

Abstract
It has been well-characterized that the renin-angiotensin system (RAS) physiologically regulates systemic arterial pressure. However, RAS signaling has also been shown to increase cell proliferation during malignancy, and angiotensin receptor blockers (ARBs) are able to decrease pro-survival signaling by inhibiting anti-apoptotic molecules and suppressing caspase activity. In this study, the apoptotic effects of telmisartan, a type of ARB, was evaluated using a non-cancerous human renal cell line (HEK) and a human renal cell carcinoma (RCC) cell line (786). Both types of cells were treated with telmisartan for 4 h, 24 h, and 48 h, and then were assayed for levels of apoptosis, caspase-3, and Bcl-2 using MTT assays, flow cytometry, and immunostaining studies. Analysis of variance was used to identify significant differences between these data (P < 0.05). Following the treatment of 786 cells with 100 µM and 200 µM telmisartan, a marked inhibition of cell proliferation was observed. 50 µM cisplatin also caused high inhibition of these cells. Moreover, these inhibitions were both concentration- and time-dependent (P < 0.05). Various apoptotic effects were also observed compared with control cells at the 24 h and 48 h timepoints assayed (P < 0.001). Furthermore, positive caspase-3 staining and down-regulation of Bcl-2 were detected, consistent with induction of cell death. In contrast, treatment of HEK cells with telmisartan did not produce an apoptotic effect compared with control cells at the 24 h timepoint (P > 0.05). Treatment with cisplatin promoted in HEK cells high index of apoptosis (P < 0.001). Taken together, these results suggest that telmisartan induces apoptosis via down-regulation of Bcl-2 and involvement of caspase-3 in human RCC cells.
Keywords: Telmisartan, human cancer cells, caspase-3, Bcl-2, apoptosis
Introduction
Renal cell carcinoma (RCC) is the most common pathological pattern for 3% of the malignant tumors detected in adults. Its worldwide incidence also continues to steadily increase each year,1 and the 5-year survival rate for pT1 and pT2 stage RCC currently ranges from 80% to 90%. However, for patients with metastatic disease, this rate is only 10%.2,3 Therefore, the ability to reduce risk factors associated with RCC, and the ability to identify novel therapeutics that target RCC, are extremely important.4
Angiotensin II (Ang II) is a multifunctional, bioactive octapeptide of the renin-angiotensin system (RAS) that serves as a vasoconstrictor to control cardiovascular function and renal homeostasis. Ang II binds two subtypes of receptors to mediate its biological effects, the Ang II type 1 receptor (AT1R) and the Ang II type 2 receptor (AT2R). The former mediates the major functions of Ang II in tumor growth and angiogenesis.5,6 In addition, stimulation of AT1R has been shown to activate anti-apoptotic molecules such as survivin and Bcl-XL,7 and to inactivate pro-apoptotic proteins.8
Activation of AT1R in malignant cells can also enhance pro-survival signaling by activating: (1) nuclear factor-κB (NF-κB) and the production of anti-apoptotic molecules such as Bcl-2 and survivin, and (2) the PI3K-Akt pathway which leads to the suppression of caspases.8 Conversely, signaling through the AT2R has been associated with the promotion of apoptosis. For example, in prostate cancer cells, overexpression of AT2R was found to induce apoptosis independent of Ang II. However, this pathway was dependent on p38 mitogen-activated protein kinase (MAPK), caspase 8, and caspase 3, and was mediated via an extrinsic cell death-signaling pathway that was partially dependent on TP53 rather than P21 activation.9 Correspondingly, AT2R overexpression in a human lung adenocarcinoma cell line was found to reduce cell viability and to induce apoptosis, which was associated with a significant reduction in procaspase-3 levels. Thus, a disruption in the stoichiometry of AT1R and AT2R, and/or alterations in their signaling pathways, can potentially influence whether cancer cells undergo apoptosis or survive in response to activation of the RAS.9
It is also possible that angiotensin receptor blockers (ARBs) modulate the development and progression of cancer.10 ARBs selectively block the activation of AT1Rs, thereby suppressing the RAS. Ang II has also been characterized as an anti-apoptotic agent and growth factor, which stimulates cell replication through the epidermal growth factor receptor (EGFR) transactivation/extracellular signal-regulated kinase (ERK) signaling pathway.11
In the present study, the objective was to evaluate the inhibitory effect of telmisartan on human renal cell carcinoma (RCC) cells and to determine whether telmisartan induces apoptosis.
Materials and methods
Reagents
The following reagents were purchased as indicated: Dulbecco’s modified Eagle’s medium (Life Technologies, Grand Island, NY, USA), 10% (v/v) heat-inactivated fetal bovine serum (CULTILAB LTDA/Brazil), trypsin/EDTA (Gibco BRL, Life Technologies, Grand Island, NY, USA), cisplatin (citoplax, 50 mg, BergamoTaboão da Serra, SP, Brazil), and telmisartan (Micardis, 80 mg, BoehringerIngelheim, SP, Brazil).
Cell culture
A human non-cancerous renal cell line (HEK) and a human RCC cell line (786) were purchased from the Culture Collection of the Federal University of Rio de Janeiro (RJCB Collection, Rio de Janeiro, RJ). HEK cells were maintained in Dulbecco's modified Eagle's medium and 786 cells were maintained in RPMI 1640 medium, both supplemented with 10% (v/v) heat-inactivated fetal bovine serum. Telmisartan and cisplatin solutions were filtered using a 0.22 µm minipore membrane, then were aliquoted and stored at –20℃.
Cell proliferation studies
Approximately 1.0 × 104 cells were plated on 8 × 8 mm2 multichamber slides (Nunc, Copenhagen, Denmark) and were treated with telmisartan (0–400 µM) and cisplatin (0–100 µM). Cell viability was subsequently measured at various timepoints using a modified 3-(4,5-dimethylthiazol-2- thiazolyl)-2,5-diphenyltetrazolium bromide (MTT) assay (WST-1 assay; Dojindo, Kumamoto, Japan) and a microplate reader. The results are presented as a percentage of the proliferation detected for control cells.
Annexin V and propidium iodide staining
Cells were plated in 6-well plates (5 × 104 cells/well) with 2 mL medium/well. After 24 h, concentrations of telmisartan (100 microMolar and 200 microMolar) and cisplatin (50 microMolar) were added for 4 h, 24 h, and 48 h. In parallel, control cells were maintained in culture medium without telmisartan and cisplatin. The cells were then assayed using the Annexin V-FITC Apoptosis Detection kit I (Biosciences Pharmingen, San Diego, USA). Annexin V-FITC and propidium iodide (PI) were added to the cellular suspension according to the manufacturer's instructions. A total of 1.0 × 106 cells from each sample were then analyzed using a FACSCalibur cytometer (BD Bioscience, Franklin Lakes, NJ, USA) and FlowJo software (BD Biosciences). Annexin V-FITC-positive/PI-negative cells were identified as cells in the early stages of apoptosis, while Annexin V-FITC-positive/PI-positive cells were identified as cells in the late stages of apoptosis, or cells that were undergoing necrosis.
Caspase-3 and Bcl-2 activities
After 24 h, the cells (5 × 104 cells/well) were treated with telmisartan (100 µM and 200 µM) and cisplatin (50 µM) in 24-well plates. After 24 h and 48 h, cells were washed, fixed with paraformaldehyde, permeabilized with Triton-X, and incubated with rabbit polyclonal anti-caspase-3 antibody (Abcam, San Francisco, CA, USA) or rat polyclonal anti-Bcl-2 antibody (Abcam). Both antibody solutions were diluted 1:500 in PBS containing bovine serum albumin (5%; Life Technologies do Brasil LTDA, São Paulo, Brazil). After an incubation at RT in a humid atmosphere for 1 h, the coverslips were incubated with AlexaFluor 488 anti-rabbit or anti-rat secondary antibody (Abcam). In addition, nuclei were stained with 4,6-diamidino-2-phenylindole (DAPI, Life Technologies do Brasil LTDA). Slides were subsequently examined using a binocular optical microscope (Nikon Eclipse 80i, Tokyo, Japan) using a 60 × oil objective, and imaged using Neurolucida software (MBF Bioscience, Williston, VT, USA).
Statistical analysis
All experiments were performed at least in triplicate. Significant differences between groups were calculated by applying analysis of variance and Bonferroni’s test, as indicated. A P value less than 0.05 was considered statistically significant.
Results
Telmisartan inhibits the growth of 786 RCC cells
To investigate the effects of telmisartan on RCC cell proliferation, a modified MTT assay was used to analyze cell viability in vitro. Following the treatment of RCC cell lines with telmisartan reduced cell viability was observed. Moreover, the half-maximal concentration that inhibited the growth of RCC cells ranged from 100 to 400 µM (Figure 1). When cells were counted 24 h after the addition of telmisartan, a marked inhibition of cell proliferation was especially observed for concentrations of 100 µM and 200 µM telmisartan (Figure 1).
Figure 1.

Telmisartan reduced cell viability in a concentration-dependent manner. Half-maximal inhibition of RCC growth was observed for concentrations of telmisartan ranging from 25 to 400 µM. ***P < 0.001
When cells were treated with cisplatin, a reduction in cell viability was also observed, with the half-maximal concentration to achieve growth inhibition ranging from 25 to 100 µΜ (Figure 2). Moreover, although cisplatin was found to inhibit the growth of RCC cells at all of the concentrations tested, a marked inhibition of cell proliferation was observed at a concentration of 50 µM cisplatin at the 24 h timepoint (Figure 2).
Figure 2.
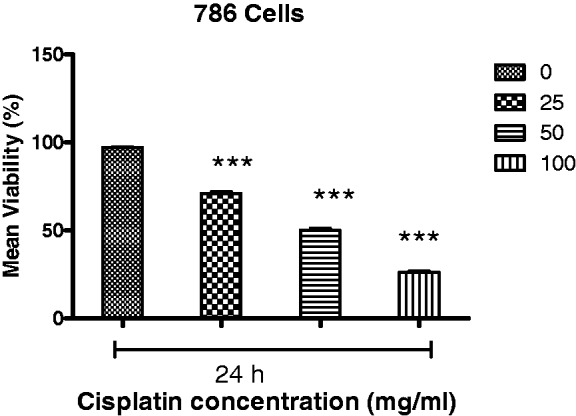
Cisplatin reduced cell viability in a concentration-dependent manner. Half-maximal inhibition of 786 RCC cell growth was observed for concentrations of telmisartan ranging from 25 to 400 µM. ***P < 0.001
Induction of apoptosis by telmisartan was evaluated using flow cytometry
To determine whether cell death induced by telmisartan was achieved through apoptosis, cells were treated with telmisartan and stained with Annexin-V-FITC and PI. Using flow cytometry, early and late stages of apoptosis were detected based on the percentage of Annexin V-FITC-positive cells/PI-negative cells and the percentage of Annexin V-FITC-positive/PI-positive cells that were present, respectively (Figures 3 and 4, lower right quadrant data versus top left quadrant data, respectively). For 786 RCC cells, treatment with 100 µM and 200 µM telmisartan induced early and late apoptosis both 24 h and 48 h after treatment, yet not after 4 h of treatment (Figure 3(a) and (b); Figure 4(b), (c), (e), (f), (h), and (i)). In contrast, treatment of HEK cells with 200 µM telmisartan for 24 h did not induce apoptosis (Figures 3(e) and 4(q)). However, late stages of apoptosis were detected 48 h after telmisartan treatment (Figures 3(f) and 4(s)). When 786 RCC cells were treated with 50 µM cisplatin, both early and late stage apoptosis were detected 4 h, 24 h, and 48 h after treatment. Although, a higher percentage of cells in the late stages of apoptosis were observed (Figure 3(c) and (d); Figure 4(k), (m), and (o)). When HEK cells were treated with 50 µM cisplatin, both early-stage and late-stage apoptosis were observed 24 h and 48 h after treatment, with the latter being more predominant (Figure 3(g) and (h); Figure 4(u) and (x)).
Figure 3.

Apoptosis induced by telmisartan and cisplatin in 786 RCC cells was detected using flow cytometry. Levels of early apoptosis (EA) (a) and late apoptosis (LA) (b) were detected for cells treated with 100 µM and 200 µM telmisartan for 4 h, 24 h, and 48 h. Levels of EA (c) and LA (d) were also detected for cells treated with 50 µM cisplatin for 4 h, 24 h, and 48 h. Apoptosis induced by telmisartan and cisplatin in HEK cells was detected using flow cytometry. Levels of EA (e) and LA (f) were detected for cells treated with 200 µM telmisartan for 24 h and 48 h. Levels of EA (g) and LA (h) were detected for cells treated with 50 µM cisplatin for 24 h and 48 h. ***P < 0.001; **P < 0.01; *P < 0.05
Figure 4.



Effects of telmisartan and cisplatin on early and late apoptosis as detected using flow cytometry. Treatment of 786 RCC cells with 100 µM and 200 µM telmisartan induced early- or late-stage apoptosis after 24 h (e, f) and 48 h (h, i), but not at the 4 h timepoint (b, c). In contrast, treatment of normal HEK cells with 200 µM telmisartan did not induce apoptosis after 24 h (q), yet did after 48 h (s). The lower right quadrants represent the Annexin V-FITC-positive/PI-negative cells in the early stages of apoptosis, while the top right quadrants include Annexin V-FITC-positive/PI-positive cells in the late stages of apoptosis. Treatment with 50 µM cisplatin induced either early or late apoptosis in 786 RCC cells at the 4 h (k), 24 h (m), and 48 h (o) timepoints, and a greater number of late stage cells were observed. Treatment with 50 µM cisplatin also induced early and late apoptosis in HEK cells 24 h (u) and 48 h (x) after treatment, with the latter being more predominant. (A color version of this figure is available in the online journal.)
Caspase-3 and Bcl-2 activities
To study the mechanism(s) that mediate telmisartan-induced apoptosis, activation of caspase-3 and Bcl-2 activities were examined in telmisartan-treated cells using immunofluorescence microscopy. In Figure 5, representative images of 786 RCC cells and normal HEK cells treated with telmisartan and cisplatin treatment are shown. The absence of caspase-3 staining indicates that the death of HEK cells 24 h after treatment with telmisartan is not mediated by an apoptotic process (Figure 5(j)). Conversely, positive staining for caspase-3 that was observed for 786 cells following treatment with telmisartan indicates that the cell death induced is mediated by an apoptotic process (Figure 5(b) and (c)). Positive staining for caspase-3 with DAPI staining is also shown (Figure 5(b.1) and (b.2)).
Figure 5.


Detection of caspase-3 and Bcl-2 in 786 RCC cells. Cells were stained with DAPI (blue) and anti-Bcl-2 and anti-caspase-3 antibodies (green) to detect caspase-3 activation and Bcl-2 expression as described in the ‘Materials and methods’ section. Untreated 786 cells (a, e), and HEK control cells (i) are shown. Following exposure of 786 RCC cells to 100 µM and 200 µM telmisartan for 24 h and 48 h, positive staining for active caspase-3 (b, c) and negative staining for Bcl-2 were observed (f, g). In contrast, positive staining for caspase-3 was observed at the 24 h and 48 h timepoints following treatment with 50 µM cisplatin (d), while Bcl-2 expression remained negative (h). B.1 and C.1 show the staining of positive markers for caspase-3 without an overlay of DAPI staining. Exposure of HEK cells to dose of telmisartan (200 µM) for 24 h and 48 h did not activate caspase-3 (j). In panel (k), positive staining for caspase-3 is shown for HEK cells treated with 50 µM cisplatin for 24 h and 48 h. (A color version of this figure is available in the online journal.)
Negative staining for Bcl-2 was observed for 786 cells following treatment with telmisartan, and these results indicate that this treatment blocked the activity of Bcl-2 (Figure 5(f) and (g)). Furthermore, treatment of 786 cells with 50 µM cisplatin resulted in apoptosis that was accompanied by caspase-3 activation (Figure 5(d)) and down-regulation of Bcl-2 (Figure 5(h)). Similar results were obtained for HEK cells treated with cisplatin (Figure 5(k)).
Discussion
Internal stimuli, such as irreparable genetic damage, hypoxia, extremely high concentrations of cytosolic Ca2+, and severe oxidative stress, can trigger the intrinsic mitochondrial pathway of apoptosis.12 Moreover, regardless of stimuli, this pathway leads to increased mitochondrial permeability and the release of pro-apoptotic molecules, such as cytochrome c, into the cytoplasm.12 This pathway is closely regulated by a group of proteins belonging to the Bcl-2 family,13 which includes pro-apoptotic proteins (e.g. Bax, Bak, Bad, Bcl-Xs, Bid, Bik, Bim, and Hrk) and anti-apoptotic proteins (e.g. Bcl-2, Bcl-XL, Bcl-W, Bfl-1, and Mcl-1).14 While the latter regulate apoptosis by blocking the mitochondrial release of cytochrome c, the former promote the release of cytochrome c.15 Anti-apoptotic proteins have also been found to be up-regulated in a variety of tumor types.16 Both the release of cytochrome c into the cytoplasm during the early stages of apoptosis, prior to caspase activation, DNA fragmentation, and loss of membrane potential,9 and the blockade of anti-apoptotic Bcl-2 proteins integrated into the outer mitochondrial membrane, are mediated during the early stages of apoptosis.10
The execution phase of apoptosis involves the activation of a series of caspases. The upstream caspase for the intrinsic pathway is caspase 9, which can activate caspase-3. Caspase-3 then cleaves the inhibitor of the caspase-activated deoxyribonuclease which is responsible for nuclear apoptosis.17,18 Downstream caspases induce cleavage of protein kinases, cytoskeletal proteins, DNA repair proteins, and inhibitory subunits of endonuclease families. Correspondingly, they affect the cytoskeleton, cell cycle progression, and various signaling pathways, thereby mediating the morphological changes that accompany apoptosis.18 Events involving caspase activation and DNA fragmentation are also related to the late stages of apoptosis.19
In the present study, the pro-apoptotic activity of telmisartan was investigated using 786 RCC cells. It was observed that telmisartan significantly inhibited the growth of this cell line in a dose-dependent manner (Figure 1). Moreover, both 100 µM and 200 µM concentrations of telmisartan induced either early- or late-stage apoptosis within 24 h or 48 h of treatment (Figure 3). These results are consistent with previous studies.20 Telmisartan has also been shown to mediate potent early apoptotic effects against prostate cancer cells and urological cancer cells.21,22 In the present study, while treatment with 100 µM telmisartan did not induce apoptosis in normal HEK cells up to 24 h after treatment, a distinct increase in late stage apoptosis was observed by the 48 h timepoint. These results were further confirmed by an absence of caspase-3 labeling at the 24 h timepoint (Figure 5(i)), and were also consistent with previous results.22
Apoptosis was observed when levels of Bcl-2 family proteins decreased and caspase-3 was activated, both of which are well-characterized regulators of apoptosis. Accordingly, high levels of caspase-3 expression were detected in 786 RCC cells in the present study (Figure 5(b) and (c)), which corresponded with the late stages of apoptosis that were detected following treatment with telmisartan. Apoptotic activity has also been detected following the treatment of cells with other ARBs, and this was accompanied by increased expression of caspase-3.23 In particular, blockage of AT1R has been found to stimulate apoptosis in cancer cells.24 Furthermore, in a transgenic rat model of adenocarcinoma of prostate (TRAP), telmisartan and candesartan were found to attenuate prostate carcinogenesis by enhancing the apoptosis induced by the activation of various caspases.25
In the present study, expression of Bcl-2 in 786 RCC cells treated with 100 µM or 200 µM telmisartan decreased within 24 h, indicating that telmisartan induces early apoptosis by down-regulating the expression of Bcl-2 genes. Similarly, losartan, another type of ARB, was found to mediate a dose-dependent decrease in cell survival, an increase in levels of p53, p21, p27, and Bax, and a reduction in Bcl-2 and Bcl-xl levels in human pancreatic cells.26
Ang II is a vasoconstrictor that controls cardiovascular function and renal homeostasis. Recently, it has been shown that Ang II stimulates tumor growth and angiogenesis, especially in chorio-carcinomas,27 breast cancer,28 ovarian cancer,29 and pancreatic cancer.7 Taken together, these observations suggest that Ang II may have a role in cancer development. It has been shown that the major functions of Ang II in tumor growth and angiogenesis are mediated through AT1R.5,6 Moreover, it was recently demonstrated that stimulation of AT1R can trigger the activation of phosphatidylinositol 3 (PI3)-kinase and Akt,30 with the latter phosphorylating pro-apoptotic proteins including Bad, caspase-9, and forkhead transcription factors31–33 to inhibit apoptosis. Ang II has also been reported to significantly prevent cisplatin-induced apoptosis via activation of NF-κB and the subsequent production of anti-apoptotic molecules.34 In the present study, a greater number of cells treated with cisplatin were detected in the later stage of apoptosis than in the early stage of apoptosis in both cancer and normal cells (P < 0.001).
Drugs or treatment strategies that restore the normal function of apoptotic signaling pathways have the potential to eliminate cancer cells which otherwise depend on these defects for survival. While research is ongoing to identify new classes of anticancer drugs, ARBs are considered a significant anticancer, anti-angiogenesis, and anti-inflammatory therapeutic option.35–37
In conclusion, treatment of 786 RCC cells resulted in a significant increase in the number of cells undergoing apoptosis. Furthermore, this apoptosis was accompanied by down-regulation of Bcl-2 and activation of caspase-3-dependent signaling. Based on these results, telmisartan appears to represent a potential therapeutic candidate for the treatment of cancer.
Acknowledgement
The immunofluorescence method used was developed by the Department of Morphology, Universidade Federal do Rio Grande do Norte and the capture of images was performed at The Edmond and Lily Safra International Institute of Neuroscience of Natal, Brazil (ELS-IINN) by Pedro de Franca Cavalcanti who was funded by a scholarship (CNPq).
The authors thank the support given by International Institute of Neuroscience at Natal, RN, Brazil.
Author contributions
RFAJ and AAA participated in the design of the studies; RFAJ, ALSLO, RFMS, HAOR and PFC did the experimental study of cancer cells; RFAJ and AAA analyzed the statistical datas; RFAJ and AAA wrote and corrected the paper.
REFERENCES
- 1.Sourbier C, Massfelder T. Parathyroid hormone-related protein in human renal cell carcinoma. Cancer Lett 2006; 240(2): 170–82. [DOI] [PubMed] [Google Scholar]
- 2.Schrader AJ, Varga Z, Hegele A, Pfoertner S, Olbert P, Hofmann R. Second-line strategies for metastatic renal cell carcinoma: Classics and novel approaches. J Cancer Res Clin Oncol 2006; 132(3): 137–49. [DOI] [PubMed] [Google Scholar]
- 3.Tolle A, Jung M, Lein M, Johannsen M, Miller K, Moch H, Jung K, Kristiansen G. Brain-type and liver-type fatty acid-binding proteins: New tumor markers for renal cancer? BMC Cancer 2009; 9: 248–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Escudier B. Advanced renal cell carcinoma: Current and emerging management strategies. Drugs 2007; 67(9): 1257–64. [DOI] [PubMed] [Google Scholar]
- 5.Kosugi M, Miyajima A, Kikuchi E, Horiguchi Y, Murai M. Angiotensin II type 1 receptor antagonist candesartan as an angiogenic inhibitor in a xenograft model of bladder cancer. Clin Cancer Res 2006; 12(9): 2888–93. [DOI] [PubMed] [Google Scholar]
- 6.Fujimoto Y, Sasaki T, Tsuchida A, Chayama K. Angiotensin II type 1 receptor expression in human pancreatic cancer and growth inhibition by angiotensin II type 1 receptor antagonist. FEBS Lett 2001; 495(3): 197–200. [DOI] [PubMed] [Google Scholar]
- 7.Amaya K, Ohta T, Kitagawa H, Kayahara M, Takamura H, Fujimura T, Nishimura G, Shimizu K, Miwa K. Angiotensin II activates MAP kinase and NF-kappaB through angiotensin II type I receptor in human pancreatic cancer cells. Int J Oncol 2004; 25(4): 849–56. [PubMed] [Google Scholar]
- 8.Cardone MH, Roy N, Stennicke HR, Salvesen GS, Franke TF, Stanbridge E, Frisch S, Reed JC. Regulation of cell death protease caspase-9 by phosphorylation. Science 1998; 282(5392): 1318–21. [DOI] [PubMed] [Google Scholar]
- 9.George AJ, Thomas WG, Hannan RD. The renin-angiotensin system and cancer: Old dog, new tricks. Nat Rev Cancer 2010; 10(11): 745–59. [DOI] [PubMed] [Google Scholar]
- 10.Li H, Qi Y, Li C, Braseth LN, Gao Y, Shabashvili AE, Katovich MJ, Sumners C. Angiotensin type 2 receptor-mediated apoptosis of human prostate cancer cells. Mol Cancer Ther 2009; 8(12): 3255–65. [DOI] [PubMed] [Google Scholar]
- 11.Danial NN, Korsmeyer SJ. Cell death: Critical control points. Cell 2004; 116(2): 205–19. [DOI] [PubMed] [Google Scholar]
- 12.Tsujimoto Y, Finger LR, Yunis J, Nowell PC, Croce CM. Cloning of the chromosome breakpoint of neoplastic B cells with the t(14;18) chromosome translocation. Science 1984; 226(4678): 1097–9. [DOI] [PubMed] [Google Scholar]
- 13.Reed JC. Bcl-2 family proteins: Regulators of apoptosis and chemoresistance in hematologic malignancies. Semin Hematol 1997; 34(4): 9–19. [PubMed] [Google Scholar]
- 14.Kroemer G, Galluzzi L, Brenner C. Mitochondrial membrane permeabilization in cell death. Physiol Rev 2007; 87(1): 99–163. [DOI] [PubMed] [Google Scholar]
- 15.Kirkin V, Joos S, Zornig M. The role of Bcl-2 family members in tumorigenesis. Biochim Biophys Acta 2004; 1644(2–3): 229–49. [DOI] [PubMed] [Google Scholar]
- 16.Fan J, Zhang N, Yin GY, Zhang ZT, Cheng G, Qian WW, Long H, Cai W. Edaravone protects cortical neurons from apoptosis by inhibiting the translocation of BAX and increasing the interaction between 14-3-3 and p-BAD. Int J Neurosci 2012; 122(11): 665–74. [DOI] [PubMed] [Google Scholar]
- 17.Chipuk JE, Green DR. How do BCL-2 proteins induce mitochondrial outer membrane permeabilization? Trends Cell Biol 2008; 18(4): 157–64.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ghobrial IM, Witzig TE, Adjei AA. Targeting apoptosis pathways in cancer therapy. Cancer J Clin 2005; 55(3): 178–94. [DOI] [PubMed] [Google Scholar]
- 19.Elmore S. Apoptosis: A review of programmed cell death. Toxicol Pathol 2007; 35(4): 495–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Funao K, Matsuyama M, Naganuma T, Kawahito Y, Sano H, Nakatani T, Yoshimura R. The cysteiny lLT1 receptor in human renal cell carcinoma. Mol Med Rep 2008; 1(2): 185–9. [PubMed] [Google Scholar]
- 21.Funao K, Matsuyama M, Kawahito Y, Sano H, Chargui J, Touraine JL, Nakatani T, Yoshimura R. Telmisartan is a potent target for prevention and treatment in human prostate cancer. Oncol Rep 2008; 20(2): 295–300. [PubMed] [Google Scholar]
- 22.Matsuyama M, Funao K, Kuratsukuri K, Tanaka T, Kawahito Y, Sano H, Chargui J, Touraine JL, Yoshimura N, Yoshimura R. Telmisartan inhibits human urological cancer cell growth through early apoptosis. Exp Ther Med 2010; 1(2): 301–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Papp M, Li XP, Zhuang JJ, Wang RQ, Uhal BD. Angiotensin receptor subtype AT(1) mediates alveolar epithelial cell apoptosis in response to ANG II. Am J Physiol-Lung C 2002; 282(4): L713–L8. [DOI] [PubMed] [Google Scholar]
- 24.Arrieta O, Guevara P, Escobar E, Garcia-Navarrete R, Pineda B, Sotelo J. Blockage of angiotensin II type I receptor decreases the synthesis of growth factors and induces apoptosis in C6 cultured cells and C6 rat glioma. Br J Cancer 2005; 92(7): 1247–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Takahashi S, Uemura H, Seeni A, Tang MX, Komiya M, Long N, Ishiguro H, Kubota Y, Shirai T. Therapeutic targeting of angiotensin II receptor type 1 to regulate androgen receptor in prostate cancer. Prostate 2012; 72(14): 1559–72. [DOI] [PubMed] [Google Scholar]
- 26.Gong QK, Davis M, Chipitsyna G, Yeo CJ, Arafat HA. Blocking angiotensin II type 1 receptor triggers apoptotic cell death in human pancreatic cancer cells. Pancreas 2010; 39(5): 581–94. [DOI] [PubMed] [Google Scholar]
- 27.Ino K, Uehara C, Kikkawa F, Kajiyama H, Shibata K, Suzuki T, Khin EE, Ito M, Takeuchi M, Itakura A, Mizutani S. Enhancement of aminopeptidase A expression during angiotensin II-induced choriocarcinoma cell proliferation through AT(1) receptor involving protein kinase C- and mitogen-activated protein kinase-dependent signaling pathway. J Clin Endocr Metab 2003; 88(8): 3973–82. [DOI] [PubMed] [Google Scholar]
- 28.Muscella A, Greco S, Elia MG, Storelli C, Marsigliante S. Angiotensin II stimulation of Na+/K(+)ATPase activity and cell growth by calcium-independent pathway in MCF-7 breast cancer cells. J Endocrinol 2002; 173(2): 315–23. [DOI] [PubMed] [Google Scholar]
- 29.Suganuma T, Ino K, Shibata K, Kajiyama H, Nagasaka T, Mizutani S, Kikkawa F. Functional expression of the angiotensin II type 1 receptor in human ovarian carcinoma cells and its blockade therapy resulting in suppression of tumor invasion, angiogenesis, and peritoneal dissemination. Clin Cancer Res 2005; 11(7): 2686–94. [DOI] [PubMed] [Google Scholar]
- 30.Takahashi T, Taniguchi T, Konishi H, Kikkawa U, Ishikawa Y, Yokoyama M. Activation of Akt protein kinase B after stimulation with angiotensin II in vascular smooth muscle cells. Am J Physiol-Heart C 1999; 276(6): H1927–H34. [DOI] [PubMed] [Google Scholar]
- 31.Cardone M, Roy N, Stennicke H, Stanbridge E, Franke T, Salvesen G, Franke TF, Stanbridge SF, Reed JC. Regulation of cell death protease caspase-9 by Akt-mediated protein phosphorylation. Mol Biol Cell 1998; 9: 246–246. [Google Scholar]
- 32.Datta SR, Dudek H, Tao X, Masters S, Fu HA, Gotoh Y, Gotoh Y, Greenberg ME. Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell 1997; 91(2): 231–41. [DOI] [PubMed] [Google Scholar]
- 33.Brunet A, Bonni A, Zigmond MJ, Lin MZ, Juo P, Hu LS, Anderson MJ, Arden KC, Blenis J, Greenberg ME. Akt promotes cell survival by phosphorylating and inhibiting a forkhead transcription factor. Cell 1999; 96(6): 857–68. [DOI] [PubMed] [Google Scholar]
- 34.Zhao Y, Chen X, Cai L, Yang Y, Sui G, Wu J. Angiotensin II suppresses adriamycin-induced apoptosis through activation of phosphatidylinositol 3-kinase/Akt signaling in human breast cancer cells. Acta Biochim Biophys Sin 2008; 40(4): 304–10. [DOI] [PubMed] [Google Scholar]
- 35.Bangalore S, Kumar S, Kjeldsen SE, Makani H, Grossman E, Wetterslev J, Gupta AK, Sever PS, Gluud C, Messerli FH. Antihypertensive drugs and risk of cancer: Network meta-analyses and trial sequential analyses of 324,168 participants from randomised trials. Lancet Oncol 2011; 12(1): 65–82. [DOI] [PubMed] [Google Scholar]
- 36.Araujo AA, Lopes de Souza G, Souza TO, de Castro Brito GA, Saboia Aragao K, Xavier de Medeiros CA, Lourenço Y, Alves MSCF, Araújo RF., Jr Olmesartan decreases IL-1beta and TNF-alpha levels; downregulates MMP-2, MMP-9, COX-2, and RANKL; and upregulates OPG in experimental periodontitis. Naunyn Schmiedebergs Arch Pharmacol 2013; 386(10): 875–84. [DOI] [PubMed] [Google Scholar]
- 37.Araujo AA, Souza TO, Moura LM, Brito GA, Aragao KS, Araujo LS, Medeiros CA, Alves MS, Araújo RF., Jr Effect of telmisartan on levels of IL-1, TNF-alpha, down-regulated COX-2, MMP-2, MMP-9 and RANKL/RANK in an experimental periodontitis model. J Clin Periodontol 2013; 40(12): 1104–11. [DOI] [PMC free article] [PubMed] [Google Scholar]