

Abstract
Hypercholesterolemia impairs the quantity and function of endothelial progenitor cell. We hypothesized that glycogen synthase kinase 3β activity is involved in regulating biological function of endothelial progenitor cells in hypercholesterolemia microenvironment. For study, endothelial progenitor cells derived from apolipoprotein E-deficient mice fed with high-fat diet were used. Glycogen synthase kinase 3β activity was interfered with glycogen synthase kinase 3β inhibitor lithium chloride or transduced with replication defective adenovirus vector expressing catalytically inactive glycogen synthase kinase 3β (GSK3β-KM). Functions of endothelial progenitor cells, proliferation, migration, secretion and network formation of endothelial progenitor cells were assessed in vitro. The expression of phospho-glycogen synthase kinase 3β, β-catenin and cyclinD1 in endothelial progenitor cells was detected by Western blot. The in vivo function re-endothelialization and vasodilation were also analyzed by artery injury model transplanted with glycogen synthase kinase 3β-inhibited endothelial progenitor cells. We demonstrated that while the proliferation, migration, network formation as well as VEGF and NO secretion were impaired in apolipoprotein E-deficient endothelial progenitor cells, glycogen synthase kinase 3β inhibition significantly improved all these functions. Apolipoprotein E-deficient endothelial progenitor cells showed decreased phospho-glycogen synthase kinase 3β, β-catenin and cyclinD1 expression, whereas these signals were enhanced by glycogen synthase kinase 3β inhibition and accompanied with β-catenin nuclear translocation. Our in vivo model showed that glycogen synthase kinase 3β inhibition remarkably increased re-endothelial and vasodilation. Taken together, our data suggest that inhibition of glycogen synthase kinase 3β is associated with endothelial progenitor cell biological functions both in vitro and in vivo. It might be an important interference target in hypercholesterolemia microenvironment.
Keywords: Endothelial progenitor cell, glycogen synthase kinase, hypercholesterolemia, lithium, endothelium
Introduction
Endothelium injury is the initial factor of atherosclerosis and coronary artery disease (CAD). Delayed re-endothelialization accelerates the progression of coronary heart disease and restenosis after percutaneous coronary intervention. Recently, researchers have been attempting to repair damaged endothelium and improve endothelial function. Asahara et al.1 first found that circulating endothelial progenitor cells (EPCs) could differentiate into endothelial cells (ECs) and maintain normal endothelial function. Increasing evidences have shown that circulating EPCs derived from bone marrow are mobilized and home to the injury site and play an important role in re-endothelialization and neovascularization.2–6 However, insufficient number and impaired function of circulating EPCs in CAD patients become critical limitations for the therapeutic application of postnatal EPCs. Studies have shown that cardiovascular risk factors such as dyslipidemia, hypertension, diabetes mellitus, smoking and hyperhomocysteinemia are related with impaired EPCs.7–11 Therefore, researchers have been exploring possible mechanisms of regulation on impaired EPCs.
Glycogen synthase kinase 3β (GSK3β) is a serine/threonine kinase initially identified as the final enzyme involved in the glycogen synthesis metabolic pathway. Recently, the roles of GSK3β have expanded to regulation of cell proliferation, differentiation, apoptosis and signal transduction.12,13 As a crucial factor in many cellular signaling pathways, GSK-3β regulates several transcription factors in cell survival and function including β-catenin, heat shock factor-1 and cyclinD1.14 In resting cells, GSK3β is constitutively active and inactivated by phosphorylation of Ser9 residue in the N-terminal domain of GSK-3β. Inactivated GSK-3β leads to cytoplasmic accumulation of β-catenin and subsequent nuclear translocation on binding of Wnt to the frizzled receptor. In the nucleus, stabilized β-catenin forms complexes with members of the lymphoid enhancer factor/T-cell factor (LEF/TCF) family of transcription factors to activate gene expression.12,15
Recently, many studies have found that Wnt signaling pathway including GSK-3β and β-catenin is involved in regulation of the self-renewal and function of stem cell population.16–18 Hibbert19 and Choi20 further studied and reported that GSK3β was involved into regulating EPCs-mediated re-endothelialization and therapeutic angiogenesis. The influence and mechanism of hypercholesterolemia that is a crucial risk factor of CAD on EPC biological function need more attention. Accordingly, we performed the present study with apolipoprotein E-deficient (ApoE−/−) mice to evaluate the role of GSK3β on EPC biological function and elucidated some possible regulating mechanisms in hypercholesterolemia microenvironment.
Materials and methods
Animals
Six-week-old male homozygous ApoE−/− mice and their genetic background wild-type (WT) C57BL/6 mice were used in this study, which were fed in the same light- and temperature-controlled environment for 8 weeks. WT mice were fed on normal chow (0.5% total fat and 0.022% cholesterol). Homozygous ApoE−/− mice were fed with high-fat diet containing 1% cholesterol. Thirty ApoE−/− mice and 18 WT mice were used in the study. All of the animals were maintained in accordance with the National Institutes of Health Guidelines for the Care and Use of Laboratory Animals.
EPC culture and characterization
EPCs were cultured according to previously described techniques in our laboratory.21 Briefly, mononuclear cells derived from bone marrow were cultured in Dulbecco's minimum essential medium (DMEM) supplemented with vascular endothelial growth factor (VEGF, 50 ng/mL), basic fibroblast growth factor (5 ng/mL), Epidermal growth factor (EGF) (10 ng/mL) and cholesterol (300 mg/dL). Double-positive cells for acetylated low-density lipoprotein cholesterol (acLDL) uptake and Ulex Europeus Agglutinin-1 (UEA-1) binding were characterized as differentiating EPCs. Additionally, immunofluorescence assay was performed using rabbit polyclonal antibodies against CD31, endothelial nitric oxide synthase (eNOS) and Von Willebrand factor (VWF).
Inhibition of GSK3β activity
Inhibition of GSK3β activity was achieved using a GSK3β inhibitor lithium chloride (LiCl) or infected with replication defective adenovirus vector expressing catalytically inactive GSK3β (GSK3β-KM) where lysine residues at positions 85 and 86 were mutated to methionine and alanine. After 10 days cultured, EPCs were incubated with different concentrations of LiCl (5 mmol/L, 10 mmol/L, 20 mmol/L and 40 mmol/L) or transduced with GSK3β-KM (GSKi) or control adenoviral vector expressing green fluorescent protein (control-GFP). Effective gene transfer was confirmed by achieving more than 90% of green fluorescence-positive EPCs under fluorescent microscope.
Western blot analysis
Western blot analysis was performed as described previously.22 In brief, cultured cells proteins were separated by the electrophoresis on the Sodium dodecyl sulfate poly-acrylamide gel electrophoresis and transferred to the polyvinylidene fluoride (PVDF) membrane (Roche, Basel, Switzerland). The membrane was immunoblotted with the primary antibodies against phospho-ser9-GSK3β (pGSK3β, 1:1000 dilution; Cell Signaling Technology), β-catenin (1:1000 dilution; Cell Signaling Technology), cyclinD1 (1:500 dilution, Santa Cruz) or GAPDH, respectively, at 4℃ overnight. Protein bands were visualized by chemiluminescent detection (Amersham Biosciences) and quantified by gel image analysis system (Gel Doc XR, Bio-rad).
EPC proliferation assay
EPC proliferation was assessed by 3-{4,5-dimethylthiazol-2yl}-2,5-diphenyltetrazolium bromide (MTT) assays according to the protocol of the manufacturer. Briefly, EPCs were seeded onto 96-well plate and incubated with different concentrations of LiCl for 24 h. Alternatively, EPCs transduced with GSK3β-KM were reseeded onto 96-well plate and cultured for 24 h. Before the optical density measurement (490 nm) was performed, 15 µL MTT solution and 200 µL Dimethyl sulfoxide (DMSO) were successively added to each well. All groups of experiments were performed in quintuplicate.
EPC cell cycle study
Cell cycle studies were performed as described.23 EPCs were cultured for 24 h. Then cells were harvested, fixed and permeabilized with 70% ice-cold ethanol for 30 min, washed, resuspended and incubated in fluorescence-activated cell sorting (FACS) buffer containing 5 mg/mL propidium iodide, 100 mmol/L sodium citrate, pH 7.3 and 0.05 mg of RNase A for 30 min at 4℃. Cell fluorescence was measured by FACS. All groups of experiments were performed in quintuplicate.
EPC migration assay
EPC migration was evaluated by using a modified Boyden’s chamber assay. EPCs (1 × 105) suspended in 100 µL serum-free DMEM were reseeded in the upper chamber. The lower chamber contained endothelial-conditioned medium. After 6 h incubation, the lower side of the filter was washed with PBS and fixed with 2% paraformaldehyde. EPCs were stained with hematoxylin stain. Migration activity was evaluated as the mean number of migrated cells in random high-power fields. All groups of experiments were performed in quintuplicate.
Secretion of VEGF and nitric oxide assay
To measure the secretion of VEGF and nitric oxide (NO) by EPCs, cells were cultured with the growth factor-free basal medium for 48 h. EPCs supernatants were collected, centrifuged and assayed. The secretion of VEGF was measured by VEGF ELISA kit (R&D Systems) and NO level was assayed by the method of nitrate reductase, according to the manufacturer’s instructions. Samples were measured in quintuplicate.
Analysis of β-catenin nuclear translocation
The subcellular localization of β-catenin was analyzed as previously mentioned.24 EPCs were washed in Phosphate buffered solution (PBS), fixed with methanol for 30 min at −20℃, and washed with ice-chilled T-PBS. Anti-β-catenin antibody was used (1:100 dilution; Cell Signaling Technology). Nuclei were considered β-catenin-positive if staining intensity was greater than that seen in the cytoplasm.
Network formation assay
Matrigel network formation assay was performed.25 According to the manufacturer’s instructions, 24-well plate was coated with Matrigel (BD Biosciences) at 37℃ for 1 h to allow solidification. EPCs with or without treatment were seeded on the matrigel. After incubation at 37℃ for 6 h, network formation was observed by inverted phase contrast microscope. The degree of network formation was quantified by computer-assisted analysis using Image pro plus (Media cybernetics).
Model of carotid artery injury and re-endothelialization assay
To evaluate the role of GSK3β inhibition on EPC re-endothelialization capability, the model of carotid artery injury was performed as described previously.19 Briefly, a 32-gauge blunt needle was inserted through arterial incision and passed three times for endothelium damage. EPCs (1 × 106) of untreated (WT group/ApoE−/− group), LiCl treated (LiCl group) or GSK3β-KM transduced (GSKi group) were injected intravenously. An equivalent volume of saline was injected into un-transplanted mice (non-transfused group). Re-endothelialization was assessed by Evans blue staining via tail vein injection. The total area and re-endothelialization area were measured by computer-assisted morphometric analysis 96 h after EPC transplantation. All groups of experiments were performed in triplicate.
Endothelium-dependent vasodilatation function analysis
Endothelium-dependent vasodilatation (EDVD) was analyzed as previously mentioned.21 Briefly, carotid arteries were acquired and cut into about 3-mm vascular rings after 96 h EPC transplantation. Vascular rings were dipped into ice-cold Krebs-Henseleit (K-H) solution and hung with two micropipettes in a cold K-H solution perfusion chamber. Initial pre-tension was set as 1.0 g. Segments were bathed at 37℃ with 95% oxygen and 5% carbon dioxide for 2 h. After tension curve of vascular ring was equilibrated, EDVD in response to acetylcholine was measured. All groups of experiments were performed in triplicate.
Statistical analysis
Data from independent experiments were presented as mean ± standard error of mean from at least three experiments. Significance of the difference between two groups was determined by unpaired Student’s t-test, and multiple comparisons were evaluated by one-way analysis of variance. A probability value < 0.05 was considered statistically significant.
Results
EPC functions were impaired in ApoE−/− mice
After 3–4 days culture, bone marrow-derived EPCs appeared to be spindle-shaped and gradually proliferated. These cells took up acLDL and showed UEA-1 binding. Immunofluorescence confirmed that the cultured EPCs expressed endothelial phenotypes CD31, eNOS and VWF (data not shown). We found that the proliferation and migration of EPC were obviously impaired in ApoE−/− mice in comparison with WT mice (Figure 1(a) and (b), P < 0.01, n = 5). In addition, the secretion of angiogenic cytokines from EPCs in ApoE−/− mice was damaged. The concentrations of NO and VEGF were notably lower in the supernatant of ApoE−/− EPCs than that of WT EPCs (Figure 1(c) and (d), P < 0.01, n = 5).
Figure 1.

EPC functions including proliferation, migration and secretion were obviously impaired in ApoE−/− mice. (a, b) EPC proliferation and migration was significantly lower in ApoE−/− mice than that in WT mice. (c, d) Secretion of NO and VEGF from EPC was damaged in ApoE−/− mice in comparison with WT EPCs. *P < 0.01 versus WT group, n = 5
GSK3β inhibition improved EPC proliferation in ApoE−/− mice
To confirm the effects of GSK3β inhibitor on EPC proliferation in ApoE−/− mice, EPCs were pretreated with different doses of LiCl. Compared with control group (ApoE−/− EPCs used as controls), EPC proliferation was significantly enhanced in a dose-dependent manner (Figure 2(a), P < 0.01, n = 5). However, EPCs were obviously reduced and dead at 40 mmol/L of LiCl. In order to observe the role of a catalytically inactive GSK3β on EPCs proliferation, ApoE−/− EPCs were transduced with GSK3β-KM. Similar to LiCl, transduction of GSK3β-KM increased EPCs proliferation. There was no significant difference between GSKi group and LiCl group (20 mmol/L). Moreover, combination treatment with LiCl (20 mmol/L) and GSK3β-KM transduction could not further improve EPC proliferation (Figure 2(b), P > 0.05, n = 5). FACS analyses revealed that ApoE−/− EPCs treated with LiCl or GSK3β-KM transduction had a greater accumulation in the S phase than control group (Figure 2(c), n = 5).
Figure 2.

GSK3β inhibition improved EPCs proliferation in ApoE−/− mice. (a) LiCl promoted ApoE−/− EPCs proliferation in a dose-dependent manner. (b) Compared with control-GFP group, the proliferation of ApoE−/− EPCs was increased significantly in GSK3β-KM group. (c) FACS analysis showed that ApoE−/− EPCs treated with GSK3β inhibition had a greater accumulation in the S phase than control group. *P < 0.01, versus control group, n = 5
GSK3β inhibition improved ApoE−/− EPC migration
The effect of GSK3β inhibition on EPC migration was measured by using in vitro migration assay. Figure 3(a) showed that treatment with LiCl resulted in a progressive increase in migration in a dose-dependent manner (P < 0.01, n = 5). Figure 3(b) displayed that EPC migration in GSKi group was notably greater than that in control-GFP group (P < 0.01, n = 5). However, there was no difference among drug (LiCl 20 mmol/L), gene transfer (GSK3β-KM transduction) and combination treatment in EPC migration (Figure 3(b), P > 0.05, n = 5).
Figure 3.

GSK3β inhibition enhanced EPC migration in ApoE−/− mice. (a). LiCl accelerated ApoE−/− EPCs migration in vitro in a dose-dependent manner. (b) Compared with control-GFP group, EPC migration was increased significantly in GSK3β-KM group. Combination treatment with LiCl (20 mmol/L) and GSK3β-KM transduction had no predominant effect on EPC migration compared with only drug pretreatment or gene transfer. *P < 0.01,versus control, n = 5
GSK3β inhibition increased VEGF and NO secretion from ApoE−/− EPC
We investigated the effect of inhibiting GSK3β activity on the secretion of angiogenic cytokines in ApoE−/− EPCs. Compared with control group, the concentrations of NO and VEGF were significantly higher in the supernatant of ApoE−/− EPCs with LiCl treatment (Figure 4(a) and (b), P < 0.05, n = 5). Similar results were obtained from the treatment with GSK3β-KM transduction (Figure 5(c) and (d), n = 5). Dual treatment with LiCl (20 mmol/L) and GSK3β-KM transduction did not synergistically improve secretion of NO and VEGF from ApoE−/− EPCs (P > 0.05, n = 5).
Figure 4.

GSK3β inhibition increased VEGF and NO secretion from ApoE−/− EPCs. (a and b) LiCl increased the secretion of NO and VEGF in a dose-dependent manner. (c and d) GSK3β-KM gene transfer augmented the NO and VEGF secretion from ApoE−/− EPCs in comparison with control group. Dual treatment with LiCl (20 mmol/L) and GSK3β-KM transduction did not synergistically improve the secretion of angiogenic cytokines from ApoE−/− EPCs. #P < 0.05, *P < 0.01, versus control, n = 5
Figure 5.

GSK3β inhibition enhanced network formation in vitro. Matrigel network formation assay was performed to study the role of GSK3β inhibition on the angiogenic function of EPCs. ApoE−/− EPCs had little ability of the network formation. EPCs in WT mice showed more angiogenic function than that in ApoE−/− mice. GSK3β-KM gene transfer or LiCl pretreatment significantly augmented the network formation of ApoE−/− EPCs. *P < 0.01 versus control, n = 3
GSK3β inhibition improved network formation of AopE−/− EPCs in vitro
For evaluating the ability of EPCs to form tubule-like structure, matrigel network formation assay was performed. In comparison with WT mice, the network formation ability of EPCs was significantly impaired in ApoE−/− mice (Figure 5, P < 0.01, n = 3). It is obvious that inhibition of GSK3β activity increases network formation of ApoE−/− EPCs in vitro. Compared with ApoE−/− group, the ability of network formation was increased remarkably in LiCl (20 mmol/L) group and GSK3β-KM group (Figure 6, P < 0.01, n = 3).
Figure 6.

GSK3β inactivation plays an important effect in GSK3β signaling pathway in EPCs. (a) The expression of pGSK3β, β-catenin and cyclinD1 was lower in ApoE−/− EPCs than that in WT EPCs. LiCl treatment or GSK3β-KM transduction significantly increased the expression of pGSK3β, β-catenin and cyclinD1 in ApoE−/− EPCs. (b) GSK3β inhibition promoted nuclear accumulation of β-catenin. ApoE−/− EPCs have the same nuclear localization of β-catenin as WT EPCs. LiCl treatment or GSK3β-KM transduction increased significantly the nuclear accumulation of β-catenin. #P < 0.05, * P < 0.01, versus ApoE−/− group, n = 3. (A color version of this figure is available in the online journal.)
Effect of GSK3β inhibition on GSK3β signaling pathway in ApoE−/− EPCs
In order to investigate the regulation of GSK3β signaling pathway on EPCs, pGSK3β, β-catenin and cyclin D1 were measured by Western blot. Western blot analysis revealed that the expression of pGSK3β, β-catenin and cyclinD1 in ApoE−/− EPCs was lower than that in WT EPCs (P < 0.01, n = 3). GSK3β inhibition significantly increased the expression of pGSK3β (LiCl, 2.92-fold; GSK3β-KM, 3.06-fold), β-catenin (LiCl, 2.04-fold; GSK3β-KM, 2.15-fold) and cyclinD1 (LiCl, 2.16-fold; GSK3β-KM 2.41-fold) (Figure 6(a), n = 3). To study further the nuclear translocation of β-catenin at the subcellular level, β-catenin immunofluorescence staining was performed. Nuclear localization of β-catenin had no prominence in ApoE−/− EPC or WT EPC. LiCl treatment or GSK3β-KM transduction increased significantly the nuclear translocation of β-catenin in ApoE−/− EPCs (Figure 6(b), n = 3).
GSK3β inhibition accelerated functional re-endothelialization of EPCs in ApoE−/− mice
Endothelial generation is an important step in the repair process of a denudated endothelium. We determined that capacity of EPCs transplantation on re-endothelialization of injured artery. Computer-based morphometric analysis showed that AopE−/− EPCs transplantation increased re-endothelialization area in comparison with non-transfused group. However, the re-endothelialization capacity of EPCs in AopE−/− group was obviously lower than that in WT group. Inhibition of GSK3β activity improved impaired capability of EPCs from AopE−/− mice on endothelial regeneration (Figure 7(a), P < 0.05, n = 3). There was no obvious difference in re-endothelialization between LiCl group and GSKi group (Figure 7(a), P > 0.05, n = 3). The recovery of endothelial function is considered as important as endothelial generation. We further assessed whether GSK3β inhibition ameliorated endothelial dysfunction of damaged vessels during the process of re-endothelialization. Acetylcholine-mediated EDVD was lower in ApoE−/− group than that in WT group. GSK3β inhibition increased the capability of AopE−/− EPCs on restore of endothelial function (Figure 7(b), P < 0.05, n = 3). There was no statistical difference between LiCl group and GSKi group.
Figure 7.
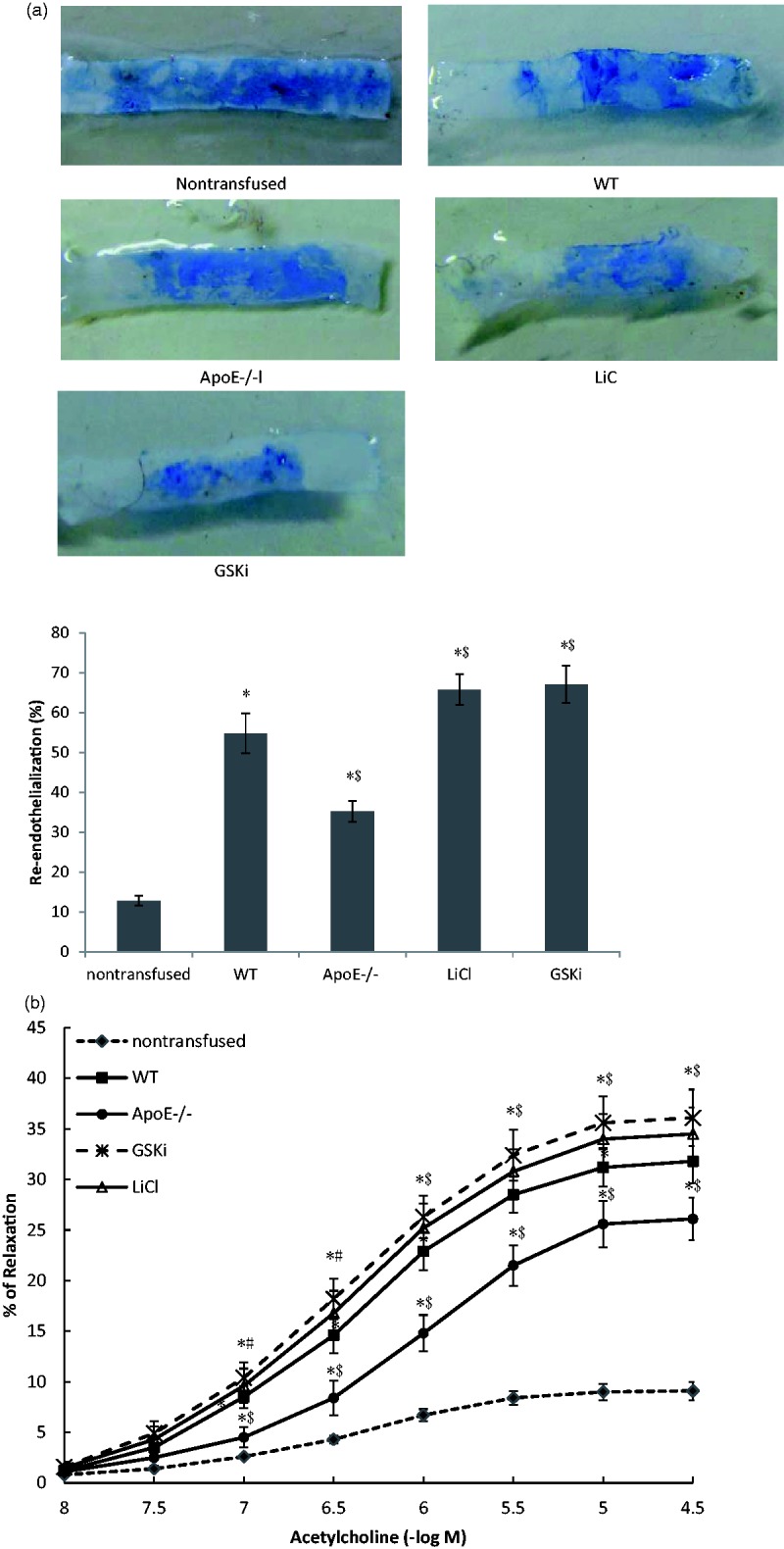
GSK3β inhibition accelerated functional re-endothelialization of EPCs in ApoE−/− mice. (a) Re-endothelialization area of injured arteries was measured by even’s blue dye after 96 h after carotid denudation and EPCs transplantation. (b) The effect of GSK3β inhibition on the recovery of endothelial function was measured by endothelium-dependent vasodilation (EDVD). *P < 0.01, versus non-transfused group; # P < 0.05, $P < 0.01, versus WT group; n = 3. (A color version of this figure is available in the online journal.)
Discussion
Hypercholesterolemia is a critical pathogenic factor of cardiovascular disease. Lipid accumulation within arterial wall participates in atherogenesis. Endothelial injury and dysfunction is an initial step in the progression of atherosclerosis.26 The recovery of damaged endothelium as soon as possible will help delay the development of CAD. EPCs have EC features and play an important role for the maintenance of vascular wall homeostasis. Recently, studies showed that dyslipidemia affected EPC survival and function. Low-density lipoprotein cholesterol was associated with EPC dysfunction,27 while increased high-density lipoprotein cholesterol enhanced EPC function and inhibited neointima formation.28 Therefore, the possible mechanism of hypercholesterolemia microenvironment on EPC function has been further investigating.
In the current study, we observed that EPC functions including proliferation, migration and network formation were obviously impaired in ApoE−/− mice in comparison with WT mice. The biological dysfunctions might weaken the beneficial effect of EPCs on endothelium repair. In further study, we found that cholesterol decreased the expression of pGSK3β protein in ApoE−/− EPCs. We conjectured that GSK3β activation might be involved with impaired functions of EPC in hypercholesterolemia microenvironment. Recently, GSK3β has been shown to play pleiotropic roles in cell proliferation, apoptosis and migration. Thus, we investigated the role of GSK3β inhibition in the regulation of EPC functions in ApoE−/− mice.
In the present study, GSK3β inhibition improved significantly EPC functions including proliferation, migration and network formation. GSK3β inhibition increased obviously phospho-GSK3β, β-catenin and cyclinD1 expression and β-catenin nuclear translocation. Stabilized β-catenin forms complexes with LEF/TCF to activate gene expression. CyclinD1, a downstream regulator of GSK3β signaling pathway, regulates the propagation of the cell cycle from the G1 phase to the S phase.29 The increased expression of cyclinD1 might contribute to ApoE−/− EPCs proliferation. Clinical studies suggest that the functional character of EPCs is as important as their abundance.30 The enhanced abilities of migration and network formation facilitated EPCs to participate in the process of re-endothelialization for cardiovascular disease.
Interestingly, we observed that the maximal beneficial effect of GSK3β inhibitor was similar to GSK3β-KM gene transfer. Gene transfection neither further augment the helpful role of GSK3β inhibitor on EPCs functions. Hibbert19 reported that combination treatment with different GSK3β inhibitor was unable to improve further EPCs yield. The maximal effect with one or the other treatment did not alter in a synergistic manner due to an off-target effect of one of the drugs. We found that this phenomenon existed not only between different drugs but also between gene transduction and drugs. Therefore, we speculated that drug treatment might play the beneficial role on EPCs functions instead of gene transfection while avoiding transgenic side effect and ethical issues. The above phenomenon might provide a new therapeutic idea for improving EPCs dysfunction in clinic pharmacology.
The endothelium plays a protective effect on the vascular homeostasis. Denude endothelium leads to vasomotor disorder, platelet aggregation, vascular smooth muscle cells abnormal proliferation and migration. Rapid re-endothelialization inhibits effectively neointimal hyperplasia and stent thrombosis after percutaneous coronary intervention. Our previous study showed that EPC transplantation accelerates the process of injured endothelial repair. In this study, EPC re-endothelialization was obviously impaired in hypercholesterolemia microenvironment. However, inhibition of GSK3β activity in ApoE−/− EPCs facilitated to restore intact endothelium as soon as possible. Unlike the previous studies, we valued the functional improvement more than morphological restoration after EPC transplantation. Further study showed that GSK3β inhibition improved acetylcholine-induced EDVD of damaged artery in hypercholesterolemia microenvironment. We speculated that rapidly functional re-endothelialization depends not only on the amount and functions of EPCs but also on the interactions between EPCs and their microenvironment.
EPCs secrete various cytokines and growth factors, which regulate EPCs biological functions and exert effects on their surrounding microenvironment through autocrine or paracrine manner.31 VEGF,32 hepatocyte growth factor,33 stromal-derived factor-134 and NO5 play important roles on EPCs in re-endothelialization. We observed that AopE−/− mice have impaired secretion from EPCs. Compared with WT mice, the concentrations of VEGF and NO were significantly reduced in the supernatant of ApoE−/− EPCs. Further studies showed that GSK3β inhibition enhanced VEGF and NO secretion by AopE−/− EPCs. As an endothelial growth factor, VEGF has been verified to stimulate EPCs and ECs proliferation and migration. In addition, VEGF up-regulates the expression of eNOS and stimulates EPCs to secrete NO. NO plays important roles in regulating vascular tone, cell proliferation and adhesion. We previously reported that NO enhances EPCs partial biological functions.21 In the present study, NO secreted by EPCs might participate into regulating EDVD. EPCs that incorporated into the vascular injury site influence surrounding cells growth status and regulate vasomotion. Therefore, we speculated that GSK3β inhibition augmented the functional re-endothelialization EPCs in AopE−/− mice partly through autocrine/paracrine manner.
Conclusion
This study provided novel insights into the potential importance of GSK3β signaling for the regulation of EPC functions and vascular homeostasis in hypercholesterolemia microenvironment. GSK3β inhibition effectively enhanced EPC biological functions in vitro and increased re-endothelial and vasodilation in vivo. Further studies are ongoing to elucidate the complex mechanism and beneficial potential of GSK3β signaling pathway in the process of functional re-endothelialization.
ACKNOWLEDGEMENTS
We are grateful to Professor Zhang JH for critical reading of the manuscript. This work was supported by the National Natural Science Foundation of China (No. 81100149; No. 81000134; No. 81100110).
Author contributions
Each author has participated sufficiently in the work to take public responsibility for appropriate portions of the content. BC has made a substantial contribution to the concept and design, acquisition, analysis and drafted the manuscript. JJ has made a substantial contribution to the concept and design and revised the manuscript. XD has made a substantial contribution to analysis of data. MD, MS and YY have made a substantial contribution to acquisition and analysis of data. SY, XZ and JC have made a substantial contribution to analysis and interpretation of data. LH has made a substantial contribution to the concept and design and revised critically the manuscript. All the authors approved the version to be published.
Conflict of interest
We declare that we have no financial and personal relationships with other people or organizations that can inappropriately influence our works. There is no professional or other personal interest of any nature or kind in any product, service and/or company that could be construed influencing the position presented in.
References
- 1.Asahara T, Murohara T, Sullivan A, Silver M, van-der-Zee R, Li T, Witzenbichler B, Schatteman G, Isner JM. Isolation of putative endothelial progenitor cells for angiogenesis. Science 1997; 275: 964–67. [DOI] [PubMed] [Google Scholar]
- 2.Kawamoto A, Gwon HC, Iwaguro H, Yamaguchi JI, Uchida S, Masuda H, Silver M, Ma H, Kearney M, Isner JM, Asahara T. Therapeutic potential of ex vivo expanded endothelial progenitor cells for myocardial ischemia. Circulation 2001; 103: 634–37. [DOI] [PubMed] [Google Scholar]
- 3.Burlacu A, Grigorescu G, Rosca AM, Preda MB, Simionescu M. Factors secreted by mesenchymal stem cells and endothelial progenitor cells have complementary effects on angiogenesis in vitro. Stem Cells Dev 2013; 22: 643–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chen L, Wu F, Xia WH, Zhang YY, Xu SY, Cheng F, Liu X, Zhang XY, Wang SM, Tao J. CXCR4 gene transfer contributes to in vivo reendothelialization capacity of endothelial progenitor cells. Cardiovasc Res 2010; 88: 462–70. [DOI] [PubMed] [Google Scholar]
- 5.Kong D, Melo LG, Mangi AA, Zhang L, Lopez-Ilasaca M, Perrella MA, Liew CC, Pratt RE, Dzau VJ. Enhanced inhibition of neointimal hyperplasia by genetically engineered endothelial progenitor cells. Circulation 2004; 13: 1769–75. [DOI] [PubMed] [Google Scholar]
- 6.Murohara T, Ikeda H, Duan J, Shintani S, Saski K, Eguchi H, Onitsuka K, Matsui K, Imaizumi T. Transplanted cord blood-derived endothelial precursor cells augment postnatal neovascularization. J Clin Invest 2000; 105: 1527–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hill JM, Zaloss G, Halcox JP, SchenkeWH, Waclawiw MA, Quyyumi AA, Finkel T. Circulating endothelial progenitor cells, vascular function, and cardiovascular risk. N Engl J Med 2003; 348: 593–600. [DOI] [PubMed] [Google Scholar]
- 8.Jialal I, Devaraj S, Singh U, Huet BA. Decreased number and impaired functionality of endothelial progenitor cells in subjects with metabolic syndrome: implications for increased cardiovascular risk. Atherosclerosis 2010; 211: 297–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kim S, Kim NH, Kim YK, Yoo JH, Shin SN, Ko JS, Kim YK, Rhee SJ, Yun KH, Lee EM, Yoo NJ, Oh SK, Jeong JW. The number of endothelial progenitor cells is decreased in patients with non-dipper hypertension. Korean Circ J 2012; 42: 329–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vasa M, Fichtlscherer S, Aicher A, Adler K, Urbich C, Martin H, Zeiher AM, Dimmeler S. Number and migratory activity of circulating endothelial progenitor cells inversely correlate with risk factors for coronary artery disease. Circ Res 2001; 89: e1–7. [DOI] [PubMed] [Google Scholar]
- 11.Tepper OM, Galiano RD, Capla JM, Kalka C, Gagne PJ, Jacobowitz GR, Levine JP, Gurtner GC. Human endothelial progenitor cells from type II diabetics exhibit impaired proliferation, adhension, and incorportation into vascular structures. Circulation 2002; 106: 2781–6. [DOI] [PubMed] [Google Scholar]
- 12.Gregory MA, Qi Y, Hann SR. Phosphorylation by glycogen synthase kinase-3 controls c-myc proteolysis and subnuclear localization. J Biol Chem 2003; 278: 51606–12. [DOI] [PubMed] [Google Scholar]
- 13.Shin SH, Lee EJ, Chun J, Hyun S, Kim YI, Kang SS. The nuclear localization of glycogen synthase kinase 3β is required its putative PY-nuclear localization sequences. Mol Cells 2012; 34: 375–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Park KW, Yang HM, Youn SW, Yang HJ, Chae IH, Oh BH, Lee MM, Park YB, Choi YS, Kim HS, Walsh K. Constitutively active glycogen synthase kinase-3β gene transfer sustains apoptosis, inhibits proliferation of vascular smooth muscle cells, and reduces neointima formation after balloon injury in rats. Arterioscler Thromb Vasc Biol 2003; 23: 1364–9. [DOI] [PubMed] [Google Scholar]
- 15.Diehl JA, Cheng M, Roussel MF, Sherr CJ. Glycogen synthase kinase-3β regulates cyclin D1 proteolysis and subcellular localization. Genes Dev 1998; 12: 3499–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Etheridge SL, Spencer GJ, Heath DJ, Genever PG. Expression profiling and functional analysis of Wnt signaling mechanisms in mesenchymal stem cells. Stem Cells 2004; 22: 849–60. [DOI] [PubMed] [Google Scholar]
- 17.Umehara H, Kimura T, Ohtsuka S, Nakamura T, Kitajima K, Ikawa M, Okabe M, Niwa H, Nakano T. Efficient derivation of embryonic stem cells by inhibition of glycogen synthase kinase-3. Stem Cells 2007; 25: 2705–11. [DOI] [PubMed] [Google Scholar]
- 18.Bechard M, Dalton S. Subcellular localization of glycogen synthase kinase 3beta controls embryonic stem cell self-renewal. Mol Cell Biol 2009; 29: 2092–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hibbert B, Ma X, Pourdjabbar A, Holm E, Rayner K, Chen YX, Sun J, Filion L, O’Brien ER. Inhibition of endothelial progenitor cell glycogen synthase kinase-3β results in attenuated neointima formation and enhanced re-endothelialization after arterial injury. Cardiovasc Res 2009; 83: 16–23. [DOI] [PubMed] [Google Scholar]
- 20.Choi JH, Hur J, Yoon CH, Kim JH, Lee CS, Youn SW, Oh IY, Skurk C, Murohara T, Park YB, Walsh K, Kim HS. Augmentation of therapeutic angiogenesis using genetically modified human endothelial progenitor cells with altered glycogen synthase kinase-3β activity. J Biol Chem 2004; 27: 49430–8. [DOI] [PubMed] [Google Scholar]
- 21.Cui B, Huang L, Fang Y, Guo R, Yin Y, Zhao X. Transplantation of endothelial progenitor cells overexpressing endothelial nitric oxide synthase enhances inhibition of neointimal hyperplasia and restores endothelium-dependent vasodilatation. Microvasc Res 2011; 81: 143–50. [DOI] [PubMed] [Google Scholar]
- 22.Woulfe KC, Gao E, Lal H, Harris D, Fan Q, Vagnozzi R, DeCaul M, Shang X, Patel S, Woodgett JR, Force T, Zhou J. Glycogen synthase kinase-3β regulates post–myocardial infarction remodeling and stress-induced cardiomyocyte proliferation in vivo. Circ Res 2010; 106: 1635–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wegiel B, Gallo DJ, Raman KG, Karlsson JM, Ozanich B, Chin BY, Tzeng E, Ahmad S, Ahmed A, Baty CJ, Otterbein LE. Nitric oxide–dependent bone marrow progenitor mobilization by carbon monoxide enhances endothelial repair after vascular injury. Circulation 2010; 121: 537–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wong MM, Winkler B, Karamariti E, Wang X, Yu B, Simpson R, Chen T, Margariti A, Xu Q. Sirolimus stimulates vascular stem/progenitor cell migration and differentiation into smooth muscle cells via epidermal growth factor receptor/extracellular signal–regulated kinase/β-catenin signaling pathway. Arterioscler Thromb Vasc Biol 2013; 33: 2397–406. [DOI] [PubMed] [Google Scholar]
- 25.Di Santo S, Seiler S, Fuchs AL, Staudigl J, Widmer HR. The secretome of endothelial progenitor cells promotes brain endothelial cell activity through PI3-kinase and MAP-kinase. PLoS One 2014; 9: e95731–e95731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Perrault LP, Mahlberg F, Breugnot C, Bidouard JP, Villeneuve N, Vilaine JP, Vanhoutte PM. Hypercholesterolemia increases coronary endothelial dysfunction, lipid content, and accelerated atherosclerosis after heart transplantation. Arterioscler Thromb Vasc Biol 2000; 20: 728–36. [DOI] [PubMed] [Google Scholar]
- 27.Yu X, Song M, Chen J, Zhu G, Zhao G, Wang H, Hunag L. Hepatocyte growth factor protects endothelial progenitor cell from damage of low-density lipoprotein cholesterol via the PI3K/Akt signaling pathway. Mol Biol Rep 2010; 37: 2423–9. [DOI] [PubMed] [Google Scholar]
- 28.Feng Y, Jacobs F, Van Craeyveld E, Brunaud C, Snoeys J, Tjwa M, Van Linthout S, De Geest B. Human apoA-I transfer attenuates transplant arteriosclerosis via enhanced incorporation of bone marrow–derived endothelial progenitor cells. Arterioscler Thromb Vasc Biol 2008; 28: 278–83. [DOI] [PubMed] [Google Scholar]
- 29.Kim KI, Cho HJ, Hahn JY, Kim TY, Park KW, Koo BK, Shin CS, Kim CH, Oh BH, Lee MM, Parkand YB, Kim HS. β-catenin overexpression augments angiogenesis and skeletal muscle regeneration through dual mechanism of vascular endothelial growth factor -mediated endothelial cell proliferation and progenitor cell mobilization. Arterioscler Thromb Vasc Biol 2006; 26: 91–8. [DOI] [PubMed] [Google Scholar]
- 30.Seeger FH, Tonn T, Krzossok N, Zeiher AM, Dimmeler S. Cell isolation proceduresmatter: a comparison of different isolation protocols of bone marrow mononuclear cells used for cell therapy in patients with acute myocardial infarction. Eur Heart J 2007; 28: 766–72. [DOI] [PubMed] [Google Scholar]
- 31.Im W, Kim M. Cell therapy strategies vs. paracrine effect in Huntington’s disease. J Mov Disord 2014; 7: 1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Iwaguro H, Yamaguchi JI, Kalka C, Murasawa S, Masuda H, Hayashi SI, Silver M, Li T, Isner JM, Asahara T. Endothelial progenitor cell vascular endothelial growth factor gene transfer for vascular regeneration. Circulation 2002; 105: 732–8. [DOI] [PubMed] [Google Scholar]
- 33.Jiang M, Wang B, Wang C, He B, Fan H, Guo TB, Shao Q, Gao L, Liu Y. Angiogenesis by transplantation of HIF-1 alpha modified EPCs into ischemic limbs. J Cell Biochem 2008; 103: 321–34. [DOI] [PubMed] [Google Scholar]
- 34.Yamaguchi J, Kusano KF, Masuo O, Kawamoto A, Silver M, Murasawa S, Bosch-Marce M, Masuda H, Losordo DW, Isner JM, Asahara T. Stromal cell-derived factor-1 effects on ex vivo expanded endothelial progenitor cell recruitment for ischemic neovascularization. Circulation 2003; 107: 1322–8. [DOI] [PubMed] [Google Scholar]