

Abstract
The important regulator of cardiac function, cAMP, is hydrolyzed by different cyclic nucleotide phosphodiesterases (PDEs), whose expression and activity are not uniform throughout the heart. Of these enzymes, PDE2 shapes β1 adrenoceptor-dependent cardiac cAMP signaling, both in the right and left ventricular myocardium, but its role in regulating β2 adrenoceptor-mediated responses is less well known. Our aim was to investigate possible differences in PDE2 transcription and activity between right (RV) and left (LV) rat ventricular myocardium, as well as its role in regulating β2 adrenoceptor effects. The free walls of the RV and the LV were obtained from Sprague–Dawley rat hearts. Relative mRNA for PDE2 (quantified by qPCR) and PDE2 activity (evaluated by a colorimetric procedure and using the PDE2 inhibitor EHNA) were determined in RV and LV. Also, β2 adrenoceptor-mediated effects (β2-adrenoceptor agonist salbutamol + β1 adrenoceptor antagonist CGP-20712A) on contractility and cAMP concentrations, in the absence or presence of EHNA, were studied in the RV and LV. PDE2 transcript levels were less abundant in RV than in LV and the contribution of PDE2 to the total PDE activity was around 25% lower in the microsomal fraction of the RV compared with the LV. β2 adrenoceptor activation increased inotropy and cAMP levels in the LV when measured in the presence of EHNA, but no such effects were observed in the RV, either in the presence or absence of EHNA. These results indicate interventricular differences in PDE2 transcript and activity levels, which may distinctly regulate β2 adrenoceptor-mediated contractility and cAMP concentrations in the RV and in the LV of the rat heart.
Keywords: PDE2 activity, interventricular differences, β2 adrenoceptor, inotropy, cAMP levels
Introduction
The heart can be considered as two independent pumps, one delivering blood to the lungs and other to the rest of the body. Because the contractile work performed by each pump is different, the anatomy as well as the mechanical and biochemical properties of the right and left ventricle (RV and LV, respectively) is also different.1,2
The second messenger cAMP is an important regulator of cardiac contractility and its cellular levels are determined by a balance between synthesis and degradation. Activation of β adrenoceptors by the sympathetic nervous system or circulating catecholamines is one of the major neurohormonal mechanisms controlling myocardial cAMP production. Stimulation of both β1 and β2 adrenoceptors can induce the activation of the stimulatory G protein (Gαs)/adenylylcyclase (AC)/cAMP/cAMP-dependent protein kinase A (PKA) signaling pathway that leads to the phosphorylation of several target proteins regulating excitation–contraction coupling within the cardiac myocyte.3 The β1 adrenoceptor is the most abundant subtype present in the mammalian heart, where it is expressed in a β1:β2 ratio of approximately 75:25%4,5 and it was initially thought to be the only one responsible for the inotropic response to catecholamines.6,7 However, recent studies indicate that β2 adrenoceptors can also provide inotropic support and are involved in the regulation of cardiac metabolism as well as in the pathogenesis of a number of diseases, including pathological hypertrophy, arrhythmia, and heart failure.8
Degradation of cAMP is regulated by the activity of different cyclic nucleotide phosphodiesterases (PDEs) that break down cAMP into 5′-AMP.9 On the basis of the structure, kinetics, and substrate specificity, PDEs can be grouped into different families,9 of which at least four (PDE1–4) are present in the heart of a variety of animal species, including humans.9 Among these families, PDE3 and PDE4 provide the major PDE activity in the heart, and their role in regulating cardiac cAMP signaling has been extensively studied.10–12 There is evidence that PDE2 may also play a significant role in modulating cardiac function. Indeed, a PDE2-dependent decrease of cAMP has been reported to be responsible for attenuating β-adrenergic signaling in rabbit atrioventricular nodal cells.13 In human cardiac myocytes, PDE2 regulates L-type calcium current, which is involved both in heart rate and contractility.14 Accordingly, β1 adrenoceptor activation, along with PDE2 inhibition, results in a greater production of cAMP in left ventricular myocytes15 as well as in the right ventricular free wall of rat heart.16 These data imply that PDE2 attenuates cAMP production induced by β1 adrenoceptor agonists both in the right and left ventricular myocardium, but the regulation of cardiac β2 adrenoceptor-mediated responses by PDE2 is less well known. Although PDE2 is expressed in a wide variety of tissues, including ventricular myocardium,9 it is unclear whether PDE2 activity and expression are equally distributed in right and left ventricular myocardium. Therefore, the aim of the present study was to determine the differences in PDE2 transcription and activity in RV and LV, in addition to determining its role in regulating β2 adrenoceptor effects on cardiac function.
Materials and methods
Animals
The study was performed in accordance with the European Union Council Directive of 22 September 2010 (2010/63/EU) and reviewed and approved by the Ethical Committee of the University of Murcia.
Sprague–Dawley rats (250–350 g) were kept under standardized conditions: 12 h-light/dark circle, 22℃, and 70% humidity. Food and water were available ad libitum. Animals were stunned by a blow to the head and exsanguinated. The chest was opened and the heart rapidly removed and placed in Tyrode solution of the following composition (mmol/L): NaCl 136.9, KCl 5.0, CaCl2 1.8, MgCl2 1.5, NaH2PO4 0.4, NaHCO3 11.9, and dextrose 5.0.
RNA isolation and quantitative PCR (qPCR)
To determine PDE2A transcripts, which is the only PDE2 variant expressed in rat and human cardiac tissues,17 RV and LV were powdered on dry ice and RNA was isolated using TRI Reagent® according to the manufacturer’s instructions. RNA concentration was measured with a NanoDrop 1000 spectrophotometer (Thermo Fisher Scientific, Rockford, IL, USA). One microgram of RNA was treated with DNase I (Invitrogen™, Thermo Fisher Scientific) and then reversed transcribed using hexanucleotide mix (Roche Diagnostics, Hoffmann-La Roche, Basel, Switzerland) and SuperScript™ II reverse transcriptase (Invitrogen™, Thermo Fisher Scientific). Relative mRNA was quantified by qPCR on a StepOnePlus system (Applied Biosystems®, Thermo Fisher Scientific) using Power SYBR® Green PCR Master Mix (Applied Biosystems®, Thermo Fisher Scientific). Analysis was performed by the ΔΔCT method using TATA binding protein (TBP) as endogenous control. TBP transcript levels did not differ between RV and LV. Primer sequences were as follows:
PDE2A forward CGATCCAACACTGCTTCCACTAC,
PDE2A reverse CTTCTGGAAGGCCAAGGTACTG,
TBP forward ATGAGAATAAGAGAGCCACGAACA
TBP reverse TGCACACCATTTTCCCAGAA.
Subcellular fractionation and protein content
Right or left ventricle free walls (∼3 g wet weight) were trimmed of connective tissue, pooled and quick-frozen in liquid nitrogen. Cytosolic and microsomal fractions were obtained as previously described.18 Briefly, samples were homogenized on ice in a sucrose-containing buffer by three 10-s bursts using a Polytron homogenizer (Brinkmann Instruments, Westbury, NY, USA). Next, samples were centrifuged at 5900 g for 10 min at 4℃ and then the supernatant was collected and subsequently centrifuged at 51,000 g for 60 min. The supernatant was saved as the cytosolic fraction, i.e. right ventricle cytosol (RVC) or left ventricle cytosol (LVC), and the pellet was resuspended in a medium containing 0.6 mol/L KCl and resedimented at 100,000 g for 40 min to obtain the microsomal fraction, i.e., right ventricle microsomes (RVM) or left ventricle microsomes (LVM). To remove nucleotides and phosphate from the cytosolic extracts, samples were centrifuged at 800 g for 1 min using chromatography minicolumns (38 × 100 mm) filled with BioGel P-6DG (BioRad, Hercules, CA, USA) and pre-equilibrated with 10 mmol/L Tris–HCl buffer (pH 7.4).19 Isolated samples were aliquotted and stored at −80℃ until use.
Protein concentration was measured by the bicinchoninic acid method20 using the BCA Protein Assay Kit from Pierce® (Thermo Fisher Scientific). When the protein content was referred to as g of wet tissue, the yield of protein was 9.1 ± 1.0 mg/g in RVM and 8.5 ± 0.8 mg/g in LVM. Similarly, 24.9 ± 1.3 mg/g and 23.3 ± 0.5 mg/g were the protein yields in RVC and LVC, respectively.
PDE activity
A two-step colorimetric procedure adapted to a microplate format was used.21 In this assay, the 5′-AMP (Enzo Life Sciences, Farmingdale, NY, USA) released after cleavage of the substrate cAMP was further hydrolyzed into adenosine and inorganic phosphate (Pi) by the presence of excess 5′-nucleotidase (Enzo Life Sciences). The time-dependent Pi accumulation was quantified by the malachite green reagent.22 The linear dependence between standard 5′-AMP and Pi was first determined. The reaction was carried out in a 96-well plate containing 10 mmol/L Tris-HCl (pH 7.4), 25 mmol/L NaCl, 0.2 mmol/L MgCl2, 0–50 µmol/L 5′-AMP, and 5′-nucleotidase from Crotalus atrox venom at 50 U/µL in final volumes of 100 µL. The reaction was allowed to proceed at 37℃ for 30 min and stopped by adding 200 µL malachite green reagent. After 1 min at room temperature, 10 µL of 34% sodium citrate was added and the plate was shaken at room temperature for another 30 min for colour development. The 5′-AMP standard curve was constructed by plotting nmol of 5′-AMP versus absorbance at 660 nm. The PDE activity was also measured in the same microplate format. The final composition in each well was 10 mmol/L Tris–HCl (pH 7.4), 25 mmol/L NaCl, 0.2 mmol/L MgCl2, 50 U/µL 5′-nucleotidase and the corresponding aliquot of microsomal or cytosolic extract. After equilibration at 37℃, the reaction was started by adding 100 µmol/L cAMP. The enzymatic reaction was stopped at different times by adding 200 µL malachite green reagent and the samples were processed as described above. The time course of cAMP hydrolysis was evaluated by plotting absorbance at 660 nm versus time. The blank was performed by adding malachite green reagent before cAMP. PDE2 activity in the subcellular fractions was deduced by subtraction when the PDE activity was measured in the absence or presence of 30 µmol/L erythro-9-[2-hydroxy-3-nonyl] adenine (EHNA). Specific activities are expressed as nmol Pi/min/mg protein, which is equivalent to nmol 5′-AMP/min/mg protein.
Paced rat ventricular tissues
Right ventricular strips (1 mm wide, 10 mm long and 0.5 mm thick) and left ventricular papillary muscles were mounted longitudinally between two platinum electrodes, in Tyrode solution maintained at 37℃, pH 7.4, and gassed with 95% O2–5% CO2. The preparations were electrically stimulated (Grass SD-9 stimulator, Quincy, MA, USA) at a frequency of 1 Hz, 1 ms of duration and supramaximal voltage (threshold + 25%). A length–force curve was obtained and the tissues left at the length associated with the maximal developed force.23 Contractions were measured using a force–displacement transducer (Grass FT-03, Quincy, MA, USA) and displayed on a computer screen using a Stemtech amplifier (Stemtech Inc., Houston, TX, USA) and ACODAS software (Dataq Instruments, Inc., Akron, OH, USA). Tissues were allowed to equilibrate for 45–60 min before drug challenge.
To investigate β2 adrenoceptor-mediated inotropic effects, concentration–response curves for salbutamol were performed in the presence of the β1 adrenoceptor antagonist (±)-2-hydroxy-5-(2-((2-hydroxy-3-(4-(1-methyl-4-(trifluoromethyl)-1H-imidazol-2-l)phenoxy)propyl)amino)ethoxy)-benzamide methanesulfonate (CGP-20712A). Salbutamol concentrations were increased stepwise by 0.5 log unit as soon as the response to the previous concentration had stabilized. Cumulative concentration–response curves for β2 adrenoceptor-mediated effects of salbutamol in the presence of 300 nmol/L CGP-20712A were determined in the absence or 15 min after the addition of 10 µmol/L EHNA, which effectively inhibits PDE2 activity in rat myocardium.10 EHNA is also an inhibitor of adenosine deaminase, which catalyzes the metabolism of adenosine.9 To investigate the possible involvement of adenosine in the results obtained with EHNA, we also obtained concentration–response curves for salbutamol and CGP-20712A in the presence of 10 µmol/L 2′-deoxycoformycin (DCF), which inhibits adenosine deaminase but does not affect PDE2.24 Only one concentration–response curve for salbutamol and CGP-20712A in the absence or presence of the above mentioned drugs was determined in the same ventricular tissue. Drugs were added to a 30 mL organ bath in a volume smaller than or equal to 0.1 mL. Inotropic responses were expressed as percentages of the basal force. The relaxation phase of the isometric twitch was also assessed by measuring the time-to-half relaxation (t1/2), a standard index of isometric relaxation in mammalian myocardium.25
Measurement of cAMP
The levels of cAMP were measured by radioimmunoassay [125I] TME-S-cAMP (Diagnostic Pasteur, France) according to the manufacturer’s instruction, using strips of RV or LV (50–70 mg). CGP-20712A at 300 nmol/L was present in the media to produce an effective blockade of β1 adrenoceptors, and cAMP was measured in the absence or 15 min after exposure to 10 µmol/L EHNA. The incubation time was similar to that previously used for studying the effect of this PDE inhibitor on intracellular cAMP levels.10,16 To investigate the effects of β2 adrenoceptor activation on cAMP levels, salbutamol was added and the effect was evaluated after 3 min in the absence or presence of EHNA. The effect of salbutamol in the presence of DCF on cAMP levels was also determined. After incubation with the drugs, the tissues were immediately frozen. The preparation was weighed and homogenized in 1.5 mL of cold perchloric acid (0.3 mol/L) using a Polytron homogenizer and was then centrifuged at 10,000 g at 4℃ for 15 min. The supernatants were treated with potassium phosphate until pH of 6.2 was reached. The sensitivity of the assay was 2 pmol/mL. Intra- and inter-assay coefficients of variation were 7.7% and 8.2%, respectively. The antibody cross-reacted 100% with 3′,5′-cAMP and less than 0.3% with other nucleotides.
Chemicals
Chemicals used for this study, unless otherwise noted, were purchased from Sigma-Aldrich (St Louis, MO, USA). DCF was a gift from Pfizer S.A. Madrid, Spain.
Statistical analysis
Results are expressed as mean ± S.E.M. Student’s t-test or one-way analysis of variance followed by the Fisher LSD post hoc test were used for multiple comparisons. The criterion for significance was P < 0.05.
Results
PDE2A mRNA levels and activity
Quantitative PCR analysis performed on rat ventricular tissue indicated that transcript levels of PDE2A were less abundant in RV than in LV (Figure 1). Moreover, when the right or left ventricle free walls were subjected to subcellular fractionation, total PDE activity was lower in the RVM fraction (0.73 ± 0.07 nmol Pi/min/mg protein) than in the LVM fraction (1.09 ± 0.09 nmol Pi/min/mg protein) (n = 4–6 experiments, P < 0.05) (Figure 2a). However, total PDE activity was 2.14 ± 0.14 nmol Pi/min/mg protein in RVC and 2.50 ± 0.17 nmol Pi/min/mg protein in LVC (n = 5–9 experiments). In this case, the difference between the two values did not reach statistical significance.
Figure 1.
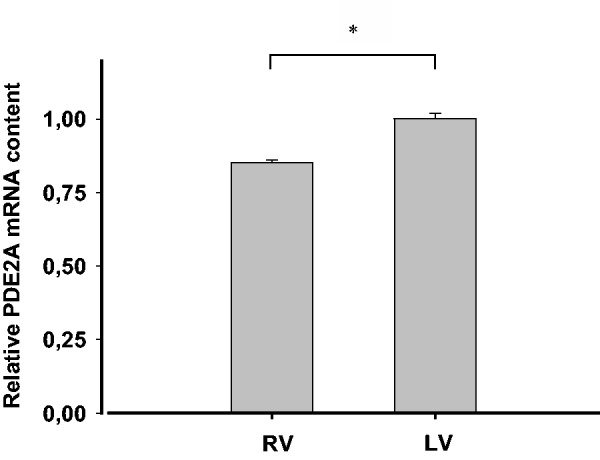
PDE2A (the only PDE2 variant expressed in both rat and human myocardium) is differentially transcribed in the right and left ventricles. Relative quantification of PDE2A mRNA levels in RV or LV, respectively (n = 6 per group). Values are expressed as mean ± SEM., *P < 0.05
Figure 2.
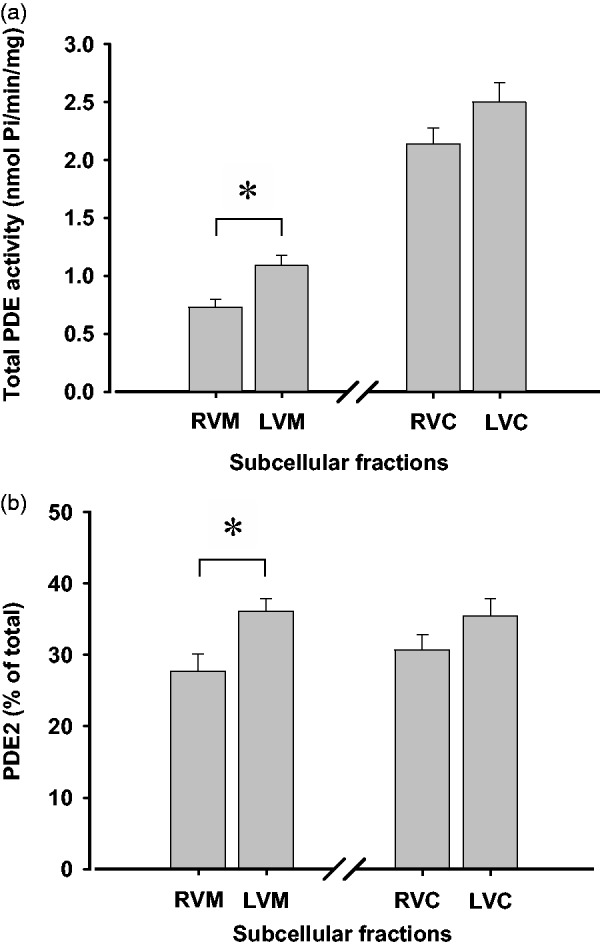
Total PDE activity and percentage of PDE2 activity in subcellular fractions of right and left ventricle of rat heart. (a) Total PDE activity expressed as nmol Pi/min/mg protein was measured in RVM, LVM, RVC, and LVC using 100 µmol/L cAMP as a substrate. Values are the mean ± S.E.M., *P < 0.05. (b) PDE2 activity was expressed as percentage with respect to the corresponding total PDE activity displayed in panel (a). PDE2 activity in nmol Pi/min/mg protein was 0.20 ± 0.02 in RVM, 0.39 ± 0.02 in LVM, 0.66 ± 0.04 in RVC, and 0.89 ± 0.06 in LVC. Values are the mean ± S.E.M., *P < 0.05
The next aim was to evaluate the inhibitory effect of EHNA and thus estimate the contribution of PDE2 to the total PDE activity. PDE2 activity was also lower in RVM (0.20 ± 0.02 nmol Pi/min/mg protein) than in LVM (0.39 ± 0.02 nmol Pi/min/mg protein) (n = 4–9 experiments, P < 0.05). Similar experiments revealed that PDE2 activity was 0.66 ± 0.04 nmol Pi/min/mg protein in RVC and 0.89 ± 0.06 nmol Pi/min/mg protein in LVC (n = 4–9 experiments), but no significant difference between the two values was found. When expressed as percentage of total PDE activity in the corresponding fraction, PDE2 activity was 27.4% in RVM, 35.8% in LVM, 30.8% in RVC, and 35.6% in LVC (Figure 2b). Therefore, the contribution of PDE2 was around 25% lower in RVM than in LVM, whereas the difference between RVC and LVC was not statistically significant.
Contractility
When used alone, the PDE2 inhibitor EHNA had no significant effect in our preparations of RV (3.3 ± 0.5 mN versus 3.5 ± 0.3 mN in the absence or presence of 10 µmol/L EHNA, respectively; n = 4) and LV (3.9 ± 0.3 mN versus 4.1 ± 0.4 mN in the absence or presence of 10 µmol/L EHNA, respectively; n = 5). Moreover, β2 adrenoceptor stimulation (salbutamol + 300 nmol/mL CGP-20712A) in LV papillary muscle was devoid of inotropic effect, but in the presence of 10 µmol/L EHNA it produced a concentration-dependent increase in force generation (Emax: 35.2 ± 5.7%; −log EC50: 6.2 ± 0.03, n = 5) (Figure 3). In contrast, salbutamol plus CGP-20712A did not increase contractility in the absence or presence of EHNA when assayed in RV samples (Figure 3). We also studied whether EHNA affects the β2 adrenoceptor-mediated effects of salbutamol on relaxation time. Salbutamol and CGP-20712A in RV did not affect the relaxation time in the absence or presence of EHNA (Figure 4a). In contrast, 3 µmol/L salbutamol, in the presence of CGP-20712A, which produced a maximal inotropic response in LV, also reduced the relaxation time from 50 ± 1.9 ms to 40 ± 1.7 ms (n = 6, P < 0.05) when applied in the presence of 10 µmol/L EHNA in this tissue (Figure 4b). The combination of salbutamol and CGP-20712A failed to produce any effect on contractility or relaxation, either in LV or RV, when studied in the presence of DCF (data not shown).
Figure 3.

β2 adrenoceptor-mediated effects of salbutamol on the basal force of contraction in rat ventricular myocardium. β1 adrenoceptors were blocked in all cases by the presence of 300 nmol/L CGP-20712A. Representative traces, in electrically driven strips, taken from the free wall of right ventricle (a and b) or left ventricle (c and d) showing that in the right ventricular myocardium, salbutamol was devoid of contractile effect both in the absence (a) or in the presence of 10 µmol/L EHNA (b). However, in the left ventricular myocardium, salbutamol did not increase contractility alone (c), but produced a concentration-dependent inotropic effect in the presence of EHNA (d). (e) Cumulative concentration–response curves for the effect of salbutamol alone in the right (♦) or left (▴) ventricular myocardium and also in the right (▪) or left (•) ventricular myocardium when 10 µmol/L EHNA was present. Each point represents the mean ± S.E.M. of 5–6 experiments
Figure 4.

β2 adrenoceptor-mediated lusitropic responses to salbutamol (SAL) in right and left ventricular myocardium. Strips of rat right ventricle (a) or left ventricular papillary muscle (b) were exposed to 3 µmol/L salbutamol plus 300 nmol/L CGP-20712A to block β1 adrenoceptors either in the absence (dotted lines) or in the presence (solid lines) of the PDE2 inhibitor EHNA (10 µmol/L). Top: original registration. Bottom: time required to half relaxation time. Data are expressed as means ± S.E.M. of six experiments. *P < 0.05
Effects of salbutamol, in the presence of the β1 adrenoceptor antagonist CGP-20712A, and EHNA on cAMP levels
To investigate whether inotropy correlated with cAMP levels, we studied the effect of 1 µmol/L salbutamol, a concentration similar to its EC50 for the β2 adrenoceptor-mediated inotropic effect (see above), and 300 nmol/L CGP-20712A on cAMP concentrations when measured in the absence or presence of EHNA in rat RV and LV.
In RV, 10 µmol/L EHNA tended to increase cAMP levels, although this effect was not statistically significant when compared to the control (Figure 5a). In the same tissue, salbutamol failed to increase cAMP concentrations, alone or combined with EHNA (Figure 5a). However, EHNA and salbutamol in LV were devoid of any effect when used separately, but their combination produced an increase in cAMP levels (Figure 5b). DCF did not alter the basal cAMP content in RV (462 ± 53 pmol/g, control, n = 6 versus 457 ± 37 pmol/g, in the presence of DCF, n = 4, P > 0.05) or LV (463 ± 43 pmol/g, control, n = 6 versus 473 ± 34 pmol/g, in the presence of DCF, n = 4, P > 0.05). Neither did it modify the effect of salbutamol in RV (428 ± 41 pmol/g, control, n = 6 versus 443 ± 38 pmol/g, in the presence of DCF, n = 4, P > 0.05) or in LV (506 ± 37 pmol/g, control, n = 6 versus 492 ± 47 pmol/g, in the presence of DCF, n = 4, P > 0.05).
Figure 5.
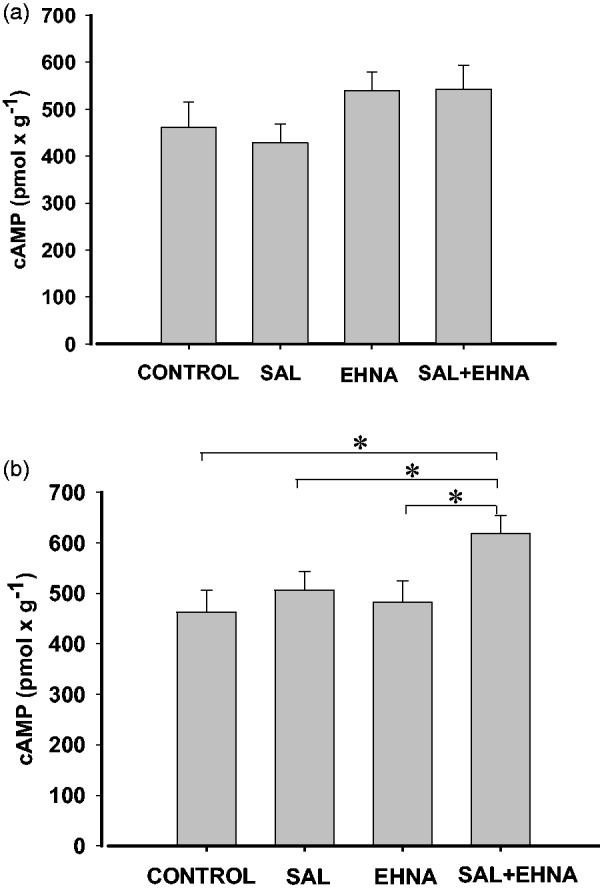
β2 adrenoceptor-mediated effects of salbutamol, in the presence of the β1 adrenoceptor antagonist CGP-20712A, on cAMP production, in rat right (a) or left (b) ventricular myocardium. cAMP production was induced by 1 µmol/L salbutamol (SAL) plus 300 nmol/L CGP-20712A in the absence or in the presence of 10 µmol/L EHNA. Values are ± S.E.M. of six experiments. *P < 0.05
Discussion
In the present study, we report for the first time, different expression and activity levels of PDE2 in RV and LV. Importantly, we also found that PDE2 differentially regulates β2 adrenoceptor-mediated cAMP production and contractility in the right and left ventricular myocardium.
Transcripts of the PDE2A gene have previously been identified in rat and human myocardium17 and now we show that PDE2A mRNA levels are significantly lower in RV compared to LV. The microsomal fraction of heart muscle is enriched in sarcoplasmic reticulum membrane and contains some key proteins involved in cardiac contractility, including PDE2.9 Our data on enzyme activity indicate that PDE2 activity and the contribution of PDE2 to the total PDE activity were significantly lower in the microsomal fraction of the RV with respect to the LV. It should be noted that PDE2 in the microsomal and cytosolic fractions was approximately one-third of the corresponding total PDE activity and differ from previous results in rat myocardium indicating that the PDE2 contribution was minimal15,26 or null.17 This discrepancy may be due to different assay conditions in which the catalytic activity was estimated. Assessment of catalytic activity depends on substrate concentration and the enzyme Michaelis constant (Km). Previous studies on PDE2 activity were performed in the presence of 0.5 µmol/L cAMP17 or 1 µmol/L cAMP.26,27 Given that Km for PDE2 is 30 µmol/L,28 a low substrate concentration will not reveal the maximal enzymatic activity, so we used 100 µmol/L cAMP for the evaluation of the PDE2 activity.
We also studied the interventricular regulation of β2 adrenoceptor-mediated contractile responses by PDE2. β2 adrenoceptors are present in rat ventricular myocardium, but their inotropic effects are blunted by PDE activity. In fact, a global PDE inhibition unmasks a β2 adrenoceptor-mediated inotropic effect in this tissue.8,29 In the present study, β2 adrenoceptor stimulation was devoid of inotropic effect both in RV and LV, which is consistent with previous results showing a lack of β2 adrenoceptor agonist-mediated inotropic effects in rodent myocardium.6,7 However, in the presence of EHNA, stimulation of the β2 adrenoceptor with salbutamol increased contractility in LV. The inhibitory effect of EHNA on adenosine deaminase9 does not seem to play any role in the response because no β2 adrenoceptor-mediated inotropic effect was observed in the presence of DCF, which also inhibits adenosine deaminase but is devoid of PDE2 inhibitory activity.24 β2 adrenoceptor activation also hastened relaxation in the presence of EHNA, in LV. This agrees with previous results in adult rat ventricular myocytes showing that the inhibition of PDE2 with EHNA slightly, but significantly enhanced the effect of β2 adrenoceptor stimulation on relaxation kinetics.30 In contrast, we observed that β2 adrenoceptor stimulation in RV failed to produce any inotropic or lusitropic effect in the absence or presence of EHNA.
To test whether inotropic or lusitropic responses correlate with cAMP levels, we measured the interventricular cAMP production induced by activation of β2 adrenoceptors in the absence or presence of EHNA. β2 adrenoceptor activation did not increase cAMP concentration, which is consistent with previous results whereby β2 adrenoceptor agonists failed to enhance intracellular cAMP levels in rat ventricular myocardium.29,31,32 In contrast, β1 adrenoceptor activation has been shown to raise cAMP levels in rat ventricular myocardium.16,32,33 It is known that β2 adrenoceptor-mediated signaling is locally confined, unlike the generalized cAMP production generated by β1 adrenoceptor activation.34 This is likely due to specific localization of β2 adrenoceptors inside the caveolae, whereas β1 adrenoceptors are also present in other cell fractions.35 Inside the caveolae, β2 adrenoceptors are colocalized with G protein, AC isoforms, PKA and PDEs, the last limiting the diffusion of cAMP produced by β2 adrenoceptor stimulation.31 In fact, the inhibition of PDE activity unmasks a β2 adrenoceptor-mediated increase in cAMP production in rat ventricular myocardium.29 The results of the present study showing that salbutamol + CGP-20712A in the presence of EHNA produces an increase in cAMP concentration in the left ventricular myocardium is consistent with this and indicates that PDE2 limits β2 adrenoceptor-mediated cAMP production in this tissue. In RV, however, we failed to detect any effect of salbutamol + CGP-20712A on cAMP levels, in the absence or presence of EHNA.
Taken together, our results demonstrate a differential interventricular PDE2 mRNA transcription and activity, being higher in LV. They also show that PDE2 inhibition unmasks a β2 adrenoceptor-mediated increase in cAMP concentrations in LV. It is known that the enhancement of cAMP levels activates PKA leading to the phosphorylation of different PKA substrates such as phospholamban, L-type calcium channel or ryanodine receptor which result in increased contractility and relaxation.3,25 This is consistent with the β2 adrenoceptor-mediated inotropic and lusitropic effects that we observed in LV when PDE2 was inhibited. A disparity in β2 adrenoceptor density does not seem to be responsible for this difference since it is virtually the same both in RV and LV of rat heart.36–38 A likely explanation is that different interventricular PDE2 activity distinctly modulates β2 adrenoceptor-mediated effects in the RV and LV. However, the possibility that interventricular variations in PDE2 distribution in intracellular microdomains also contribute to these effects cannot be excluded.
Right and left ventricles have different functions and consequently perform different contractile work, meaning that there are important anatomical and electrophysiological disparities between both ventricles.2,39 Importantly, proteomic analysis of both RV and LV has revealed that sarcomeric proteins such as myosin light chain 2, α-actin and myosin heavy chain, or metabolic proteins such as peroxiredoxin 3 which elicit an antioxidant effect at mitochondrial level, are differentially expressed.1 Some evidence also indicates that PDE activity may be different in right and left ventricular myocardium. Indeed, variations of PDE activities in different regions of rodent heart40,41 and higher PDE5 activity in RV than in LV of feline heart42 have been reported. Our results, showing a higher PDE2 mRNA transcripts and activity in left than in right rat ventricular myocardium, further support this view and suggest a different interventricular shaping of cAMP signals. This is likely to be of physiological significance and may contribute to better protecting the function of the LV (the most important heart chamber). Indeed, β2 adrenoceptors enhance cAMP tissue levels by activating the Gαs/AC pathway, which increases cardiac contractility but may also induce some deleterious cardiac effects such as arrhythmias, apoptosis, or pathological hypertrophy.8 β2-adrenoceptor also regulates an alternative signaling pathway through activation of the inhibitory G protein (Gαi) and the heterodimer formed by the β and γ subunits of the G protein (Gβγ).8 Besides the inhibition of AC, the main signaling pathway regulated by β2-adrenoceptors through Gαi/Gβγ appears to be the phosphatidylinositol 3-kinase (PI3K) signaling cascade which improves energy metabolism and protects against apoptosis in cardiac myocytes. Hydrolysis of cAMP by PDE2 may improve the benefit/risk profile of β2-adrenoceptors by reducing their cAMP-mediated deleterious effects without affecting their PI3K-mediated beneficial effects. This may be particularly true in the case of an increase in cAMP production, whether compartmentalized or throughout the cell.
It is known that the Km of PDEs is a key factor in shaping cAMP cell signaling,43 therefore PDE2 with a Km of 30 µmol/L28 is practically inactive at ∼1.2 µmol/L of cAMP, which is the basal level detected in adult ventricular myocytes.44 Other PDEs with lower Km, such as PDE3 and PDE4, are already acting at or close to saturation at the basal cAMP concentration. However, when transient increases of cAMP are elicited, as occurs after adrenergic stimulation, PDE2 may come into play and take on a relevant role in limiting cAMP enhancement. Interestingly, a marked PDE2 upregulation has recently been reported in failing rat and human hearts. This has been considered to be an important defence mechanism during cardiac stress, and the pharmacological activation of PDE2 has been proposed as a novel therapeutic strategy in heart failure.45
In conclusion, to the best of our knowledge, this study is the first to show differential interventricular expression and activity levels of PDE2. Our results also indicate that PDE2 differentially regulates β2 adrenoceptor-mediated cAMP production, inotropy, and lusitropy in RV and LV. These results suggest a different interventricular shaping of the cAMP signals, while the higher PDE2 activity in LV may contribute to better protecting this heart chamber from β2 adrenoceptor/cAMP-mediated cardiac toxicity. Further understanding of the detailed molecular mechanism of the chamber-specific regulation of PDE2 expression and its functional consequences will help us to understand the pathophysiological significance of these findings.
Authors’ contributions
JHC initiated the study and performed the experiments assessing cardiac contractility. FS and FFB carried out subcellular fractionation and PDE activity. JPS and CH determined PDE2A transcripts and TF measured cAMP tissue levels. All authors have contributed to writing this manuscript.
Acknowledgements
This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
References
- 1.Cadete VJ, Lin HB, Sawicka J, Wozniak M, Sawicki G. Proteomic analysis of right and left cardiac ventricles under aerobic conditions and after ischemia/reperfusion. Proteomics 2012; 12: 2366–77. [DOI] [PubMed] [Google Scholar]
- 2.Pandit SV, Kaur K, Zlochiver S, Noujaim SF, Furspan P, Mironov S, Shibayama J, Anumonwo J, Jalife J. Left-to-right ventricular differences in IKATP underlie epicardial repolarization gradient during global ischemia. Heart Rhythm 2011; 8: 1732–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Woo AY, Xiao RP. Beta-Adrenergic receptor subtype signaling in heart: from bench to bedside. Acta Pharmacol Sin 2012; 33: 335–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Steinberg SF. The molecular basis for distinct beta-adrenergic receptor subtype actions in cardiomyocytes. Circ Res 1999; 85: 1101–11. [DOI] [PubMed] [Google Scholar]
- 5.Brodde OE, Bruck H, Leineweber K. Cardiac adrenoceptors: physiological and pathophysiological relevance. J Pharmacol Sci 2006; 100: 323–37. [DOI] [PubMed] [Google Scholar]
- 6.Freyss-Beguin M, Griffaton G, Lechat P, Picken D, Quennedey MC, Rouot B, Schwartz J. Comparison of the chronotropic effect and the cyclic AMP accumulation induced by beta 2-agonists in rat heart cell culture. Br J Pharmacol 1983; 78: 717–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Juberg EN, Minneman KP, Abel PW. Beta 1- and beta 2-adrenoceptor binding and functional response in right and left atria of rat heart. Naunyn Schmiedeberg’s Arch Pharmacol 1985; 330: 193–202. [DOI] [PubMed] [Google Scholar]
- 8.Pérez-Schindler J, Philp A, Hernandez-Cascales J. Pathophysiological relevance of cardiac β2-adrenergic receptor and its potential as therapeutic target to improve cardiac function. Eur J Pharmacol 2013; 698: 39–47. [DOI] [PubMed] [Google Scholar]
- 9.Bender AT, Beavo JA. Cyclic nucleotide phosphodiesterases: molecular regulation to clinical use. Pharmacol Rev 2006; 58: 488–520. [DOI] [PubMed] [Google Scholar]
- 10.Verde I, Vandecasteele G, Lezoualc’h F, Fischmeister R. Characterization of the cyclic nucleotide phosphodiesterase subtypes involved in the regulation of the L-type Ca2+ current in rat ventricular myocytes. Br J Pharmacol 1999; 127: 65–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mongillo M, McSorley T, Evellin S, Sood A, Lissandron V, Terrin A, Huston E, Hannawacker A, Lohse MJ, Pozzan T, Houslay MD, Zaccolo M. Fluorescence resonance energy transfer-based analysis of cAMP dynamics in live neonatal rat cardiac myocytes reveals distinct functions of compartmentalized phosphodiesterases. Circ Res 2004; 95: 67–75. [DOI] [PubMed] [Google Scholar]
- 12.Vandecasteele G, Rochais F, Abi-Gerges A, Fischmeister R. Functional localization of cAMP signalling in cardiac myocytes. Biochem Soc Transac 2006; 34: 484–8. [DOI] [PubMed] [Google Scholar]
- 13.Han X, Kobzik L, Balligand JL, Kelly RA, Smith TW. Nitric oxide synthase (NOS3)-mediated cholinergic modulation of Ca2+ current in adult rabbit atrioventricular nodal cells. Circ Res 1996; 78: 998–1008. [DOI] [PubMed] [Google Scholar]
- 14.Fischmeister R, Castro L, Abi-Gerges A, Rochais F, Vandecasteele G. Species- and tissue-dependent effects of NO and cyclic GMP on cardiac ion channels. Comp Biochem Phys A 2005; 142: 136–43. [DOI] [PubMed] [Google Scholar]
- 15.Mongillo M, Tocchetti CG, Terrin A, Lissandron V, Cheung YF, Dostmann WR, Pozzan T, Kass DA, Paolocci N, Houslay MD, Zaccolo M. Compartmentalized phosphodiesterase-2 activity blunts beta-adrenergic cardiac inotropy via an NO/cGMP-dependent pathway. Circ Res 2006; 98: 226–34. [DOI] [PubMed] [Google Scholar]
- 16.Juan-Fita MJ, Vargas ML, Hernandez J. Comparative actions of diazepam and other phosphodiesterase inhibitors on the effects of noradrenaline in rat myocardium. Pharmacol Toxicol 2003; 93: 23–8. [DOI] [PubMed] [Google Scholar]
- 17.Johnson WB, Katugampola S, Able S, Napier C, Harding SE. Profiling of cAMP and cGMP phosphodiesterases in isolated ventricular cardiomyocytes from human hearts: comparison with rat and guinea pig. Life Sci 2012; 90: 328–36. [DOI] [PubMed] [Google Scholar]
- 18.Smith CJ, Huang R, Sun D, Ricketts S, Hoegler C, Ding JZ, Moggio RA, Hintze TH. Development of decompensated dilated cardiomyopathy is associated with decreased gene expression and activity of the milrinone-sensitive cAMP phosphodiesterase PDE3A. Circulation 1997; 96: 3116–23. [DOI] [PubMed] [Google Scholar]
- 19.Penefsky HS. Reversible binding of Pi by beef heart mitochondrial adenosine triphosphatase. J Biol Chem 1977; 252: 2891–9. [PubMed] [Google Scholar]
- 20.Smith PK, Krohn RI, Hermanson GT, Mallia AK, Gartner FH, Provenzano MD, et al. Measurement of protein using bicinchoninic acid. Anal Biochem 1985; 150: 76–85. [DOI] [PubMed] [Google Scholar]
- 21.Harder KW, Owen P, Wong LKH, Aebersold R, Clark-Lewis I, Jirik FR. Characterization and kinetic analysis of the intracellular domain of human protein tyrosine phosphatase β (HPTPβ) using synthetic phosphopeptides. Biochem J 1994; 298: 395–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lanzetta PA, Alvarez LJ, Reinach PS, Candia OA. An improved assay for nanomole amounts of inorganic phosphate. Anal Biochem 1979; 100: 95–7. [DOI] [PubMed] [Google Scholar]
- 23.Gonzalez-Muñoz C, Nieto-Ceron S, Cabezas-Herrera J, Hernández-Cascales J. Glucagon increases contractility in ventricle but not in atrium of the rat heart. Eur J Pharmacol 2008; 587: 243–7. [DOI] [PubMed] [Google Scholar]
- 24.Rivet-Bastide M, Vandecasteele G, Hatem S, Verde I, Bénardeau A, Mercadier JJ, Fischmeister R. cGMP-stimulated cyclic nucleotide phosphodiesterase regulates the basal calcium current in human atrial myocytes. J Clin Invest 1997; 99: 2710–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Brutsaert DL, Sys SU. Relaxation and diastole of the heart. Physiol Rev 1989; 69: 1228–315. [DOI] [PubMed] [Google Scholar]
- 26.Rochais F, Abi-Gerges A, Horner K, Lefebvre F, Cooper DM, Conti M, Fischmeister R, Vandecasteele G. A specific pattern of phosphodiesterases controls the cAMP signals generated by different Gs-coupled receptors in adult rat ventricular myocytes. Circ Res 2006; 98: 1081–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Richter W, Xie M, Scheitrum C, Krall J, Movsesian MA, Conti M. Conserved expression and functions of PDE4 in rodent and human heart. Basic Res Cardiol 2011; 106: 249–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Martins TJ, Mumby MC, Beavo JA. Purification and characterization of a cyclic GMP-stimulated cyclic nucleotide phosphodiesterase from bovine tissues. J Biol Chem 1982; 257: 1973–9. [PubMed] [Google Scholar]
- 29.Gonzalez-Muñoz C, Fuente T, Hernandez-Cascales J. Phosphodiesterases inhibition unmask a positive inotropic effect mediated by beta2-adrenoceptors in rat ventricular myocardium. Eur J Pharmacol 2009; 607: 151–5. [DOI] [PubMed] [Google Scholar]
- 30.Macdougall DA, Agarwal SR, Stopford EA, Chu H, Collins JA, Longster AL, Colyer J, Harvey RD, Calaghan S. Caveolae compartmentalise β2-adrenoceptor signals by curtailing cAMP production and maintaining phosphatase activity in the sarcoplasmic reticulum of the adult ventricular myocyte. J Mol Cell Cardiol 2012; 52: 388–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Steinberg SF. Beta(2)-Adrenergic receptor signaling complexes in cardiomyocyte caveolae/lipid rafts. J Mol Cell Cardiol 2004; 37: 407–15. [DOI] [PubMed] [Google Scholar]
- 32.Rybin VO, Pak E, Alcott S, Steinberg SF. Developmental changes in beta2-adrenergic receptor signaling in ventricular myocytes: the role of Gi proteins and caveolae microdomains. Mol Pharmacol 2003; 63: 1338–48. [DOI] [PubMed] [Google Scholar]
- 33.Juan-Fita MJ, Vargas ML, Hernandez J. The phosphodiesterase 3 inhibitor cilostamide enhances inotropic responses to glucagon but not to dobutamine in rat ventricular myocardium. Eur J Pharmacol 2005; 512: 207–13. [DOI] [PubMed] [Google Scholar]
- 34.Nikolaev VO, Moshkov A, Lyon AR, Miragoli M, Novak P, Paur H, Lohse MJ, Korchev YE, Harding SE, Gorelik J. Beta2-adrenergic receptor redistribution in heart failure changes cAMP compartmentation. Science 2010; 327: 1653–7. [DOI] [PubMed] [Google Scholar]
- 35.Rybin VO, Xu X, Lisanti MP, Steinberg SF. Differential targeting of beta –adrenergic receptor subtypes and adenylyl cyclase to cardiomyocyte caveolae. A mechanism to functionally regulate the cAMP signaling pathway. J Biol Chem 2000; 275: 41447–57. [DOI] [PubMed] [Google Scholar]
- 36.Horinouchi T, Morishima S, Tanaka T, Suzuki F, Tanaka Y, Koike K, Miwa S, Muramatsu I. Pharmacological evaluation of plasma membrane beta-adrenoceptors in rat hearts using the tissue segment binding method. Life Sci 2006; 79: 941–8. [DOI] [PubMed] [Google Scholar]
- 37.Myslivecek J, Nováková M, Palkovits M, Krizanová O, Kvetnanský R. Distribution of mRNA and binding sites of adrenoceptors and muscarinic receptors in the rat heart. Life Sci 2006; 79: 112–20. [DOI] [PubMed] [Google Scholar]
- 38.Hardouin S, Bourgeois F, Toraasson M, Oubenaissa A, Elalouf JM, Fellman D, Dakhli T, Swynghedauw B, Moalic JM. β-Adrenergic and muscarinic receptor mRNA accumulation in the sinoatrial node area of adult and senescent rat hearts. Mech Age Dev 1998; 100: 277–97. [DOI] [PubMed] [Google Scholar]
- 39.Brunet S, Aimond F, Li H, Guo W, Eldstrom J, Fedida D, Yamada KA, Nerbonne JM. Heterogeneous expression of repolarizing, voltage-gated K+ currents in adult mouse ventricles. J Physiol 2004; 559: 103–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Demirel-Yilmaz E, Cenik B, Ozcan G, Derici M.K. Various phosphodiesterase activities in different regions of the heart alter the cardiac effects of nitric oxide. J Cardiovasc Pharmacol 2012; 60: 283–92. [DOI] [PubMed] [Google Scholar]
- 41.Hua R, Adamczyk A, Robbins C, Ray G, Rose RA. Distinct patterns of constitutive phosphodiesterase activity in mouse sinoatrial node and atrial myocardium. PLoS ONE 2012; 7: e47652–e47652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shan X, Margulies KB. Differential regulation of PDE5 expression in left and right ventricles of feline hypertrophy models. PLoS ONE 2011; 6: e19922–e19922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Matthiesen K, Nielsen J. Cyclic AMP control measured in two compartments in HEK293 cells: phosphodiesterase KM is more important than phosphodiesterase localization. PLoS ONE 2011; 6: e24392–e24392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Iancu RV, Ramamurthy G, Warrier S, Nikolaev VO, Lohse MJ, Jones SW, Harvey RD. Cytoplasmic cAMP concentrations in intact cardiac myocytes. Am J Physiol Cell Physiol 2008; 295: C414–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mehel H, Emons J, Vettel C, Wittköpper K, Seppelt D, Dewenter M, Lutz S, Sossalla S, Maier LS, Lechene P, Leroy J, Lefevre F, Varin A, Eschenhagen T, Nattel S, Dobrev D, Zimmermann WH, Nikolaev VO, Vandecasteele G, Fischmeister R, El-Armouche A. Phosphodiesterase-2 is upregulated in human failing hearts and blunts β-adrenergic responses in cardiomyocytes. J Am Coll Cardiol 2013; 62: 1596–606. [DOI] [PubMed] [Google Scholar]