regulation of glucose and lipid metabolism. During the fed state, fatty acids are stored in adipose tissue and glucose in liver as glycogen. In response to fasting, the release of these stores in addition to gluconeogenesis prevents hypoglycemia. Transcriptome profiling studies have identified ∼10% of transcripts in the liver to exhibit circadian expression (1, 19). Deletion of an essential clock gene, Bmal1 (brain and muscle arnt-like protein-1), from the liver has been demonstrated to mute the rhythmic expression of genes involved in glucose transport and gluconeogenesis resulting in hypoglycemia in the fasted state but has no major effects on obesity (15). However, deletion of Bmal1 from adipose tissue has been shown to alter feeding behavior, resulting in obesity in mice (20). Given the interplay of adipose tissue and the liver, we hypothesized that the disruption of circadian clocks in both tissues would potentiate the development of metabolic disease.
METHODS
Animals.
Mice with loxP sites flanking exon 8 (Bmal1f/f) were generously donated by Dr. Karyn Esser of the University of Kentucky, and transgenic mice hemizygous for a Cre recombinase transgene driven by the adipocyte fatty acid-binding protein promoter (aP2-Cre+/0) were purchased from The Jackson Laboratory (Bar Harbor, ME). Female Bmal1f/f were bred to male Bmal1f/f-aP2-Cre+/0 mice using trio breeding to obtain adipose-Bmal1 deletion. Recombinant adenoviral vectors containing either no insert (Ad-Empty) or cDNA for Cre recombinase (Ad-Cre) were amplified in HEK293Q cells, purified on cesium chloride gradients, and dialyzed against PBS. Mice were injected with 4 × 1012 particles/kg via tail vein injection. Bmal1f/f mice without the aP2-Cre allele were injected with Ad-Empty virus (control). Bmal1f/f mice with the aP2-Cre allele were injected with either Ad-empty virus or Ad-Cre virus to generate adipose-Bmal1 knockout (ABKO) and liver- and adipose-Bmal1 knockout (LABKO) mice, respectively.
Mice were maintained on a C57BL/6J background and housed two to four mice per cage in individually ventilated cages in a temperature-controlled room with access to standard rodent chow (Teklad 2918). For breeding, mice were maintained on a 14:10-h light-dark cycle. Following adenoviral injection at 7 wk of age, mice were transferred to a 12:12-h light-dark cycle for the duration of the study.
All animal procedures were conducted in conformity with Public Health Service policies for the humane care and use of laboratory animals and were approved by the University of Kentucky Institutional Animal Care and Use Committee.
Indirect calorimetry.
Measurements for indirect calorimetry were performed on the TSE System (Chesterfield, MO). This system includes 24 chambers plus a reference chamber. O2 (%), CO2 (%), food intake, and locomotor activity were measured directly from each chamber in sequence in 30-min intervals. Reference measurements from room air were also taken in 30-min intervals. The length of each measurement and reference measurement was determined by the measurement interval divided by the number of active chambers. Flow rate to the chambers was 0.45 l/min. Respiratory exchange ratio and energy expenditure were calculated by the system using measured values [%CO2, %O2, and flow (l/min)]. Resting energy expenditure was defined as energy expenditure during the resting period (1000 to 1800), with a total activity count of <150 beam breaks/30 min. Locomotor activity was defined as the number of times a mouse broke two adjacent beams in succession or the same beam twice. Mice were acclimated to chambers for 7 days then transferred to clean chambers immediately prior to the start of measurements. Data from the first and last days of measurements were not included in the analyses. Data across 3 days were averaged for each mouse.
Diet and physiological measurements.
For 24-h blood glucose measurements, mice 13–24 wk old on standard rodent chow in 12:12-h light-dark conditions were provided food only during the dark cycle for a 24-h period. Blood glucose from tail vein pricks was measured every 4 h for 24 h beginning at circadian time 0, defined as the beginning of lights-on.
For the diet intervention study, male mice 8 wk of age were fed either a control diet (CD) comprised of 63% carbohydrate, 16% fat, and 21% protein (Bio-Serv F4031) or a diabetogenic diet (DD) comprised of 26% carbohydrate, 59% fat, and 15% protein (Bio-Serve F3282). Body weight was measured weekly, and body composition was determined by EchoMRI-100 (Echo Medical Systems) during weeks 8, 12, 16, 20, and 24. Adiposity was calculated by dividing fat mass by total body mass.
During weeks 12, 16, 20, and 24, fasting glucose was measured following a 4-h fast beginning at lights-on. Glucose tolerance and insulin sensitivity tests were conducted during weeks 16 and 24. For the glucose tolerance test, 10 μl/g body wt of a 20% glucose solution was injected intraperitoneally following a 4-h fast beginning at lights-on. For the insulin sensitivity test, 10 μl/g body wt of insulin (0.2 IU/ml) was injected intraperitoneally following a 1-h fast beginning at lights-on. Blood glucose levels were measured before and 30, 60, 90, and 120 min after the injection from blood obtained from a tail vein prick. All blood glucose measurements were conducted using a standard glucometer.
At the termination of the study, mice were administered ketamine-xylazine solution for sedation and euthanized by exsanguination. For the 24-h gene expression, mice were euthanized every 4 h for 24 h beginning at circadian time 0. For the diet intervention study, mice were euthanized at circadian time 0. Blood was stored at 4°C for 4 h and centrifuged at high speed for 10 min, and then serum was collected and stored at −80°C. Tissues were dissected and immediately frozen in liquid nitrogen. For histological analysis, pieces of liver and adipose tissue were fixed in 10% formalin for 24 h at room temperature and then transferred to 70% ethyl-alcohol for long-term storage at 4°C.
Histology.
Liver and epididymal fat sections were embedded in paraffin and cut into 5-μm-thick sections. Sections stained with hematoxylin and eosin (H & E) and picrosirius red were imaged at ×10 magnification. Adipocyte size and number were quantified using the NIS Elements software (Nikon Instruments, Tokyo, Japan). The image threshold and object count features were used to quantify three 700 × 700 μm areas to represent each section (2 sections/mouse).
Hepatic lipid extraction and lipid measurements.
Hepatic lipids were extracted from 100 μg of tissue using the Folch method. Extracts were solubilized in 1% Triton in water, as previously described (30). Total cholesterol, triglycerides, and nonesterified fatty acids were measured using enzymatic, colorimetric assays (Wako Chemicals).
Real-time PCR.
RNA was isolated from frozen tissues using RNA STAT-60 (Tel-Test) and the RNeasy Mini Kit (Qiagen). cDNA was synthesized using the iScript cDNA Synthesis Kit (Bio-Rad). Quantitative real-time PCR was conducted using SYBR Green and the 7900HT Fast Real-Time PCR System (Applied Biosystems). For markers of lipolysis, TaqMan (Applied Biosystems) assays were used. Threshold cycle (CT) values of measured transcripts were normalized to that of Gapdh and expressed relative to control mice on the CD using the ΔΔCT method. Primer sequences used for real-time PCR are listed in Table 1.
Table 1.
Primer sequences for quantitative real-time PCR
| Gene | Primer Sequence |
|---|---|
| Bmal1 | |
| Forward | 5′-ACACCAAGGAAGGATCAAGA-3′ |
| Reverse | 5′-GGTACCAAAGAAGCCAATTCA-3′ |
| Dbp | |
| Forward | 5′-ACCGGCCAGCTGTCTCCTGA-3′ |
| Reverse | 5′-CCACAGCAGCGGCGCAAAAA-3′ |
| Pparg | |
| Forward | 5′-CACAATGCCATCAGGTTTGG-3′ |
| Reverse | 5′-GCTGGTCGATATCACTGGAGATC-3′ |
| C/ebpa | |
| Forward | 5′-GACATCAGCGCCTACATCGA-3′ |
| Reverse | 5′-TCGGCTGTGCTGGAAGAG-3′ |
| C/ebpb | |
| Forward | 5′-ATTTCTATGAGAAAAGAGGCGTATGT-3′ |
| Reverse | 5′-AAATGTCTTCACTTTAATGCTCGAA-3′ |
| Zfp423 | |
| Forward | 5′-TGGCCTGGGATTCCTCTGT-3′ |
| Reverse | 5′-CTCTTGACTTGTCACGCTGTT-3′ |
| Hsl | |
| Forward | 5′-GGAAAGAATTGATGGAGCCGGC-3′ |
| Reverse | 5′-TCCATGCTGTGTGAGAACGCT-3′ |
| F4/80 | |
| Forward | 5′-CTTTGGCTATGGGCTTCCAGTC-3′ |
| Reverse | 5′-GCAAGGAGGACAGAGTTTATCGTG-3′ |
| Mcp-1 | |
| Forward | 5′-TTAAAAACCTGGATCGGAACCAA-3′ |
| Reverse | 5′-GCATTAGCTTCAGATTTACGGGT-3′ |
| Hmgcr | |
| Forward | 5′-CTTGTGGAATGCCTTGTGATTG-3′ |
| Reverse | 5′-AGCCGAAGCAGCACATGAT-3′ |
| Hmgcs | |
| Forward | 5′-GCCGTGAACTGGGTCGAA-3′ |
| Reverse | 5′-GCATATATAGCAATGTCTCCTGCAA-3′ |
| Abcg5 | |
| Forward | 5′-TGGATCCAACACCTCTATGCTAAA-3′ |
| Reverse | 5′-GGCAGGTTTTCTCGATGAACTG-3′ |
| Abcg8 | |
| Forward | 5′-TGCCCACCTTCCACATGTC-3′ |
| Reverse | 5′-ATGAAGCCGGCAGTAAGGTAGA-3′ |
| Ldlr | |
| Forward | 5′-AGGCTGTGGGCTCCATAGG-3′ |
| Reverse | 5′-TGCGGTCCAGGGTCATCT-3′ |
| Srb1 | |
| αForward | 5′-TCCCCATGAACTGTTCTGTGAA-3′ |
| Reverse | 5′-TGCCCGATGCCCTTGA-3′ |
| Cyp7a1 | |
| Forward | 5′-TAGTGGCGGGCGTCCCTATT-3′ |
| Reverse | 5′-GCCCAGAGGATCACGAGGTG-3′ |
| Cyp8b1 | |
| Forward | 5′-GCCTTCAAGTATGATCGGTTCCT-3′ |
| Reverse | 5′-GATCTTCTTGCCCGACTTGTAGA-3′ |
| Gapdh | |
| Forward | 5′-TGTGTCCGTCGTGGATCTGA-3′ |
| Reverse | 5′-CCTGCTTCACCACCTTCTTGAT-3′ |
Bmal1, brain and muscle arnt-like protein-1; Dbp, D site albumin promoter-binding protein; Pparg, peroxisome proliferator-activated receptor-γ; C/ebpa and C/ebpb, CCAAT enhancer-binding protein a and b, respectively; Zfp423, zinc finger protein 423; Hsl, hormone-sensitive lipase; Mcp-1, monocyte chemoattractant protein-1; Hmgcr, 3-hydroxy-3-methylglutaryl coenzyme A reductase; Hmgcs, 3-hydroxy-3-methylglutaryl-CoA synthase; Abcg5 and -8, ATP-binding cassette transporter G5 and G8, respectively; Ldlr, LDL receptor; Srb1, scavenger receptor class B, member 1; Cyp7a1, cytochrome P -450, family 7, subfamily A, and polypeptide 1; Cyp8b1, sterol 12α-hydroxylase.
Immunoblot analysis.
SDS-PAGE and immunodetection of proteins was conducted as reported previously (24). To enrich for nuclear proteins, frozen tissues were homogenized in buffer containing 20 mM Tris hydrochloride, 2 mM magnesium chloride, 0.25 M sucrose, and protease inhibitors. The homogenate was centrifuged at 2,000 g for 10 min, and the remaining pellet was solubilized in protein sample buffer. An insulin syringe was used to shear DNA. For measuring total and phosphorylated Akt (p-Akt), the supernatant of the homogenate was collected. Protein concentrations were measured by BCA assay. The following primary antibodies were used: BMAL1 at 1:2,000 dilution (A302-616A; Bethyl Laboratories), total Akt at 1:5,000 dilution (4685; Cell Signaling Technology), p-Akt at 1:5,000 dilution (9271; Cell Signaling Technology), histone H3 at 1:2,000 dilution (4499; Cell Signaling Technology), and GAPDH at 1:200 dilution (MAB374; Chemicon Internation). The secondary antibodies used were a goat anti-rabbit IgG horseradish peroxidase conjugated antibody (Pierce Biotechnology 31460) and a horse anti-mouse IgG horseradish peroxidase conjugated antibody (7076; Cell Signaling Technology) at a 1:1,000 dilution.
Statistical analyses.
Comparisons across groups defined by one factor (e.g., genotype) were performed using a t-test or one-way ANOVA and across groups defined by two factors (e.g., genotype and diet) were performed using two-way ANOVA. For gene expression measured over a 24-h period, variances across groups differed. Comparisons across groups defined by one factor (e.g., genotype) while accounting for lean mass were performed using one-way ANCOVA. Comparisons across groups defined by two factors (e.g., genotype and diet) while accounting for 8-wk body weight were performed using two-way ANCOVA. Comparisons across groups in which time was a factor were performed using repeated-measures ANOVA. Statistical analyses were conducted using GraphPad Prism (version 6) and SAS (version 9.3). Data are summarized as means ± SE, and statistical significance is defined by P < 0.05.
RESULTS
Liver and adipose peripheral clocks mediate centrally regulated functions.
We generated adipose-Bmal1 knockout (ABKO) and liver- and adipose-Bmal1 knockout (LABKO) mice to study the role of adipose and liver peripheral clocks in metabolic disease. (Fig. 1). Peak mRNA and protein expression were blunted in adipose tissue of female ABKO and LABKO mice expressing the aP2-Cre transgene, whereas mRNA and protein expression were blunted in liver of LABKO mice injected with the Ad-Cre virus (Fig. 1, A and B). Furthermore, the diurnal mRNA expression of Dbp, a BMAL1 target gene, was disrupted in adipose of ABKO and LABKO mice and in liver of LABKO mice (Fig. 1C). To determine whether peripheral Bmal1 regulates behavioral changes related to metabolism, measurements of food intake, locomotor activity, and energy expenditure were taken in female mice maintained on a standard rodent chow diet (Fig. 2). Both ABKO and LABKO mice had increased total food intake. The increase was observed in both the light and dark phases, although the increase did not reach statistical significance in the dark phase in ABKO mice (Fig. 2, A and B). Total locomotor activity was increased in ABKO mice compared with control mice. This was due to increased activity predominantly in the light phase and tended to be increased in the dark phase (Fig. 2, C and D). Further deletion of Bmal1 in the liver reversed this effect (Fig. 2D). Other differences in activity patterns were also observed. Compared with control and LABKO mice, ABKO mice had a greater and more prolonged peak in activity following the onset of the dark phase. ABKO mice also exhibited bursts of activity midway through the light and dark phases (Fig. 2C).
Fig. 1.

Deletion of Bmal1 in white adipose tissue (WAT) and liver of 13- to 24-wk-old female mice on standard rodent chow. Hour 0 represents the beginning of lights-on; n = 3–9/group. A: Bmal1 expression in WAT, genotype F(2, 49) = 18.23, P < 0.0001; time F(5, 49) = 29.76, P < 0.0001; interaction F(10, 49) = 6.604, P < 0.0001, and in liver, genotype F(2, 55) = 8.709, P = 0.0005; time F(5, 55) = 22.75, P < 0.0001; interaction F(10, 55) = 1.860, P = 0.0714. B: Western blot and densitometry analyses of BMAL1 in WAT of mice negative or positive for the aP2-Cre transgene; T = 4.944, DF = 6, and P = 0.0026, and in liver of mice injected with Ad-empty or Ad-Cre virus T = 7.732, DF = 6, and P = 0.0002 at hour 0. C: Dbp expression in WAT, genotype F(2, 49) = 2.481, P = 0.0941; time F(5, 49) = 6.358, P = 0.0001; interaction F(10, 49) = 2.223, P = 0.0317, and in liver, genotype F(2, 55) = 0.9127, P = 0.4075; time F(5, 55) = 11.35, P < 0.0001; interaction F(10, 55) = 1.874, P = 0.0695. *P < 0.05, comparison between control and adipose-Bmal1 knockout (ABKO); #P < 0.05, comparison between control and liver- and adipose-Bmal1 knockout (LABKO); +P < 0.05, comparison between ABKO and LABKO for A and C; ** and ##P < 0.01; *** and ###P < 0.001, **** and ####P < 0.0001.
Fig. 2.

Food intake, physical activity, and energy expenditure changes in 12-wk-old female mice on standard rodent chow. Hour 0 represents the beginning of lights-on; n = 7–9/group. A: food intake over 24 h. Genotype: F(2, 3,842) = 10.28, P < 0.0001; time: F(47, 3,842) = 3.594, P < 0.0001; interaction: F(94, 3,842) = 1.361, P = 0.0124. B: food intake during the light phase, F(2, 20) = 9.791, P = 0.001; dark phase, F(2, 20) = 5.956, P = 0.0093; both phases, F(2, 20) = 16.65, P < 0.0001. C: locomotor activity defined as the sum of ambulatory (break of 2 adjacent beams in succession) and fine (break of the same beam twice) activity over 24 h. Genotype: F(2, 3,888) = 54.14, P < 0.0001; time: F(47, 3,888) = 16.03, P < 0.0001; interaction: F(94, 3,888) = 1.741, P < 0.0001. D: locomotor activity during the light phase, F(2, 20) = 7.541, P = 0.0036; dark phase, F(2, 20) = 3.555, P = 0.0477; both phases, F(2, 20) = 5.853, P = 0.01. E: resting energy expenditure relative to lean mass; slope P = 0.458, intercept P = 0.171. F: respiratory exchange ratio, F(2, 20) = 25.67, P < 0.0001. Effects of genotype are indicated by horizontal lines terminating in vertical lines. *P < 0.05, comparison between control and ABKO; #P < 0.05, comparison between control and LABKO; +P < 0.05, comparison between ABKO and LABKO in A and C; **, ##, and ++P < 0.01; ***, ###, and +++P < 0.001; **** and ++++P < 0.0001.
Resting energy expenditure was analyzed using lean mass as a covariate. After controlling for lean mass, there were no significant differences among genotypes on resting energy expenditure (Fig. 2E). The respiratory exchange ratio was slightly higher in ABKO mice, indicating a greater utilization of glucose for energy (Fig. 2F). Body weight, lean mass, fat mass, and adiposity were all increased in LABKO mice compared with control and ABKO mice (Fig. 3, A–D).
Fig. 3.

Body weight, body composition, and blood glucose of 13- to 24-wk-old female mice fed a standard rodent chow; n = 7–9/group. A: body weight, F(2, 20) = 37.99, P < 0.0001. B: lean mass, F(2, 20) = 10.63, P = 0.0007. C: fat mass, F(2, 20) = 124.9, P < 0.0001. D: %adiposity, F(2, 20) = 106.3, P < 0.0001. E: blood glucose measured beginning at circadian time 0. Genotype: F(2, 74) = 10.72, P < 0.0001; time: F(5, 370) = 65.51, P < 0.0001; interaction: F(10, 370) = 1.058, P = 0.3938 (n = 17–31/group). Effects of genotype are indicated by horizontal lines terminating in vertical lines. *P < 0.05, comparison between control and ABKO; +P < 0.05, comparison between ABKO and LABKO in E; ***P < 0.001; ****P < 0.0001.
Plasma glucose oscillates following a diurnal pattern (14, 20, 22). ABKO mice had higher blood glucose concentrations throughout the light phase, which was maintained upon feeding during the dark phase. The increase in glucose was reversed in LABKO mice, resulting in a similar blood glucose pattern between control and LABKO mice (Fig. 3E).
Disruptions in liver and adipose peripheral clocks lead to increased body weight and adiposity.
To assess the development of obesity and obesity-related phenotypes, a second cohort of male mice was fed a low-fat control diet (CD) or high-fat diabetogenic diet (DD) for 16 wk. The DD itself decreased Bmal1 mRNA expression in epididymal adipose tissue but not in the liver (Fig. 4, A and C). However, this decrease was not reflected in BMAL1 protein levels observed at hour 0 (Fig. 4B). As in the cohort of female mice, male LABKO mice were slightly heavier prior to the initiation of diet at 8 wk of age (Fig. 5A). At 24 wk, the overall analysis of variance indicated a main effect of diet and a main effect of genotype on body weight, weight gain, and adiposity (Fig. 5, B–D). Body weight at 24 wk was significantly increased in LABKO mice fed either diet (Fig. 5B). The increase in weight gain over a 16-wk period appeared to be greater in control diet-fed LABKO mice, but the difference did not reach statistical significance, whereas diabetogenic diet-fed LABKO mice had significantly increased weight gain (Fig. 5C). A two-way ANCOVA was performed for 24-wk body weight, using 8-wk body weight as the covariate and genotype and diet as the two factors to determine whether the increased body weight at 24 wk in LABKO mice was predicted by elevated body weight at 8 wk. The 8-wk body weight covariate was significantly associated with 24-wk body weight (P = 0.002), as was genotype (P = 0.003) and diet (P < 0.001), but there was no genotype by diet interaction (P = 0.149). Adiposity followed a similar pattern as 24-wk body weight for both diet groups (Fig. 5D) Within diet, as there were no differences in lean mass between genotypes (data not shown), the increase in body weight and weight gain was due to increased adiposity.
Fig. 4.
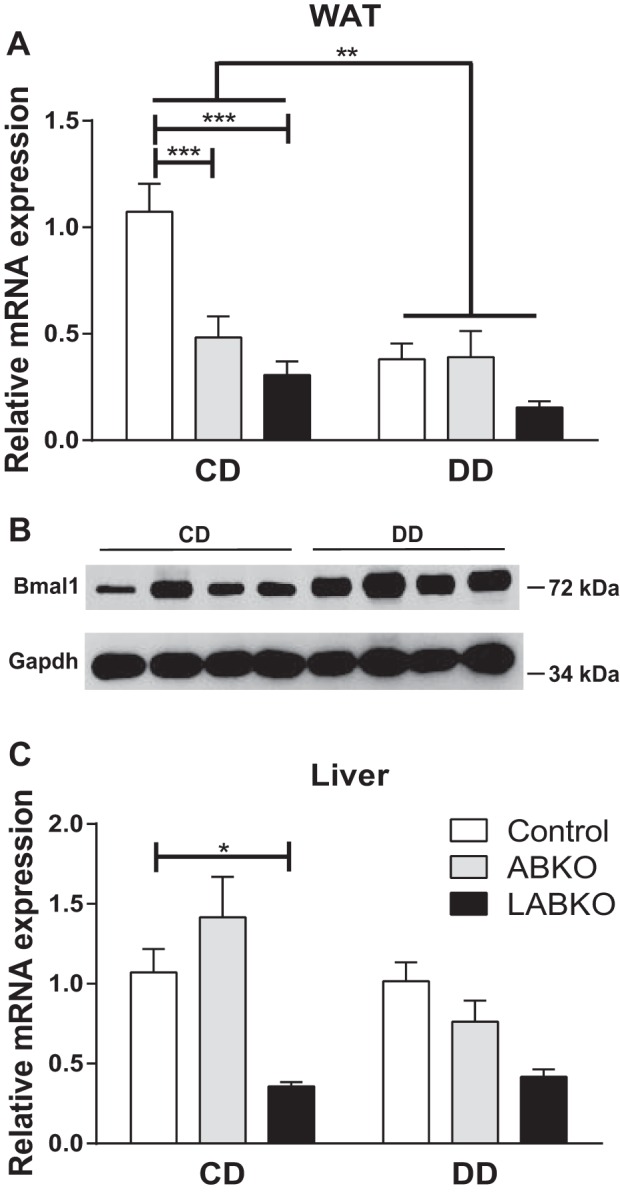
Deletion of Bmal1 in WAT and liver of 24-wk-old male mice on control diet (CD) or diabetogenic diet (DD) for 16 wk. A: Bmal1 expression at circadian time 0 in WAT. Genotype: F(2, 39) = 8.433, P = 0.0009; diet: F(1, 39) = 9.94, P = 0.0031; interaction: F(2, 39) = 4.676, P = 0.0151 (n = 4–11/group). B: Western blot analysis of BMAL1 in WAT of control mice on CD or DD. C: Bmal1 expression at circadian time 0 in liver. Genotype: F(2, 37) = 8.64, P = 0.0008; diet: F(1, 37) = 2.305, P = 0.1374; interaction: F(2, 37) = 2.611, P = 0.0869 (n = 5–9/group). Effects of genotype are indicated by horizontal lines terminating in vertical lines. Effects of diet are indicated by brackets terminating in horizontal lines. *P < 0.05; **P < 0.01; ***P < 0.001.
Fig. 5.

Body weight and composition of male mice fed a control diet (CD) or a diabetogenic diet (DD) for 16 wk; n = 5–18/group. A: body weight prior to initiation of diets at week 8; F(2, 59) = 8.981, P = 0.0004. B: body weight at the end of study at week 24. Genotype: F(2, 56) = 15.12, P < 0.0001; diet: F(1, 56) = 204.1, P < 0.0001; interaction: F(2, 56) = 2.003, P = 0.1444. C: change in body weight from week 8 to week 24. Genotype: F(2, 56) = 9.023, P = 0.0004; diet: F(1, 56) = 228.7, P < 0.0001; interaction: F(2, 56) = 2.032, P = 0.1407. D: %adiposity at week 24. Genotype: F(2, 56) = 8.202, P = 0.0008; diet: F(1, 56) = 95.17, P < 0.0001; interaction: F(2, 56) = 3.521, P = 0.0363. Effects of genotype are indicated by horizontal lines terminating in vertical lines. Effects of diet are indicated by brackets terminating in horizontal lines. *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001.
The diabetogenic diet resulted in overall increased adipocyte size and the appearance of crown-like structures (Fig. 6). Among groups fed the control diet, adipocyte area was similar between genotypes, with a tendency for increased size in LABKO mice (Fig. 6, A and B). This resulted in a greater number of larger adipocytes, reflected by the rightward shift in the histogram (Fig. 6C). However, this genetic difference was lost in mice fed the diabetogenic diet despite both ABKO and LABKO mice having increased fat mass compared with controls (Figs. 5D and 6).
Fig. 6.

Adipose histology, quantification, and measurement of adipogenesis and lipolysis in WAT at circadian time 0 in 24-week-old male mice fed a control diet (CD) or a diabetogenic diet (DD) for 16 wk. A: representative images of hematoxylin and eosin (H & E) stain from epididymal adipose. B: adipocyte area. Genotype: F(2, 14) = 1.392, P = 0.281; diet: F(1, 14) = 6.985, P = 0.0193; interaction: F(2, 14) = 1.599, P = 0.2369 (n = 2–4/group). C: histogram of adipocyte count distribution of specified adipocyte area. D: gene expression of adipogenic and lipolytic genes. For C/ebpβ, F(2, 20) = 5.893, P = 0.0097; n = 5–9/group. Effects of genotype are indicated by horizontal lines terminating in vertical lines. Effects of diet are indicated by brackets terminating in horizontal lines. *P < 0.05; **P < 0.01.
Adipose inflammation appeared to increase the presence of crown-like structures in the DD-fed mice but was not affected by genotype (Fig. 6A). Similarly, the DD increased the expression of both F4/80 [genotype: F(2, 39) = 0.4142, P = 0.6637; diet: F(1, 39) = 63.71, P < 0.0001; interaction: F(2, 39) = 0.4215, P = 0.6590] and Mcp-1 [genotype: F(2, 39) = 3.123, P = 0.0552; diet: F(1, 39) = 19.12, P < 0.0001; interaction: F(2, 39) = 1.555, P = 0.2241] (data not shown). Mcp-1 expression appeared to be higher in both ABKO and LABKO mice but was significant only in the LABKO group (control: 3.266 ± 0.291; LABKO: 8.767 ± 3.976) (data not shown).
To determine whether the increase in adiposity could be explained by increased adipogenesis or decreased lipolysis, markers of these processes were measured in control diet-fed mice. There was a significant increase in C/ebpβ expression in LABKO mice, but other measured genes of adipogenesis (Pparγ, C/ebpα, Zfp423) and lipolysis (Hsl, Pnpla2, Abhd5) were unchanged by genotype (Fig. 6D).
Loss of glycemic control does not correspond with increased body weight and adiposity.
To determine whether peripheral clocks play a role in the loss of glycemic control associated with obesity, fasting glucose, glucose tolerance, and insulin sensitivity were measured in male mice challenged with a DD. There was a main effect of diet on fasting glucose but no effect of genotype in either diet (Fig. 7A). Similarly, there was a main effect of diet on blood glucose concentrations following an intraperitoneal injection of glucose but no differences between genotypes (Fig. 7, B and C). Both ABKO and LABKO mice fed a DD had greater reductions in blood glucose following an intraperitoneal injection of insulin than control mice (Fig. 7, D and E). To determine whether adipose tissue of ABKO and LABKO mice respond better to insulin, total and p-Akt were measured. Although p-Akt was increased in white adipose tissue (WAT) of ABKO and LABKO mice, total Akt expression was increased as well. Therefore, there were no significant differences in p-Akt (Fig. 7F).
Fig. 7.

Glycemic control in 24-wk-old male mice fed a control diet (CD) or a diabetogenic diet (DD) for 16 wk; n = 5–18/group. A: fasting blood glucose concentrations following a 4-h fast. Genotype: F(2, 56) = 1.956, P = 0.1509; diet: F(1, 56) = 8.497, P = 0.0051; interaction: F(2, 56) = 0.05681, P = 0.9448. B: glucose tolerance following a 4-h fast in mice fed CD, genotype F(2, 28) = 0.8944, P = 0.4202, time F(4, 112) = 75.99, P < 0.0001, interaction F(8, 112) = 1.436, P = 0.1894; and in mice fed DD, genotype F(2, 28) = 1.29, P = 0.291, time F(4, 112) = 80.27, P < 0.0001, interaction F(8, 112) = 1.909, P = 0.0654. C: area under the curve (AUC) calculated from glucose tolerance test. Genotype: F(2, 56) = 0.3693, P = 0.6929; diet: F(1, 56) = 18.25, P < 0.0001; interaction: F(2, 56) = 1.989, P = 0.1464. D: insulin sensitivity following a 1-h fast in mice fed DD. Genotype: F(2, 28) = 7.759, P = 0.0021; time: F(4, 112) = 33.28, P < 0.0001; interaction: F(8, 112) = 3.42, P = 0.0015. E: AUC calculated from insulin sensitivity test. F(2, 28) = 7.768, P = 0.0021. F: Western blot and densitometry analyses of total and phosphorlated Akt (p-Akt) in WAT of mice on DD injected with insulin, F(2, 6) = 3.495, P = 0.0985. Effects of genotype are indicated by horizontal lines terminating in vertical lines. Effects of diet are indicated by brackets terminating in horizontal lines. *P < 0.05, comparison between control and ABKO; #P < 0.05, comparison between control and LABKO; +P < 0.05, comparison between ABKO and LABKO in B and D; **P < 0.01; ****P < 0.0001.
Disruptions in liver and adipose peripheral clocks increase hepatic cholesterol in LABKO mice.
There was a main effect of diet on serum total cholesterol but no significant differences between genotypes in either diet group. Serum triglycerides and nonesterified fatty acids were not affected by diet or genotype (data not shown). However, it should be noted that mice were not fasted for these measures.
As expected, there was a main effect of diet on liver weights. However, there were no differences in liver weights or percent liver weight between genotypes in either diet group (data not shown). Despite any lack of differences, ABKO mice appeared to have decreased lipid deposition in the liver compared with control and LABKO mice within the DD groups (Fig. 8A). There was a main effect of diet on hepatic total cholesterol and triglycerides (Fig. 8, B and C). Within mice given a DD, LABKO mice had the highest level of hepatic total cholesterol (Fig. 8B). A similar pattern was observed for triglycerides, but these differences did not reach statistical significance (Fig. 8C). In an effort to explain the increase in hepatic cholesterol in LABKO mice fed a DD, genes involved in cholesterol synthesis and regulation were measured in liver. Hmgcr and Hmgcs, which are involved in cholesterol synthesis, exhibited similar patterns of expression, with LABKO mice on CD and both ABKO and LABKO mice on DD having increased expression (Fig. 8D). Sterol transporters Abcg5 and Abcg8 were both suppressed by diet, but no significant differences between genotypes were observed (Fig. 8D). Although genes involved in cholesterol uptake, Ldlr and Srb1, had the highest expression in LABKO mice on a CD, there were no differences in Ldlr or Srb1 expression between genotypes in mice on a DD (data not shown). Bile acid synthesis genes Cyp7a1 and Cyp8b1 were unchanged by diet or genotype (data not shown).
Fig. 8.

Liver histology, hepatic lipids, and gene expression from liver isolated at circadian time 0 in 24-wk-old male mice fed a control diet (CD) or a diabetogenic diet (DD) for 16 wk. A: representative images of H & E stain. B: total cholesterol. Genotype: F(2, 26) = 5.095, P = 0.0136; diet: F(1, 26) = 25.42, P < 0.0001; interaction: F(2, 26) = 0.9054, P = 0.4167. C: triglycerides. Genotype: F(2, 26) = 2.519, P = 0.1000; diet: F(1, 26) = 44.3, P < 0.0001; interaction: F(2, 26) = 0.3547, P = 0.7047 (n = 4–7/group). D: gene expression of Hmgcr. Genotype: F(2, 39) = 6.118, P = 0.0049; diet: F(1, 39) = 1.268, P = 0.267; interaction: F(2, 39) = 3.374, P = 0.0445. E: gene expression of Hmgcs. Genotype: F(2, 39) = 3.582, P = 0.0373; diet F(1, 39) = 12.54, P = 0.001; interaction: F(2, 39) = 2.471, P = 0.0976. F: gene expression of Abcg5. Genotype F(2, 39) = 0.8746, P = 0.4251; diet: F(1, 39) = 19.89, P < 0.0001; interaction: F(2, 39) = 1.024, P = 0.3685. G: gene expression of Abcg8. Genotype: F(2, 39) = 2.004, P = 0.1484; diet: F(1, 39) = 18.92, P < 0.0001; interaction: F(2, 39) = 1.747, P = 0.1876 (n = 5–9/group). Effects of genotype are indicated by horizontal lines terminating in vertical lines. Effects of diet are indicated by brackets terminating in horizontal lines. *P < 0.05; **P < 0.01; ****P < 0.0001.
DISCUSSION
Our study confirms and extends previous findings indicating that peripheral oscillators, particularly in adipose, modulate feeding and locomotor activity. Although the loss of Bmal1 in adipose and the liver exacerbates the development of obesity, it does not have further effects on other metabolic comorbidities.
The central clock synchronizes the oscillations of peripheral clocks; however, peripheral clocks can be uncoupled from the central clock through manipulations in the feeding/fasting cycle (6, 9, 33). This can have impacts on feeding behavior and metabolic disease such that increased food intake during the inactive phase results in obesity (2, 8, 25). Alternatively, time-restricting food intake to the active phase or the beginning rather than the end of the active phase is protective (2, 3, 10, 25). Our study sought to determine the impact of directly disrupting adipose and liver oscillators on centrally regulated functions related to metabolism such as feeding behavior and locomotor activity. A previous study has shown that adipose deletion of Bmal1 results in changes in plasma polyunsaturated fatty acid concentrations, which leads to alterations in the hypothalamic regulation of feeding behavior (20). We corroborated the finding that ABKO mice exhibit an impaired feeding pattern and have increased food intake during the light phase. In both studies, the aP2-Cre transgene was used for adipose deletion. Although AP2 is abundantly expressed in adipose and is commonly used to drive the expression of Cre recombinase for adipose-specific deletion, a limitation of this line is that AP2 has also been found to be expressed in other tissues such as macrophages, embryonic tissues, and brain (16, 32). In our present study, we observed residual Bmal1 mRNA and protein expression in both adipose and liver. Although this may be due to incomplete penetrance of Cre recombinase in the targeted tissues, the presence of other cell types expressing Bmal1 in these tissues is also a possibility.
Further deletion of Bmal1 from the liver in LABKO mice maintained the increase in food intake during the light phase, with additional increases during the dark phase. Although Paschos et al. (20) did not observe changes in locomotor activity with adipose-Bmal1 deletion, we found ABKO mice tended to be more active, particularly during the light phase. This difference in observation is likely due to differences in environmental conditions or the diet used in our studies. Nonetheless, the increase in activity together with the delay in peak activity at the onset of the dark phase and the spurts of activity in ABKO mice indicate that adipose Bmal1 plays a role in modulating physical activity and movement. Interestingly, the increased pattern of activity was reversed in LABKO mice; however, liver-specific deletion of Bmal1 is insufficient to alter locomotor activity and feeding behavior (15). These findings demonstrate that disrupting the liver clock in the setting of adipose clock disruption also has an impact on physical activity, suggesting that the coordination of functional adipose and liver clocks is necessary to properly mediate centrally regulated functions. Furthermore, the differences in food intake and activity between genotypes suggest tissue-specific roles of Bmal1.
Despite ABKO mice eating more during the light phase and subsequently having increased overall food intake similar to that of LABKO mice, body weight and adiposity were not significantly increased. This is likely due to the increased activity that we observed in ABKO but not in LABKO mice on a chow diet. Although we cannot rule out a direct role of peripheral Bmal1 in regulating energy metabolism, our findings suggest that changes in feeding behavior and activity in ABKO and LABKO mice are the primary regulators of body weight and composition.
Blood glucose concentrations exhibit diurnal variations, and several models of dysfunctional circadian rhythm exhibit impaired glycemic control (14, 22, 31). When the availability of a chow diet was restricted to 12 h during the dark phase, ABKO mice tended to have slightly elevated blood glucose concentrations over 24 h, but the diurnal pattern was similar to the other genotypes. In the setting of restricted food availability to normal feeding times, disrupting adipose and liver Bmal1 was insufficient to impair the diurnal pattern of blood glucose.
Obesity is highly associated with the development of insulin resistance, type 2 diabetes, and fatty liver disease. Therefore, in the presence of a diabetogenic diet, we expected the disruption of Bmal1 in two metabolic tissues to exacerbate these conditions. Although LABKO mice were more obese than control mice, there was no further loss of glycemic control or evidence of increased hepatic inflammation and fibrosis in either ABKO or LABKO mice. There was a trend toward increased insulin sensitivity in adipose tissue of ABKO and LABKO mice fed a diabetogenic diet. Overall, the disruption of peripheral clocks from two of the primary tissues involved in maintaining energy homeostasis was insufficient to exacerbate metabolic disease beyond the development of obesity. One explanation for this observation is that a high-fat diet itself has effects on circadian rhythms. A high-fat diet has been shown to lengthen the circadian period in mice prior to any body weight effect. Furthermore, diurnal feeding behavior and clock gene expression are attenuated with short-term high-fat feeding (13). Consistent with these findings, we found that 16 wk of a diabetogenic diet results in the suppression of peak Bmal1 expression in white adipose tissue. Protein expression, however, was slightly increased with a diabetogenic diet. Because this reflects expression at a single time point, the discordance may reflect a phase shift in BMAL1 expression or posttranscriptional regulation. Interestingly, peak Bmal1 expression was not affected in the liver. Others have demonstrated slight to significant attenuations in liver Bmal1 oscillation, depending on the duration of high-fat feeding (7, 13). It is likely that although disrupting multiple peripheral clocks indeed alters central behavior, these effects seem to be overcome by the effects of a high-fat diet, thereby muting any exacerbations in metabolic disease.
In addition to the probable role of circadian rhythms in the development of obesity and insulin resistance, evidence suggests that lipid homeostasis is dependent on circadian function. The presence of a mutant form of Clock in mice increases plasma triglycerides and cholesterol and exacerbates atherosclerosis (18). Additionally, the open access bioinformatics database CircaDB has identified a number of rhythmic transcripts involved in cholesterol synthesis and metabolism. Although plasma lipid concentrations were unchanged in our study, hepatic cholesterol and triglycerides were least abundant in ABKO and most abundant in LABKO mice fed a diabetogenic diet. This suggests that liver Bmal1 is involved in the regulation of cholesterol either through direct or indirect mechanisms. Cholesterol synthesis, measured by transcript levels of Hmgcr and Hmgcs, tended to be increased in both ABKO and LABKO mice fed a diabetogenic diet. However, no additional changes in genes involved in cholesterol transport and bile acid synthesis that would explain the varying abundance of hepatic lipids were observed. It should be noted that transcript levels of Hmgcs and Cyp8b1 reach peak amplitude approximately at the beginning of the light cycle, Hmgcr, Ldlr, Cyp7a1, Abcg5, and Abcg8 at the beginning of the dark cycle, and Srb1 at the midlight cycle (11). Consequently, measuring gene expression throughout a 24-h period would enhance the capability of identifying essential differences.
A limitation of our study is the use of both male and female mice. It has been demonstrated that estradiol during development plays a role in diurnal rhythms, and estrogen receptor-β is regulated by the clock machinery (4, 5). Additionally, sexually dimorphic differences in clock gene expression in the suprachiasmatic nucleus and locomotor activity in response to light-induced phase shifts by leptin have been demonstrated in mice (17). Although both sexes of mice were used throughout the study, only females were used for indirect calorimetry, whereas only males were used for the diet intervention portion of the study.
Our study and others have demonstrated the selectivity of the clock's influence on metabolism and the potential that exists in targeting circadian factors as a potential therapeutic for metabolic disease. Here, we add knowledge to how disruptions in multiple peripheral clocks may differentially regulate behavior and influence the development of obesity.
GRANTS
This work was supported by National Institute of Diabetes and Digestive and Kidney Diseases (R01-DK-100892-02), the COBRE Pathology Core and Metabolic Core, National Center for Research Resources (5-P20-RR-021954-05), and the National Institute of General Medical Sciences (8-P20-GM-103527-05).
DISCLOSURES
The authors have no conflicts of interest, financial or otherwise, to disclose.
AUTHOR CONTRIBUTIONS
S.S.P. and G.A.G. conception and design of research; S.S.P., D.E.C., and G.A.G. performed experiments; S.S.P., D.E.C., R.J.C., and G.A.G. analyzed data; S.S.P., Y.W., J.L., and G.A.G. interpreted results of experiments; S.S.P. prepared figures; S.S.P. drafted manuscript; S.S.P., D.E.C., Y.W., J.L., R.J.C., and G.A.G. edited and revised manuscript; S.S.P., D.E.C., Y.W., J.L., R.J.C., and G.A.G. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank Dr. Karyn Esser for providing the Bmal1f/f mice and Dr. Wendy Katz for assistance in analysis of indirect calorimetry and adipose quantification data.
REFERENCES
- 1.Akhtar RA, Reddy AB, Maywood ES, Clayton JD, King VM, Smith AG, Gant TW, Hastings MH, Kyriacou CP. Circadian cycling of the mouse liver transcriptome, as revealed by cDNA microarray, is driven by the suprachiasmatic nucleus. Curr Biol 12: 540–550, 2002. [DOI] [PubMed] [Google Scholar]
- 2.Arble DM, Bass J, Laposky AD, Vitaterna MH, Turek FW. Circadian timing of food intake contributes to weight gain. Obesity (Silver Spring) 17: 2100–2102, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bray MS, Tsai JY, Villegas-Montoya C, Boland BB, Blasier Z, Egbejimi O, Kueht M, Young ME. Time-of-day-dependent dietary fat consumption influences multiple cardiometabolic syndrome parameters in mice. Int J Obes 34: 1589–1598, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brockman R, Bunick D, Mahoney MM. Estradiol deficiency during development modulates the expression of circadian and daily rhythms in male and female aromatase knockout mice. Horm Behav 60: 439–447, 2011. [DOI] [PubMed] [Google Scholar]
- 5.Cai W, Rambaud J, Teboul M, Masse I, Benoit G, Gustafsson JA, Delaunay F, Laudet V, Pongratz I. Expression levels of estrogen receptor beta are modulated by components of the molecular clock. Mol Cell Biol 28: 784–793, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Damiola F, Le Minh N, Preitner N, Kornmann B, Fleury-Olela F, Schibler U. Restricted feeding uncouples circadian oscillators in peripheral tissues from the central pacemaker in the suprachiasmatic nucleus. Genes Dev 14: 2950–2961, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Eckel-Mahan KL, Patel VR, de Mateo S, Orozco-Solis R, Ceglia NJ, Sahar S, Dilag-Penilla SA, Dyar KA, Baldi P, Sassone-Corsi P. Reprogramming of the circadian clock by nutritional challenge. Cell 155: 1464–1478, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fonken LK, Workman JL, Walton JC, Weil ZM, Morris JS, Haim A, Nelson RJ. Light at night increases body mass by shifting the time of food intake. Proc Natl Acad Sci USA 107: 18664–18669, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hara R, Wan K, Wakamatsu H, Aida R, Moriya T, Akiyama M, Shibata S. Restricted feeding entrains liver clock without participation of the suprachiasmatic nucleus. Genes Cells 6: 269–278, 2001. [DOI] [PubMed] [Google Scholar]
- 10.Hatori M, Vollmers C, Zarrinpar A, DiTacchio L, Bushong EA, Gill S, Leblanc M, Chaix A, Joens M, Fitzpatrick JA, Ellisman MH, Panda S. Time-restricted feeding without reducing caloric intake prevents metabolic diseases in mice fed a high-fat diet. Cell Metab 15: 848–860, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hughes ME, DiTacchio L, Hayes KR, Vollmers C, Pulivarthy S, Baggs JE, Panda S, Hogenesch JB. Harmonics of circadian gene transcription in mammals. PLoS Genet 5: e1000442, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Karlsson B, Knutsson A, Lindahl B. Is there an association between shift work and having a metabolic syndrome? Results from a population based study of 27,485 people. Occup Environ Med 58: 747–752, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kohsaka A, Laposky AD, Ramsey KM, Estrada C, Joshu C, Kobayashi Y, Turek FW, Bass J. High-fat diet disrupts behavioral and molecular circadian rhythms in mice. Cell Metab 6: 414–421, 2007. [DOI] [PubMed] [Google Scholar]
- 14.la Fleur SE, Kalsbeek A, Wortel J, Fekkes ML, Buijs RM. A daily rhythm in glucose tolerance: a role for the suprachiasmatic nucleus. Diabetes 50: 1237–1243, 2001. [DOI] [PubMed] [Google Scholar]
- 15.Lamia KA, Storch KF, Weitz CJ. Physiological significance of a peripheral tissue circadian clock. Proc Natl Acad Sci USA 105: 15172–15177, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lee KY, Russell SJ, Ussar S, Boucher J, Vernochet C, Mori MA, Smyth G, Rourk M, Cederquist C, Rosen ED, Kahn BB, Kahn CR. Lessons on conditional gene targeting in mouse adipose tissue. Diabetes 62: 864–874, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mendoza J, Lopez-Lopez C, Revel FG, Jeanneau K, Delerue F, Prinssen E, Challet E, Moreau JL, Grundschober C. Dimorphic effects of leptin on the circadian and hypocretinergic systems of mice. J Neuroendocrinol 23: 28–38, 2011. [DOI] [PubMed] [Google Scholar]
- 18.Pan X, Jiang XC, Hussain MM. Impaired cholesterol metabolism and enhanced atherosclerosis in clock mutant mice. Circulation 128: 1758–1769, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Panda S, Antoch MP, Miller BH, Su AI, Schook AB, Straume M, Schultz PG, Kay SA, Takahashi JS, Hogenesch JB. Coordinated transcription of key pathways in the mouse by the circadian clock. Cell 109: 307–320, 2002. [DOI] [PubMed] [Google Scholar]
- 20.Paschos GK, Ibrahim S, Song WL, Kunieda T, Grant G, Reyes TM, Bradfield CA, Vaughan CH, Eiden M, Masoodi M, Griffin JL, Wang F, Lawson JA, Fitzgerald GA. Obesity in mice with adipocyte-specific deletion of clock component Arntl. Nat Med 18: 1768–1777, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pendergast JS, Branecky KL, Yang W, Ellacott KL, Niswender KD, Yamazaki S. High-fat diet acutely affects circadian organisation and eating behavior. Eur J Neurosci 37: 1350–1356, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rudic RD, McNamara P, Curtis AM, Boston RC, Panda S, Hogenesch JB, Fitzgerald GA. BMAL1 and CLOCK, two essential components of the circadian clock, are involved in glucose homeostasis. PLoS Biol 2: e377, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rutter J, Reick M, McKnight SL. Metabolism and the control of circadian rhythms. Annu Rev Biochem 71: 307–331, 2002. [DOI] [PubMed] [Google Scholar]
- 24.Sabeva NS, Rouse EJ, Graf GA. Defects in the leptin axis reduce abundance of the ABCG5-ABCG8 sterol transporter in liver. J Biol Chem 282: 22397–22405, 2007. [DOI] [PubMed] [Google Scholar]
- 25.Salgado-Delgado R, Angeles-Castellanos M, Saderi N, Buijs RM, Escobar C. Food intake during the normal activity phase prevents obesity and circadian desynchrony in a rat model of night work. Endocrinology 151: 1019–1029, 2010. [DOI] [PubMed] [Google Scholar]
- 26.Sandoval D, Cota D, Seeley RJ. The integrative role of CNS fuel-sensing mechanisms in energy balance and glucose regulation. Annu Rev Physiol 70: 513–535, 2008. [DOI] [PubMed] [Google Scholar]
- 27.Shimba S, Ishii N, Ohta Y, Ohno T, Watabe Y, Hayashi M, Wada T, Aoyagi T, Tezuka M. Brain and muscle Arnt-like protein-1 (BMAL1), a component of the molecular clock, regulates adipogenesis. Proc Natl Acad Sci USA 102: 12071–12076, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Spiegel K, Tasali E, Leproult R, Van Cauter E. Effects of poor and short sleep on glucose metabolism and obesity risk. Nat Rev Endocrinol 5: 253–261, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Staels B. When the Clock stops ticking, metabolic syndrome explodes. Nat Med 12: 54–55; discussion 55, 2006. [DOI] [PubMed] [Google Scholar]
- 30.Su K, Sabeva NS, Liu J, Wang Y, Bhatnagar S, van der Westhuyzen DR, Graf GA. The ABCG5 ABCG8 sterol transporter opposes the development of fatty liver disease and loss of glycemic control independently of phytosterol accumulation. J Biol Chem 287: 28564–28575, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Turek FW, Joshu C, Kohsaka A, Lin E, Ivanova G, McDearmon E, Laposky A, Losee-Olson S, Easton A, Jensen DR, Eckel RH, Takahashi JS, Bass J. Obesity and metabolic syndrome in circadian Clock mutant mice. Science 308: 1043–1045, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Urs S, Harrington A, Liaw L, Small D. Selective expression of an aP2/Fatty Acid Binding Protein 4-Cre transgene in non-adipogenic tissues during embryonic development. Transgenic Res 15: 647–653, 2006. [DOI] [PubMed] [Google Scholar]
- 33.Wakamatsu H, Yoshinobu Y, Aida R, Moriya T, Akiyama M, Shibata S. Restricted-feeding-induced anticipatory activity rhythm is associated with a phase-shift of the expression of mPer1 and mPer2 mRNA in the cerebral cortex and hippocampus but not in the suprachiasmatic nucleus of mice. Eur J Neurosci 13: 1190–1196, 2001. [DOI] [PubMed] [Google Scholar]
- 34.Xu B, Kalra PS, Farmerie WG, Kalra SP. Daily changes in hypothalamic gene expression of neuropeptide Y, galanin, proopiomelanocortin, and adipocyte leptin gene expression and secretion: effects of food restriction. Endocrinology 140: 2868–2875, 1999. [DOI] [PubMed] [Google Scholar]
- 35.Yang S, Liu A, Weidenhammer A, Cooksey RC, McClain D, Kim MK, Aguilera G, Abel ED, Chung JH. The role of mPer2 clock gene in glucocorticoid and feeding rhythms. Endocrinology 150: 2153–2160, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yang X, Downes M, Yu RT, Bookout AL, He W, Straume M, Mangelsdorf DJ, Evans RM. Nuclear receptor expression links the circadian clock to metabolism. Cell 126: 801–810, 2006. [DOI] [PubMed] [Google Scholar]
- 37.Zheng B, Larkin DW, Albrecht U, Sun ZS, Sage M, Eichele G, Lee CC, Bradley A. The mPer2 gene encodes a functional component of the mammalian circadian clock. Nature 400: 169–173, 1999. [DOI] [PubMed] [Google Scholar]