

Abstract
Our recent studies indicate that the transient receptor potential vanilloid type 1 (TRPV1) channel may act as a potential regulator of monocyte/macrophage recruitment to reduce renal injury in salt-sensitive hypertension. This study tests the hypothesis that deletion of TRPV1 exaggerates salt-sensitive hypertension-induced renal injury due to enhanced inflammatory responses via monocyte chemoattractant protein-1 (MCP-1)/C-C chemokine receptor 2 (CCR2)-dependent pathways. Wild type (WT) and TRPV1-null mutant (TRPV1−/−) mice were subjected to uninephrectomy and deoxycorticosterone acetate (DOCA)-salt treatment for four weeks with or without the selective CCR2 antagonist, RS504393. DOCA-salt treatment increased systolic blood pressure (SBP) to the same degree in both strains, but increased urinary excretion of albumin and 8-isoprostane and decreased creatinine clearance with greater magnitude in TRPV1−/− mice compared to WT mice. DOCA-salt treatment also caused renal glomerulosclerosis, tubulointerstitial injury, collagen deposition, monocyte/macrophage infiltration, proinflammatory cytokine and chemokine production, and NF-κB activation in greater degree in TRPV1−/− mice compared to WT mice. Blockade of the CCR2 with RS504393 (4 mg/kg/day) had no effect on SBP in DOCA-salt-treated WT or TRPV1−/− mice compared to their respective controls. However, treatment with RS504393 ameliorated renal dysfunction and morphological damage, and prevented the increase in monocyte/macrophage infiltration, cytokine/chemokine production, and NF-κB activity in both DOCA-salt hypertensive strains with a greater effect in DOCA-salt-treated TRPV1−/− mice compared to DOCA-salt-treated WT mice. No differences in CCR2 protein expression in kidney were found between DOCA-salt-treated WT and TRPV1−/− mice with or without RS504393 treatment. Our studies for the first time indicate that deletion of TRPV1 aggravated renal injury in salt-sensitive hypertension via enhancing MCP-1/CCR2 signaling-dependent inflammatory responses.
Keywords: Transient receptor potential vanilloid type 1 channel, chemokine receptor 2, renal injury, deoxycorticosterone acetate-salt hypertension, mice
Introduction
Monocyte/macrophage-mediated inflammatory responses are involved in the progression of hypertension-induced kidney diseases.1,2 Monocyte/macrophage recruitment and overexpression of proinflammatory cytokines and chemokines contribute to the development of inflammation. Mechanistically, monocyte infiltration to the sites of inflammation is regulated by members of C–C chemokine ligands, also termed as monocyte chemoattractant proteins (MCPs). This is achieved through ligand/receptor binding with receptors expressed in the cell membrane of monocytes. Chemokines are required for monocytes being recruited to tissues, differentiating into macrophages and releasing additional chemokines. C–C chemokine receptor 2 (CCR2) bound by MCP-1 is mainly expressed in monocyte subsets and required for monocyte emigration.3 There is accumulating evidence showing that inhibition of CCR2 attenuates renal injury in renal diseases.4–6 These studies clearly indicate a role for CCR2 in the development of renal disease.
The transient receptor potential vanilloid type 1 (TRPV1, also known as VR1) channel is originally found in primary sensory neurons that innervate various tissues including the heart, blood vessel, and kidney.7,8 The activation of TRPV1 in primary sensory neurons evokes the release of sensory neurotransmitters including calcitonin gene-related peptide (CGRP) and substance P.7 Moreover, several studies have shown that TRPV1 is also expressed in endothelial cells, and its activation increases nitric oxide (NO) release via activation of endothelial NO synthase (eNOS).9,10 The TRPV1 is a ligand-gated non-selective ion channel and activated by multiple stimuli including noxious heat, low pH, and the pungent ingredient of chili peppers, capsaicin.11,12 In addition, a variety of endogenous proinflammatory mediators including lipid metabolites, bradykinin, and prostaglandins may activate or sensitize TRPV1.12
Upregulation of TRPV1 expression has been shown in several diseases such as inflammatory bowel disease and irritable bowel syndrome.13,14 Although activation of TRPV1 in various tissues results in neurogenic inflammation and nociception,15 cumulative evidence shows that the TRPV1 has an anti-inflammatory effect in various pathological conditions, including inflammatory conditions in the cardiovascular system.2,16 Indeed, the TRPV1 has been implicated in protecting against ischemia/reperfusion-induced inflammation of the heart.17 A similar action has been reported in the onset of sepsis and acute lung inflammatory injury induced by intratracheal administration of lipopolysaccharide.18,19 Although these studies show that TRPV1 has anti-inflammatory actions, the precise mechanisms underlying TRPV1 anti-inflammatory actions remain to be elucidated.
Our recent studies demonstrate that global deletion of TRPV1 gene aggravates deoxycorticosterone acetate (DOCA)-salt hypertension-induced renal injury, which is associated with increased renal MCP-1 production and monocyte/macrophage infiltration.2 These results support the notion that global TRPV1 ablation-induced aggravation of renal injury in DOCA-salt hypertension may be the result of enhanced renal monocyte/macrophage infiltration that is dependent of the MCP-1/CCR2 signaling pathway. Since it is well accepted that MCP-1 causes monocyte/macrophage infiltration via binding to CCR2 expressed in monocytes, we hypothesized that MCP-1/CCR2-meditaed monocyte/macrophage infiltration is a critical determinant of renal injury in global TRPV1-null mutant (TRPV1−/−) mice subjected to DOCA-salt hypertension. To achieve this goal, we evaluated the effects of RS504393, a selective CCR2 antagonist, on renal function, structure, inflammatory cell infiltration, and production of multiple inflammatory factors during the development of DOCA-salt hypertension in the wild-type (WT) and global TRPV1−/− mice.
Materials and methods
Animals
Experiments were conducted in 10-week-old male global TRPV1−/− and control WT (C57BL/6) mice (weighing 26–28 g; Charles River Laboratory, Beijing, China). TRPV1−/− mice were generated by deleting an exon encoding part of the fifth and all of the sixth putative transmembrane domains of the channel.2 The mice were backcrossed to C57BL/6 mice for at least six generation and shown to have impaired nociception. All mice were provided with a standard diet and water, and maintained under specific pathogen-free conditions. All protocols were approved by the Henan University of Traditional Chinese Medicine Animal Care and Use Committee.
Experimental protocol
The general procedure of a DOCA-salt hypertension model is well described before.2 In brief, mice were anesthetized with ketamine and xylazine (80 and 4 mg/kg sc, respectively), and the left kidney was removed. The DOCA pellet (24 mg/10 g body weight; Innovative Research of America, Sarasota, FL) was implanted subcutaneously in the neck area under anesthesia. The mice receiving DOCA were given 1% NaCl and 0.2% KCl to drink, and treatment with DOCA-salt continued for four weeks. Sham operation was performed in a similar manner except for DOCA-salt treatment. To evaluate the effect of MCP-1/CCR2 signaling, the mice were subcutaneously given vehicle or a selective CCR2 antagonist, RS504393 (2 mg/kg, twice a day; Tocris Bioscience, Ellisville, MO) for four weeks. The dose of RS504393 has previously been shown to specifically and effectively block CCR2 in vivo.20,21 There were no differences in parameters including blood pressure, renal function/structure, or inflammatory cells/factors between vehicle- and RS504393-treated sham-operated WT, and vehicle- and RS504393-treated sham-operated TRPV1−/− mice. Thus, sham-operated WT and TRPV1−/− mice with vehicle or RS504393 were pooled and treated as control WT and TRPV1−/− mice, respectively. Finally, mice were randomly assigned into the following groups (n = 6–8 per group): (1) control WT mice receiving sham operation (WT); (2) control TRPV1−/− mice receiving sham operation (TRPV1−/−); (3) WT mice receiving DOCA-salt with vehicle (DOCA-WT-Veh); (4) TRPV1−/− mice receiving DOCA-salt with vehicle (DOCA-TRPV1−/−-Veh); (5) WT receiving DOCA-salt with RS504393 (DOCA-WT-RS); (6) TRPV1−/− mice receiving DOCA-salt with RS504393 (DOCA-TRPV1−/−-RS).
At the end of four-week treatment, mice were placed in mouse metabolic cages for 24-h urine collection. After that, plasma and kidney were harvested, weighted, and stored at −80℃ for biochemical analysis or fixed in a 10% formaldehyde solution in phosphate buffer for histological analysis.
Blood pressure measurement
Since it has been reported that renal function and injury significantly correlate with systolic blood pressure (SBP), indirect tail-cuff SBP was routinely determined in conscious mice by use of a BP2000 Blood Pressure Analysis System (Visitech Systems, Apex, NC) as described previously.22,23 Blood pressure was measured one day before DOCA-salt treatment (day 0) and at the end of each week after the treatment. All mice were acclimated to the warming chamber (28–32℃) and the restrainer for 10–20 min/day for at least three days before the day of measurement. Mice were allowed to rest quietly for 10 min at 28–32℃. The blood pressure value for each mouse was calculated as the average of at least eight separate measurements at each session. All measurements were performed at the same time for all groups to prevent any diurnal variations.
Plasma and urine analysis
Plasma and urine creatinine levels were determined with an improved Jaffe creatinine assay kit (BioAssay Systems, Hayward, CA). Urinary albumin was assayed by the use of an ELISA kit (Exocell, Philadelphia, PA). Urinary 8-isoprostane concentrations were measured using the chemiluminescent enzyme immunoassay kit from Cayman Chemical (Ann Arbor, MI).
Renal histology
The formaldehyde-fixed kidneys were embedded in paraffin, and 4-µm sections were cut. Sections were stained with periodic acid-Schiff (PAS) and Masson’s trichrome stain. The degree of glomerulosclerosis and tubulointerstitial injury was determined in a masked fashion in tissue sections stained with PAS and Masson’s trichrome, respectively.
The degree of glomerulosclerosis was determined at x400 magnification using a semiquantitative scoring method as previously described.24 Briefly, 50 glomeruli from each mouse (n = 6–8 per group) were selected randomly and scored as follows: grade 0 represents no lesions, grade 1, 2, 3, and 4 represent mesangial matrix expansion or sclerosis, involving ≤25, 25–50, 50–75, or >75% of the glomerular tuft area, respectively. The ultimate score was obtained by multiplying the degree of changes by the percentage of glomeruli with the same degree of injury and adding up these scores.
The tubulointerstitial injury index was assessed at x 200 magnification in 20 fields selected randomly from each mouse (n = 6–8 per group) using a semiquantitative scoring method as reported previously.24 Briefly, the tubulointerstitial injury was scored as follows: grade 0 represents no lesions, grade 0.5, 1, 2, 3, and 4 represent tubular dilation or atrophy, interstitial fibrosis, or inflammatory cell infiltration, involving small focal area, <10%, 10–25%, 25–75%, or >75% of the renal cortex.
Hydroxyproline assay
The level of collagen in renal tissue was determined using hydroxyproline assay. Kidney samples were homogenized as described previously.24 Hydroxyproline content was assayed using a colorimetric assay and a standard curve of 0–5 µg of hydroxyproline (Sigma Chemical, St. Louis, MO). Data were expressed as micrograms of collagen per milligram of dry weight, assuming that collagen contains an average of 13.5% hydroxyproline.25
Renal immunohistochemistry
Immunohistochemistry for renal monocyte/macrophage was performed in 4-µm paraffin sections obtained four weeks after the treatment. Kidney sections were incubated with a primary antibody directed to F4/80 (1:200, rat anti-mouse macrophage monoclonal antibody; Serotec, Oxford, UK) overnight at 4℃. After being washed, horse anti-rat antibody conjugated with horseradish peroxidase (1:400; Vector Laboratories, Burlingame, CA) was applied and incubated for 1 h at room temperature, and visualized by incubating the sections with the substrate vector fast red (Vector Laboratories). The sections were counterstained with hematoxylin. Negative control experiments were performed by omitting the primary antibody during incubation. The quantification of macrophages in the cortex was carried out in a blind fashion under x400 magnification. The number of macrophages was counted in at least 10 fields for each mouse (n = 6–8 per group) and averaged. The number of positive cells was expressed as cells per square millimeter.
Renal cytokine assay
Levels of TNF-α, IL-6, and MCP-1 in kidneys were assayed using the corresponding ELISA kit (R&D Systems, Minneapolis, MN) according to the manufacturer’s protocol. Whole kidneys were homogenized in ice-cold phosphate buffer containing protease inhibitor cocktail (Sigma Chemical). The total proteins were extracted using NE-PER Cytoplasmic Extraction Reagents (Pierce, Rockford, IL) following the manufacturer’s protocol. The total protein level of extract was assayed using a protein assay kit (Bio-Rad Laboratories, Hercules, CA) and used for normalizing cytokine concentrations in renal tissue.
NF-κB transcription factor assay
Nuclear protein was extracted from the kidney using the nuclear extract kit (Active Motif, Carlsbad, CA) following the manufacturer’s protocol. Protein concentrations of nuclear extract were assayed with a protein assay kit. The nuclear concentration of p65 was determined at 450 nm with a TransAM NF-κB p65 assay kit (Active Motif) based on the manufacture’s instruction. The NF-κB activity was represented by the nuclear concentration of p65 and normalized per milligram of renal protein.
Western blot analysis
Protein was extracted from the whole kidney using RIPA buffer (Thermo Fisher Scientific Inc., Rockford, IL, USA) containing cocktail proteinase inhibitors (Thermo Fisher Scientific Inc.) and quantified with the Bio-Rad protein assay. Equal amounts of protein (50 µg) were separated on a 10% SDS-PAGE gel, and transferred to a polyvinylidene difluoride membrane. Blots were blocked for 1 h at room temperature in 5% skim milk washing solution (50 mmol/Tris-HCI, 100 mmol/L NaCI, and 0.1% Tween 20 at pH 7.5). Subsequently, blots were incubated overnight at 4℃ with rabbit anti-CCR2 polyclonal antibody (1:600, Abcam, Cambridge, MA) in block solution, washed, and then incubated at room temperature for 1 h with horseradish peroxidase-conjugated goat anti-rabbit IgG (1:800, Santa Cruz Biotechnology, Santa Cruz, CA). Immune blots were detected with an enhanced chemiluminescence Western blot kit (ECL, Amersham Biosciences, Piscataway, NJ). Blot intensity was densitometrically determined. Expression of β-actin was used to normalize protein loaded on blots.
Statistical analysis
All of the values are expressed as means ± SE. Comparisons between groups at each experimental time point were determined by the use of two-way ANOVA followed by a Bonferroni’s test. The differences among groups were evaluated using one-way analysis of variance followed by a Bonferroni’s adjustment for multiple comparisons. Differences were considered statistically significant at P < 0.05.
Results
Effect of RS504393 on body weight, kidney weight, and urine chemistries in WT and TRPV1−/− mice with DOCA-salt treatment
Table 1 summarizes the changes in body weight, kidney weight, and urine parameters during the study. There was no significant difference in body weight (BW) between groups at the end of the experimental period (P > 0.05). DOCA-salt treatment markedly enhanced the kidney to BW ratio in WT and TRPV1−/− mice compared to their respective controls (P < 0.05,), but no difference in the kidney to BW ratio was found between DOCA-salt-treated WT and TRPV1−/− mice (P > 0.05). Blockade of CCR2 with RS504393 did not affect the kidney to BW ratio in DOCA-salt-treated WT or TRPV1−/− mice (P > 0.05). DOCA-salt treatment significantly increased the urinary output in WT and TRPV1−/− mice compared to their respective controls (P < 0.05). There was no difference in urinary output between DOCA-salt-treated WT and TRPV1−/− mice (P > 0.05). However, urinary output was obviously increased in DOCA-TRPV1−/−-RS group compared to DOCA-TRPV1−/−-Veh group (P < 0.05). Urinary albumin and 8-isoprostane excretion were used as an indicator for renal injury and reactive oxygen species, respectively. DOCA-salt treatment significantly increased urinary albumin and 8-isoprostane excretion in WT and TRPV1−/− mice compared to their respective controls (P < 0.05). Also, deletion of TRPV1 markedly increased DOCA-salt-induced urinary albumin and 8-isoprostane excretion compared to DOCA-salt-treated WT mice (P < 0.05). Moreover, treatment with RS504393 prevented or significantly reduced urinary albumin and 8-isoprostane excretion in DOCA-salt-treated WT or TRPV1−/− mice (P < 0.05), and the degree of RS504393-induced decreases in urinary albumin and 8-isoprostane excretion was greater in DOCA-salt-treated TRPV1−/− mice compared to DOCA-salt-treated WT mice (urinary albumin, 57.9 ± 6.7 vs. 19.7 ± 3.1 µg/24 h; urinary 8-isoprostane, 2.53 ± 0.34 vs. 0.99 ± 0.21 ng/24 h, P < 0.05, respectively). In addition, DOCA-salt treatment significantly reduced creatinine clearance in WT and TRPV1−/− mice compared to their respective controls (P < 0.05). Administration of RS504393 abolished or inhibited the DOCA-salt-induced decrease in creatinine clearance in WT or TRPV1−/− mice (P < 0.05), and the magnitude of RS504393-induced increases in creatinine clearance was greater in DOCA-salt-treated TRPV1−/− mice compared to DOCA-salt-treated WT mice (182 ± 9 vs. 145 ± 8 mL/24 h, P < 0.05).
Table 1.
Effect of CCR2 inhibition on body weight, kidney weight, and urine chemistries in WT and TRPV1−/− mice with DOCA-salt treatment.a
| Group | Body weight (g) | Kidney weight (mg/10 g of BW) | Urinary output (mL/24 h) | Urinary albumin (µg/24 h) | Urinary 8-isoprostane (ng/24 h) | Creatinine clearance (mL/24 h) |
|---|---|---|---|---|---|---|
| WT | 28.3 ± 0.3 | 78.3 ± 1.2 | 1.3 ± 0.5 | 5.1 ± 0.7 | 0.48 ± 0.13 | 325 ± 18 |
| TRPV1−/− | 27.9 ± 0.5 | 76.8 ± 1.4 | 1.5 ± 0.3 | 4.6 ± 0.6 | 0.55 ± 0.16 | 332 ± 22 |
| DOCA-WT-Veh | 28.9 ± 0.4 | 115.3 ± 3.8* | 19.1 ± 3.6* | 26.5 ± 3.4*# | 1.52 ± 0.21*# | 168 ± 14*# |
| DOCA-TRPV1−/−-Veh | 29.4 ± 0.3 | 118.9 ± 4.1* | 16.8 ± 3.3*# | 89.3 ± 5.2*†# | 4.24 ± 0.45*†# | 98 ± 19*†# |
| DOCA-WT-RS | 28.8 ± 0.7 | 112.5 ± 5.5* | 21.6 ± 4.3* | 7.4 ± 1.7 | 0.62 ± 0.18 | 311 ± 16 |
| DOCA-TRPV1−/−-RS | 29.7 ± 0.5 | 114.7 ± 4.6* | 22.9 ± 4.1* | 31.6 ± 4.6* | 1.83 ± 0.24* | 279 ± 19* |
CCR2: chemokine receptor 2; WT: wild-type; TRPV1−/−: transient receptor potential vanilloid type 1 (TRPV1)-null mutant; DOCA: deoxycorticosterone acetate; Veh: vehicle; RS: RS504393; BW: body weight.
Values are mean ± SE; n = 6–8 mice.
P < 0.05 compared with control WT or TRPV1−/− mice.
P < 0.05 compared with DOCA-WT-Veh.
P < 0.05 compared with DOCA-WT-RS or DOCA-TRPV1−/−-RS.
Effect of RS504393 on blood pressure in WT and TRPV1−/− mice with DOCA-salt treatment
As demonstrated in Figure 1, all mice had similar baseline SBP determined by a non-invasive tail-cuff system. Compared with the controls, tail-cuff SBP was significantly higher beginning at week 1 after DOCA-salt treatment and continuing for the rest of the experiment in DOCA-salt-treated WT and TRPV1−/− mice with or without RS504393 (P < 0.05). There was no difference in SBP among DOCA-salt-treated mice with or without RS504393 treatment (P > 0.05).
Figure 1.
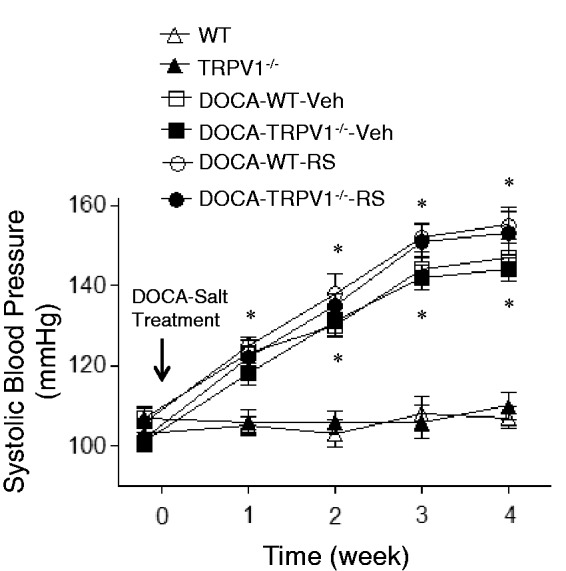
Effect of treatment with chemokine receptor 2 (CCR2) antagonist, RS504393 (RS) on systolic blood pressure in wild-type (WT) and transient receptor potential vanilloid type 1 (TRPV1)-null mutant (TRPV1−/−) mice with deoxycorticosterone acetate (DOCA)-salt treatment. Veh indicates vehicle. Values are means ± SE (n = 6–8). *P < 0.05 compared with control WT or TRPV1−/− mice
Effect of RS504393 on renal lesions in WT and TRPV1−/− mice with DOCA-salt treatment
As shown in Figure 2, DOCA-salt treatment caused more severe glomerulosclerosis determined by PAS staining in TRPV1−/− than WT mice. As illustrated in Figure 3, more severe tubular injury determined by Masson’s trichrome staining was also found in the renal cortex of DOCA-salt-treated TRPV1−/− mice compared with DOCA-salt-treated WT mice. Quantitative analysis showed that DOCA-salt treatment significantly increased glomerulosclerosis index and tubulointerstitial injury scores in WT and TRPV1−/− mice compared to their respective controls (P < 0.05), and DOCA-salt-induced renal injury was more severe in TRPV1−/− than WT mice (P < 0.05). Renal damage was not found in control WT and TRPV1−/− mice. CCR2 blockade with RS504393 prevented or significantly reduced glomerulosclerosis and tubulointerstitial injury in DOCA-salt-treated WT and TRPV1−/− mice, and the RS504393-mediated decreases in glomerulosclerosis index and tubulointerstitial injury score were greater in DOCA-salt-treated TRPV1−/− mice compared to DOCA-salt-treated WT mice (glomerulosclerosis index, 0.38 ± 0.09 vs. 0.18 ± 0.03; tubulointerstitial injury score, 2.1 ± 0.4 vs. 0.9 ± 0.3, P < 0.05, respectively).
Figure 2.

Effect of treatment with chemokine receptor 2 (CCR2) antagonist, RS504393 (RS), on renal glomerular injury in wild-type (WT) and transient receptor potential vanilloid type 1 (TRPV1)-null mutant (TRPV1−/−) mice with deoxycorticosterone acetate (DOCA)-salt treatment. (A) Histological sections stained with Periodic acid–Schiff (PAS) stain showing glomerulosclerosis in control WT (a) and TRPV1−/− (b) mice, and DOCA-salt-treated WT and TRPV1−/− mice with vehicle (Veh, c and d) or RS (e and f). Magnification, x400. (B) Bar graph showing the average of glomerulosclerosis index in all groups. Values are means ± SE (n = 6–8). *P < 0.05 compared with control WT or TRPV1−/− mice; †P < 0.05 compared with DOCA-WT-Veh; #P < 0.05 compared with DOCA-WT-RS or DOCA-TRPV1−/−-RS. (A color version of this figure is available in the online journal.)
Figure 3.

Effect of treatment with chemokine receptor 2 (CCR2) antagonist, RS504393 (RS) on renal cortical tubulointerstitial injury in wild-type (WT) and transient receptor potential vanilloid type 1 (TRPV1)-null mutant (TRPV1−/−) mice with deoxycorticosterone acetate (DOCA)-salt treatment. (A) Histological sections stained with Masson’s trichrome stain showing renal cortical tubulointerstitial injury in control WT (a) and TRPV1−/− (b) mice, and DOCA-salt-treated WT and TRPV1−/− mice with vehicle (Veh, c and d) or RS (e and f). Magnification, x200. (B) Bar graph showing the average of tubulointerstitial injury score in all groups. Values are means ± SE (n = 6–8). *P < 0.05 compared with control WT or TRPV1−/− mice; †P < 0.05 compared with DOCA-WT-Veh; #P < 0.05 compared with DOCA-WT-RS or DOCA-TRPV1−/−-RS. (A color version of this figure is available in the online journal.)
Effect of RS504393 on renal collagen levels in WT and TRPV1−/− mice with DOCA-salt treatment
Similarly to the changes in renal damage, DOCA-salt treatment led to a significant increase in renal collagen levels determined using hydroxyproline assay in WT and TRPV1−/− mice compared to control mice (P < 0.05), and the effect was found to be greater in the latter strain (Figure 4). Moreover, the DOCA-salt-induced increase in the levels of renal collagen was blocked or inhibited by RS504393 treatment in WT or TRPV1−/− mice, and the RS504393-induced decrease in renal collagen levels was more significant in DOCA-salt-treated TRPV1−/− mice compared to DOCA-salt-treated WT mice (9.2 ± 1.8 vs. 3.8 ± 0.6 µg/mg dry tissue, P < 0.05).
Figure 4.
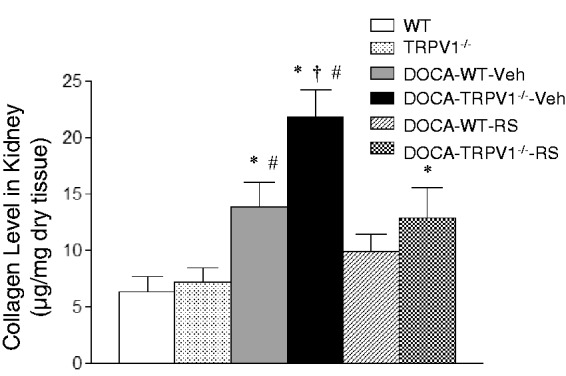
Effect of treatment with chemokine receptor 2 (CCR2) antagonist, RS504393 (RS) on renal collagen levels in wild-type (WT) and transient receptor potential vanilloid type 1 (TRPV1)-null mutant (TRPV1−/−) mice with deoxycorticosterone acetate (DOCA)-salt treatment. Veh indicates vehicle. Values are means ± SE (n = 6–8). *P < 0.05 compared with control WT or TRPV1−/− mice; †P < 0.05 compared with DOCA-WT-Veh; #P < 0.05 compared with DOCA-WT-RS or DOCA-TRPV1−/−-RS
Effect of RS504393 on renal monocyte/macrophage infiltration and proinflammatory cytokine/chemokine levels in WT and TRPV1−/− mice with DOCA-salt treatment
Monocyte/macrophage infiltration was indicated by F4/80-positive cells that stained red. As shown in Figure 5, DOCA-salt treatment significantly increased F4/80-positive cells in the renal cortex of WT and TRPV1−/− mice compared with the controls (P < 0.05), and the F4/80-positive cells were further increased in DOCA-salt-treated TRPV1−/− mice compared with DOCA-salt-treated WT mice (P < 0.05). Moreover, RS504393 treatment blocked the enhancement infiltration of monocytes/macrophages in DOCA-salt-treated WT and TRPV1−/− mice (P < 0.05), and the RS504393-induced decrease in renal monocyte/macrophage infiltration was greater in DOCA-salt-treated TRPV1−/− mice compared to DOCA-salt-treated WT mice (52 ± 5 vs. 23 ± 3 cells/mm2, P < 0.05). DOCA-salt treatment markedly increased renal protein levels of proinflammatory cytokines/chemokines including TNF-α, IL-6, and MCP-1 in WT and TRPV1−/− mice compared to their respective controls (P < 0.05), and the results are shown in Figure 6. A further elevation of renal TNF-α, IL-6, and MCP-1 levels was found in DOCA-salt-treated TRPV1−/− mice compared to DOCA-salt-treated WT mice (P < 0.05). Moreover, administration of RS504393 prevented DOCA-salt-induced increases in the levels of renal proinflammatory cytokines/chemokines in WT and TRPV1−/− mice, and the RS504393-mediated decreases in renal cytokines/chemokines were more significant in DOCA-salt-treated TRPV1−/− mice compared to DOCA-salt-treated WT mice (TNF-α, 0.49 ± 0.07 vs. 0.18 ± 0.03 pg/mg protein; IL-6, 1.41 ± 0.35 vs. 0.50 ± 0.10 pg/mg protein; MCP-1, 5.85 ± 0.53 vs. 2.53 ± 0.25 pg/mg protein, P < 0.05, respectively).
Figure 5.

Effect of treatment with chemokine receptor 2 (CCR2) antagonist, RS504393 (RS) on renal cortical interstitial monocyte/macrophage infiltration in wild-type (WT) and transient receptor potential vanilloid type 1 (TRPV1)-null mutant (TRPV1−/−) mice with deoxycorticosterone acetate (DOCA)-salt treatment. (A) Immunohistochemically stained sections showing that F4/80-positive cells (monocytes/macrophages in red) in the renal cortex of control WT (a) and TRPV1−/− (b) mice, and DOCA-salt-treated WT and TRPV1−/− mice with vehicle (Veh, c and d) or RS (e and f). The arrows indicate that F4/80-positive cells. Magnification, x400. (B) Bar graph showing the number of renal cortical F4/80-positive cells expressed as cells per square millimeter in all groups. Values are means ± SE (n = 6–8). *P < 0.05 compared with control WT or TRPV1−/− mice; †P < 0.05 compared with DOCA-WT-Veh; #P < 0.05 compared with DOCA-WT-RS or DOCA-TRPV1−/−-RS. (A color version of this figure is available in the online journal.)
Figure 6.

Effect of treatment with chemokine receptor 2 (CCR2) antagonist, RS504393 (RS) on the levels of renal TNF-α (A), IL-6 (B), and MCP-1 (C) in wild-type (WT) and transient receptor potential vanilloid type 1 (TRPV1)-null mutant (TRPV1−/−) mice with deoxycorticosterone acetate (DOCA)-salt treatment. Values are means ± SE (n = 6–8). Veh indicates vehicle. *P < 0.05 compared with control WT or TRPV1−/− mice; †P < 0.05 compared with DOCA-WT-Veh; #P < 0.05 compared with DOCA-WT-RS or DOCA-TRPV1−/−-RS
Effect of RS504393 on renal NF-κB activity in WT and TRPV1−/− mice with DOCA-salt treatment
As demonstrated in Figure 7, DOCA-salt treatment significantly increased renal NF-κB activity in WT and TRPV1−/− mice compared to their respective controls (P < 0.05), and the effect of DOCA-salt treatment was further enhanced in TRPV1−/− mice compared with WT mice (P < 0.05). Moreover, RS504393 treatment abolished the DOCA-salt-induced increase in renal NF-κB activity in WT and TRPV1−/− mice (P < 0.05), and RS504393 treatment caused greater reduction in renal NF-κB activity in DOCA-salt-treated TRPV1−/− mice than in DOCA-salt-treated WT mice (28.4 ± 3.4 vs. 12.4 ± 2.0 ng/mg protein, P < 0.05).
Figure 7.
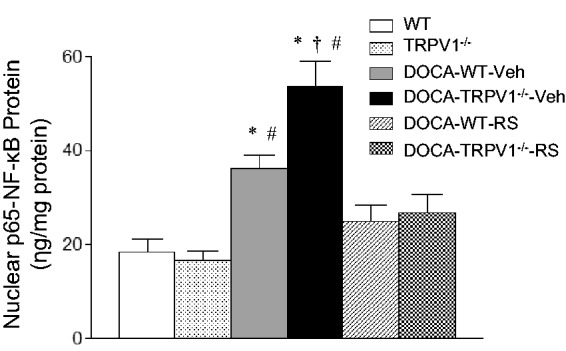
Effect of treatment with chemokine receptor 2 (CCR2) antagonist, RS504393 (RS) on renal NF-κB activity in wild-type (WT) and transient receptor potential vanilloid type 1 (TRPV1)-null mutant (TRPV1−/−) mice with deoxycorticosterone acetate (DOCA)-salt treatment. Veh indicates vehicle. Values are means ± SE (n = 6–8). *P < 0.05 compared with control WT or TRPV1−/− mice; †P < 0.05 compared with DOCA-WT-Veh; #P < 0.05 compared with DOCA-WT-RS or DOCA-TRPV1−/−-RS
Effect of RS504393 on renal CCR2 protein expression in WT and TRPV1−/− mice with DOCA-salt treatment
As shown in Figure 8, DOCA-salt treatment did not increase renal CCR2 protein expression in WT and TRPV1−/− mice compared to their respective controls (P > 0.05). Moreover, there were no differences in the protein levels of CCR2 in kidneys between DOCA-salt-treated WT and TRPV1−/− mice with or without RS504393 treatment (P > 0.05).
Figure 8.
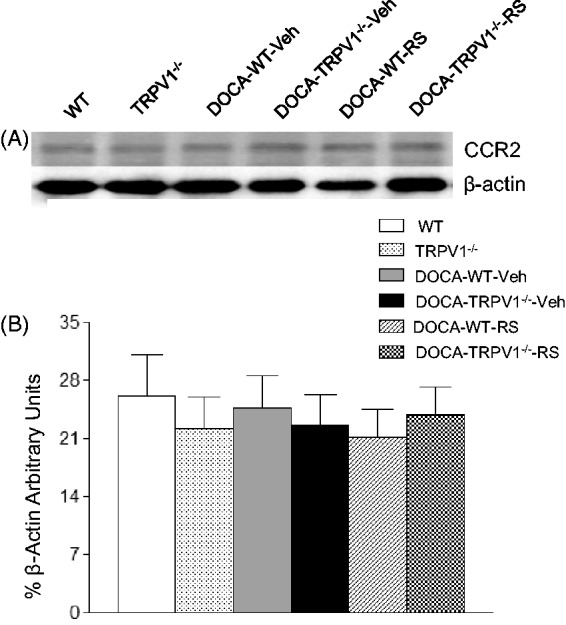
Effect of treatment with chemokine receptor 2 (CCR2) antagonist, RS504393 (RS) on renal CCR2 protein expression in wild-type (WT) and transient receptor potential vanilloid type 1 (TRPV1)-null mutant (TRPV1−/−) mice with deoxycorticosterone acetate (DOCA)-salt treatment. (A) Representative Western blot of CCR2 in kidneys from all groups. (B) Bar graphs show the relative optical density values for CCR2 in all groups. Results were normalized by the corresponding β-actin. Veh indicates vehicle. Values are means ± SE (n = 4–6)
Discussion
Our study demonstrates that pharmacological blockade of CCR2 induces renal protection as evidenced by decreased albuminuria and morphological damage associated with attenuation of monocyte/macrophage infiltration and cytokine overproduction during DOCA-salt hypertension. We also found that treatment with a CCR2 antagonist provides more potent protection against renal damage induced by DOCA-salt treatment in TRPV1−/− mice without lowering blood pressure. This is the first evidence suggesting that activation of TRPV1 may ameliorate salt-sensitive hypertension-induced renal damage via inhibiting MCP-1/CCR2-mediated monocyte/macrophage infiltration.
It is well accepted that macrophage infiltration into the kidney mediates renal damage in a number of renal diseases.5,26 The chemokine MCP-1 is a potent macrophage chemoattractant which binds to CCR2 to promote monocyte/macrophage chemotaxis and infiltration in renal diseases.27 It has been shown that MCP-1 or CCR2 deficiency attenuates the recruitment of monocytes/macrophages into the aortic wall or kidney in atherosclerosis or renal injury induced by ischemia–reperfusion or unilateral ureteral obstruction.20,21,28 These studies indicate that the MCP-1/CCR2 signaling pathway plays an important role in monocyte/macrophage infiltration. In this study, we determined the inhibitory effects of MCP-1/CCR2 signaling pathway on TRPV1 ablation-induced renal injury using a specific CCR2 antagonist, RS504393. The specificity and the efficiency of RS504393 in the blockade of CCR2 have been supported by the experiments performed in vivo and in vitro. RS504393 attenuated MCP-1-induced spleen cell or monocyte chemotaxis in a dose-dependent manner, but it did not attenuate MIP-1α-induced chemotaxis.20,29 In addition, the dose of RS504393 used in this study was shown to attenuate renal monocyte/macrophage infiltration in renal injury induced by ischemia–reperfusion or unilateral ureteral obstruction, which was similar to those observed in CCR2-deficient mice.20,21 These studies suggested that the MCP-1/CCR2 signaling pathway should be effectively blocked by the dose of RS504393 used in our studies in vivo, even though there were no changes in blood pressure. In this study, we found that renal injury associated with monocyte/macrophage infiltration was enhanced in DOCA-salt hypertensive WT mice, which was attenuated by RS504393. More importantly, the RS504393-mediated decreases in renal monocyte/macrophage infiltration and renal injury were accelerated in TRPV1−/− mice receiving DOCA-salt treatment. We also found that deletion of TRPV1 further increased DOCA-salt-induced renal MCP-1 production, but there was no evidence that TRPV1-deletion or DOCA-salt treatment increased the protein expression of CCR2 in kidneys. These data support the concept that activation of TRPV1 attenuates the MCP-1/CCR2 signaling pathway, especially MCP-1 production, to reduce the monocyte/macrophage-mediated inflammation, leading to renal protection in salt-sensitive hypertension.
It is well established that inflammatory responses are regulated at the level of transcription by the transcription factor NF-κB.30 Activated NF-κB causes numerous proinflammatory gene expression, including cytokines and chemokines, resulting in inflammatory responses. Our study showed that renal NF-κB activity and monocyte/macrophage infiltration were elevated in DOCA-salt-treated WT mice, and importantly deletion of TRPV1 further exacerbated those increases observed in DOCA-salt-treated WT. The results suggest that activated TRPV1 attenuates inflammatory responses possibly via inhibition of NF-κB activity given that NF-κB is a common inflammatory regulator. In this study, we found that CCR2 blockade reduced the production of TNF-α, IL-6, and MCP-1 in the kidney of DOCA-salt WT mice, deletion of TRPV1 accelerated these effects. The reduction of cytokines and chemokines may be proportional to the decrease in monocyte/macrophage infiltration in the kidney. In addition, the interaction between macrophages and renal epithelial cells may be important in this setting. Activated macrophages by MCP-1/CCR2 signaling produce proinflammatory cytokines and chemokines, which in turn stimulate renal tubular epithelial cells to produce cytokines and chemokines. Supporting this possibility, there is evidence showing that MCP-1 activates NF-κB, and NF-κB in tubular epithelial cells is critical for production of cytokines and chemokines.31 Moreover, our results showed that CCR2 blockade with RS504393 reduced NF-κB activity in WT and TRPV1−/− mice during DOCA-salt hypertension. Taken together, these findings support the notion that MCP-1/CCR2-dependent activation of NF-κB may amplify the production of cytokines and chemokines in the kidney, although there is evidence showing that MCP-1 is down-stream of NF-κB.32
As inflammatory responses are complicated, several mechanisms may contribute to the anti-inflammatory actions of TRPV1 beyond suppression of NF-κB activity. It is well known that TRPV1 is mainly expressed in primary sensory nerves. Denervation of sensory nerves expressing TRPV1 using capsaicin, a selective TRPV1 agonist, results in increased TNF-α levels, suggesting that TRPV1-positive sensory nerves inhibit proinflammatory mediator production.33 In addition, there is evidence showing that TRPV1 is expressed in the vascular endothelium and that activation of TRPV1 enhances NO production via activation of eNOS.9,10 Although excessive NO production is cytotoxic, NO produced by eNOS expressed in endothelial cells may attenuate production of chemokines including MCP-1.34 Consistent with these published works, we found that TRPV1 ablation further increased renal cytokine/chemokine production induced by DOCA-salt treatment. Although sympathetic nerve overactivity, endothelin-1, angiotensin II, and reactive oxygen species are involved in inflammatory responses in salt-sensitive hypertension,35,36 our data suggest that the TRPV1 dysfunction further enhances inflammatory responses in salt-sensitive hypertension. Further studies are needed to determine the detailed molecular mechanisms responsible for TRPV1-induced anti-inflammatory actions.
In this study, we found that CCR2 inhibition attenuated DOCA-salt-induced renal injury and inflammatory responses in WT and TRPV1−/− mice without lowering blood pressure. Although the tail-cuff technique does not allow us to accurately detect minor changes in blood pressure compared to the radiotelemetry, the technique is available to detect a significant change in blood pressure. In this study, we cannot completely rule out the effects of CCR2 blockade on blood pressure, but the RS504393-induced changes in blood pressure might not be very significant in DOCA-salt hypertensive WT and TRPV1−/− mice, because we did not find any significant differences in blood pressure between untreated and RS504393-treated DOCA-salt-WT and TRPV1−/− mice using the tail-cuff technique. In addition, several studies have indicated that RS504393 is a specific CCR2 antagonist, and the dose of RS504393 used in our studies can effectively block CCR2 in vivo.20,21 Based on these facts, we speculate that RS504393-induced renal protection in DOCA-salt-treated WT and TRPV1−/− mice is mainly attributed to a reduction in renal inflammation rather than blood pressure. However, it should be noted that a limitation to the present study is that blood pressure was determined by use of the tail-cuff technique. Thus, further studies are required to determine the effects of RS504393 on blood pressure and whether CCR2 blockade-mediated renal protection in salt-sensitive hypertension, especially in TRPV1-deficient mice with salt-sensitive hypertension, is secondary to the changes in blood pressure by use of the radiotelemetry.
In summary, we found that antagonism of CCR2 possesses renal anti-inflammatory and protective effects in DOCA-salt hypertension, which were empowered in DOCA-salt-treated TRPV1−/− mice in the absence of alteration in blood pressure. This is the first evidence indicating that activation of TRPV1 inhibits the MCP-1/CCR2 signaling pathway and reduces renal injury during salt-sensitive hypertension.
Perspectives and significance
Although patients with salt-sensitive hypertension are much more likely to experience renal damage, the mechanisms underlying the progression of renal injury in salt-sensitive hypertension remain undetermined. Our results suggest that activation of TRPV1 inhibits the MCP-1/CCR2 signaling pathway to reduce monocyte/macrophage infiltration, leading to renal protection during salt-sensitive hypertension. Although further studies are required to determine the mechanisms underlying TRPV1-induced renal protection, our findings suggest that improvement of TRPV1 function or blockade of MCP-1/CCR2 signaling pathway may serve as therapeutic means to alleviate renal damage occurring during salt-sensitive hypertension.
Acknowledgements
This work was supported by grants from the National Natural Science Foundation of China (No. 81170243) and Henan Provincial Science and Technology Innovative Program for Outstanding Scholarship (No. 124200510007) to Y Wang.
Author contributions
YW and MZ: conception and design of research; YW, LC, WL, XW, and SS performed experiments; YW and MZ analyzed data; YW, MZ, and HX interpreted results of experiments; YW prepared figures; YW drafted manuscript; YW, HX, and DHW edited and revised manuscript; YW, MZ, HX, LC, WL, XW, SS, and DHW approved final version of manuscript.
References
- 1.Kusunoki H, Taniyama Y, Rakugi H, Morishita R. Cardiac and renal protective effects of irbesartan via peroxisome proliferator-activated receptorγ-hepatocyte growth factor pathway independent of angiotensin II type 1a receptor blockade in mouse model of salt-sensitive hypertension. J Am Heart Assoc 2013; 2: e000103–e000103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wang Y, Wang DH. Aggravated renal inflammatory responses in TRPV1 gene knockout mice subjected to DOCA-salt hypertension. Am J Physiol 2009; 297: F1550–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zimmermann HW, Trautwein C, Tacke F. Functional role of monocytes and macrophages for the inflammatory response in acute liver injury. Front Physiol 2012; 3: 56–74.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kitagawa K, Wada T, Furuichi K, Hashimoto H, Ishiwata Y, Asano M, Takeya M, Kuziel WA, Matsushima K, Mukaida N, Yokoyama H. Blockade of CCR2 ameliorates progressive fibrosis in kidney. Am J Pathol 2004; 165: 237–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Elmarakby AA, Quigley JE, Olearczyk JJ, Sridhar A, Cook AK, Inscho EW, Pollock DM, Imig JD. Chemokine receptor 2b inhibition provides renal protection in angiotensin II-salt hypertension. Hypertension 2007; 50: 1069–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Awad AS, Kinsey GR, Khutsishvili K, Gao T, Bolton WK, Okusa MD. Monocyte/macrophage chemokine receptor CCR2 mediates diabetic renal injury. Am J Physiol 2011; 301: F1358–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wimalawansa SJ. Calcitonin gene-related peptide and its receptors: molecular genetics, physiology, pathophysiology, and therapeutic potentials. Endocr Rev 1996; 17: 533–85. [DOI] [PubMed] [Google Scholar]
- 8.Earley S. Vanilloid and melastatin transient receptor potential channels in vascular smooth muscle. Microcirculation 2010; 17: 237–49.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ching LC, Kou YR, Shyue SK, Su KH, Wei J, Cheng LC, Yu YB, Pan CC, Lee TS. Molecular mechanisms of activation of endothelial nitric oxide synthase mediated by transient receptor potential vanilloid type1. Cardiovasc Res 2011; 191: 492–501. [DOI] [PubMed] [Google Scholar]
- 10.Poblete IM, Orliac ML, Briones R, Adler-Graschinsky E, Huidobro-Toro JP. Anandamide elicits an acute release of nitric oxide through endothelial TRPV1 receptor activation in the rat arterial mesenteric bed. J Physiol 2005; 568: 539–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.O’Neill J, Brock C, Olesen AE, Andresen T, Nilsson M, Dickenson AH. Unravelling the mystery of capsaicin: a tool to understand and treat pain. Pharmacol Rev 2012; 64: 939–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Julius D, Basbaum AI. Molecular mechanisms of nociception. Nature 2001; 413: 203–10. [DOI] [PubMed] [Google Scholar]
- 13.Yiangou Y, Facer P, Dyer NH, Chan CL, Knowles C, Williams NS, Anand P. Vanilloid receptor 1 immunoreactivity in inflamed human bowel. Lancet 2001; 357: 1338–9. [DOI] [PubMed] [Google Scholar]
- 14.Chan CL, Facer P, Davis JB, Smith GD, Egerton J, Bountra C, Williams NS, Anand P. Sensory fibers expressing capsaicin receptor TRPV1 in patients with rectal hypersensitivity and faecal urgency. Lancet 2003; 361: 385–91. [DOI] [PubMed] [Google Scholar]
- 15.Veronesi B, Carter JD, Devlin RB, Simon SA, Oortgiesen M. Neuropeptides and capsaicin stimulate the release of inflammatory cytokines in a human bronchial epithelial cell line. Neuropeptides 1999; 33: 447–56. [DOI] [PubMed] [Google Scholar]
- 16.Wang Y, Wang DH. Role of the transient receptor potential vanilloid type 1 channel in renal inflammation induced by lipopolysaccharide in mice. Am J Physiol 2013; 304: R1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Huang W, Rubinstein J, Prieto AR, Thang LV, Wang DH. Transient receptor potential vanilloid gene deletion exacerbates inflammation and atypical cardiac remodeling after myocardial infarction. Hypertension 2009; 53: 243–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Clark N, Keeble J, Fernandes ES, Starr A, Liang L, Sugden D, deWinter P, Brain SD. The transient receptor potential vanilloid (TRPV1) receptor protects against the onset of sepsis after endotoxin. FASEB J 2007; 21: 2747–3755. [DOI] [PubMed] [Google Scholar]
- 19.Helyes Z, Elekes K, Németh J, Pozsgai G, Sándor K, Kereskai L, Börzsei R, Pintér E, Szabó Á, Szolcsányi J. Role of transient receptor potential vanilloid 1 receptors in endotoxin-induced airway inflammation in the mouse. Am J Physiol Lung Cell Mol 2007; 292: L1173–81. [DOI] [PubMed] [Google Scholar]
- 20.Furuichi K, Wada T, Iwata Y, Kitagawa K, Kobayashi K, Hashimoto H, Ishiwata Y, Asano M, Wang H, Matsushima K, Takeya M, Kuziel WA, Mukaida N, Yokoyama H. CC2 signaling contributes to ischemia-reperfusion injury in kidney. J Am Soc Nephrol 2003; 14: 2503–15. [DOI] [PubMed] [Google Scholar]
- 21.Kitagawa K, Wada T, Furuichi K, Hashimoto H, Ishiwata Y, Asano M, Takeya M, Kuziel WA, Matsushima K, Mukaida N, Yokoyama H. Blockade of CCR2 ameliorates progressive fibrosis in kidney. Am J Pathol 2004; 165: 237–46.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Safar ME, London GM, Plante GE. Arterial stiffness and kidney function. Hypertension 2004; 43: 163–8. [DOI] [PubMed] [Google Scholar]
- 23.Feng M, Whitesall S, Zhang Y, Beibei M, D’Alecy L, DiPetrillo K. Validation of volume-pressure recording tail-cuff blood pressure measurements. Am J Hypertens 2008; 21: 1288–91. [DOI] [PubMed] [Google Scholar]
- 24.Wang Y, Wang DH. Role of substance P in renal injury during DOCA-salt hypertension. Endocrinology 2012; 153: 5972–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chiariello M, Ambrosio G, Cappelli-Biqazzi M, Perrone-Filardi P, Briqante F, Sifola C. A biochemical method for the quantitation of myocardial scarring after experimental coronary artery occlusion. J Mol Cell Cardiol 1986; 18: 283–90. [DOI] [PubMed] [Google Scholar]
- 26.Sayyed SG, Ryu M, Kulkarni OP, Schmid H, Lichtnekert J, Gruner S, Green L, Mattei P, Hartmann G, Anders HJ. An orally active chemokine receptor CCR2 antagonist prevents glomerulosclerosis and renal failure in type 2 diabetes. Kidney Int 2011; 80: 68–78. [DOI] [PubMed] [Google Scholar]
- 27.Eardley KS, Zehnder D, Quinkler M, Lepenies J, Bates RL, Savage CO, Howie AJ, Adu D, Cockwell P. The relationship between albuminuria, MCP-1/CCL2, and interstitial macrophages in chronic kidney disease. Kidney Int 2006; 69: 1189–97. [DOI] [PubMed] [Google Scholar]
- 28.Gu L, Okada Y, Clinton SK, Gerard C, Sukhova GK, Libby P, Rollins BJ. Absence of monocyte chemoattractant protein-1 reduces atherosclerosis in low density lipoprotein receptor-deficient mice. Mol Cell 1998; 2: 275–81. [DOI] [PubMed] [Google Scholar]
- 29.Ino K, Masuya M, Tawara I, Miyata E, Oda K, Nakamori Y, Suzuki K, Ohishi K, Katayama N. Monocytes infiltrate the pancreas via the MCP-1/CCR2 pathway and differentiate into stellate cells. PLoS One 2014; 9: e84889–e84889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ivashkiv LB. Inflammatory signaling in macrophages: transitions from acute to tolerant and alternative activation states. Eur J Immunol 2011; 41: 2477–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Viedt C, Dechend R, Fei J, Hansch GM, Kreuzer J, Orth SR. MCP-1 induces inflammatory activation of human tubular epithelial cells. J Am Soc Nephrol 2002; 13: 1534–47. [DOI] [PubMed] [Google Scholar]
- 32.Jura J, Skalniak L, Koj A. Monocyte chemotactic protein-1-induced protein-1 (MCPIP1) is a novel multifunctional modulator of inflammatory reactions. Biochim Biophys Acta 2012; 1823: 1905–13. [DOI] [PubMed] [Google Scholar]
- 33.Franco-Penteado CF, De Souza IA, Camargo EA, Teixeira SA, Muscara MN, De Nucci G, Antunes E. Mechanisms involved in the enhancement of allergic airways neutrophil influx by permanent C-fiber degeneration in rats. J Pharmacol Exp Ther 2005; 313: 440–8. [DOI] [PubMed] [Google Scholar]
- 34.Shen B, Smith RS, Jr, Hsu YT, Chao L, Chao J. Kruppel-like factor 4 is a novel mediator of Kallistatin in inhibiting endothelial inflammation via increased endothelial nitric-oxide synthase expression. J Biol Chem 2009; 284: 35471–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Montecucco F, Pende A, Alessandra Q, Mach F. Inflammation in the pathophysiology of essential hypertension. J Nephrol 2011; 24: 23–34. [DOI] [PubMed] [Google Scholar]
- 36.Singh MV, Chapleau MW, Harwani SC, Abboud FM. The immune system and hypertension. Immunol Res 2014; 59: 243–53. [DOI] [PMC free article] [PubMed] [Google Scholar]