

Abstract
A feature of allergic airway disease is the observed increase of nitric oxide (NO) in exhaled breath. Gram-negative bacterial infections have also been linked with asthma exacerbations. However, the role of NO in asthma exacerbations with gram-negative bacterial infections is still unclear. In this study, we examined the role of NO in lipopolysaccharide (LPS)-induced inflammation in an ovalbumin (OVA)-challenged mouse asthma model. To determine whether NO affected the LPS-induced response, a NO donor (S-nitroso-N-acetylpenicillamine, SNAP) or a selective inhibitor of NO synthase (1400W) was injected intraperitoneally into the mice before the LPS stimulation. Decreased levels of proinflammatory cytokines were demonstrated in the bronchoalveolar lavage fluid from mice treated with SNAP, whereas increased levels of cytokines were found in the 1400W-treated mice. To further explore the molecular mechanism of NO-mediated inhibition of proinflammatory responses in macrophages, RAW 264.7 cells were treated with 1400W or SNAP before LPS stimulation. LPS-induced inflammation in the cells was attenuated by the presence of NO. The LPS-induced IκB kinase (IKK) activation and the expression of IKK were reduced by NO through attenuation of the interaction between Hsp90 and IKK in the cells. The IKK decrease in the lung immunohistopathology was verified in SNAP-treated asthma mice, whereas IKK increased in the 1400W-treated group. We report for the first time that NO attenuates the interaction between Hsp90 and IKK, decreasing the stability of IKK and causing the down-regulation of the proinflammatory response. Furthermore, the results suggest that NO may repress LPS-stimulated innate immunity to promote pulmonary bacterial infection in asthma patients.
Keywords: Nitric oxide, heat shock protein 90, asthma, IκB kinase, nuclear factor κB
Introduction
Asthma is a chronic inflammatory respiratory disease manifesting as variable airflow obstruction leading to symptoms of coughing, wheezing, and dyspnea. It has been suggested that the pathogenesis of asthma is a result of the recruitment of inflammatory cells in the airway.1 Asthma is also associated with an increased risk of serious bacterial pneumonia, especially gram-negative and atypical bacterial infections.2 These organisms have been linked with the exacerbation of asthma and suggested as a possible mechanism in asthma pathogenesis. Mechanical, immunological, and phagocytic functions play important roles in the airways to protect the host from microbial infections. However, allergic airway inflammation may suppress innate immunity and reduce pulmonary antibacterial activity.3 In asthmatic individuals, increased nitric oxide (NO) is observed in the exhaled breath. The exhaled nitric oxide (eNO) is produced endogenously in the lung by nitric oxide synthase (NOS) to elicit multiple physiologic functions, including smooth muscle relaxation, neurotransmission, vascular tone, and host defense.4 Moreover, acute asthma treated with nebulized budesonide or oral montelukast sodium improves asthmatic symptoms and reduces eNO.5 The administration of l-arginine increases the NO metabolism associated with allergic airway inflammation, leading to the alleviation of a number of features of asthma.6 However, the inhibition of airway NO synthesis does not improve asthma; therefore, the role of NO in allergic disease exacerbated by gram-negative bacterial infections remains controversial.7
NO is a potent inhibitor of the expression of cytokines involved in stimulating human macrophages.8 In addition, inhaled NO may attenuate acute lung injury via the inhibition of nuclear factor kappa-B (NF-κB) and inflammation.9 NO decreases lipopolysaccharide (LPS)-stimulated NF-κB activation in alveolar macrophages and RAW 264.7 cells.10 In the presence of NO, the expression of inducible NOS (iNOS), interleukin (IL)-1β, IL-6, and tumor necrosis factor (TNF)-α mRNAs is decreased after LPS stimulation in macrophages.11 NF-κB transcription factors are activated by a wide variety of stimuli and control key aspects of immune function and development.12 NF-κB is also known to regulate allergy-associated cytokines and chemokines during inflammation. IκB kinase (IKK) is responsible for the activation of NF-κB through the phosphorylation of IκB, leading to degradation of IκB and subsequent translocation of NF-κB into the nucleus. A previous study suggested that the IKKβ inhibitor reduces allergic airway inflammation and hyperresponsiveness in a murine model of asthma.13,14 However, the molecular mechanism of the NO-mediated inhibition of the proinflammatory response remains unexplored.
Heat shock protein 90 (Hsp90) is a molecular chaperone that is induced in response to cellular stress to stabilize client proteins involved in cell cycle control, cell proliferation, and anti-apoptotic signaling.15 Increased Hsp90 expression could be associated with asthma severity.16 The inhibition of Hsp90 exerts anti-inflammatory effects, and Hsp90 has been proposed as an essential transiently acting regulatory component in IKK signaling.17,18 The inhibition of Hsp90 reduces the expression of proinflammatory mediators by decreasing the activation of NF-κB signaling pathways.19 In this study, we used LPS, the major component of the outer membrane of gram-negative bacteria, to show that NO might down-regulate IKK, which leads to a decrease in the LPS-stimulated proinflammatory response in an allergic mouse model and murine macrophages. This down-regulation resulted from a reduction in the interaction between Hsp90 and IKK, demonstrating that Hsp90 plays an important role in the NO-mediated attenuation of LPS-stimulated proinflammatory response. Furthermore, since inflammation is an important mechanism of innate immunity against infection, our results suggest that NO-mediated attenuation of LPS-stimulated inflammation may exacerbate bacterial pneumonia in asthma patients.
Materials and methods
Materials
Penicillin, streptomycin, fetal bovine serum, trypsin-EDTA, RPMI 1640 medium, and Lipofectamine® 2000 transfection reagent were purchased from Invitrogen (Carlsbad, CA, USA). The enzyme-linked immunoadsorbent assay (ELISA) kit was purchased from R&D (Minneapolis, MN, USA). LPS, 1400W, S-nitroso-N-acetylpenicillamine (SNAP), ovalbumin (OVA), and the other chemical compounds were obtained from Sigma-Aldrich (St. Louis, MO, USA). Reverse transcriptase was from Promega (Madison, WI, USA). The anti-IKKα/β, anti-Hsp90, anti-phospho-IKKα/β (Ser180/Ser181), and peroxidase-conjugated goat anti-rabbit antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA).
Murine model of allergic asthma
Adult BALB/c female mice (6–8 weeks old) were obtained from the National Laboratory Animal Center of Taiwan (Taipei, Taiwan). The mice were bred at the specific pathogen-free Animal Center at the National Defense Medical Center (Taipei, Taiwan) in accordance with the National Institutes of Health guidelines. OVA (100 µg) was adsorbed onto 4 mg of aluminum and was resuspended in a final volume of 0.2 mL phosphate-buffered saline (PBS). To generate allergic mice, the mice were sensitized by intraperitoneal (i.p.) injection of 100 µg of OVA on experimental days 0 and 14 and were challenged with intratracheal inhalation of 25 µg of OVA on day 28. The medication was inhaled into the lungs through normal breathing.20 In addition, on day 31, the mice were subjected to intratracheal inhalation of 50 µg LPS in 50 µL PBS to elicit a proinflammatory response. The mice were sacrificed using CO2 on day 32. Bronchoalveolar lavage (BAL) fluid was collected from the sacrificed mice by instilling 2 mL of PBS into the lungs three times via an intratracheal cannula. The cells were separated from the BAL fluid by centrifugation at 1200 rpm for 5 min. The BAL fluid was stored at −80℃ for the cytokine assay. All experimental procedures were approved by the Ethics Committee of Animal Research, National Defense Medical Center, and the investigation conformed to the guidelines of the National Institutes of Health for the care and use of laboratory animals.
Immunohistochemistry
The mouse lung tissues were collected, fixed in paraformaldehyde, and embedded in paraffin. Briefly, the 4-µm-thick slides were baked at 60℃ for 2 h, deparaffinized and rehydrated, and antigen retrieval was performed by boiling in sodium citrate buffer (10 mM, pH 6.0) for 30 min and then treated with 3% hydroxylperoxide for 10 min. The tissue sections were incubated at room temperature for 60 min in anti-IKKα/β (1:75) antibody and then in Super Enhancer™ Reagent (BioGenex Laboratories, Fremont, CA, USA) for 20 min. Chromogen development was performed using 3,3′-diaminobenzidine (DAB) staining for 5 min. Counterstaining was performed using hematoxylin and eosin. The slides were rinsed with water and covered with mounting medium after dehydration (Faramount Aqueous S3025; Dako, Denmark).
Cell culture
RAW 264.7 cells were obtained from the American Type Culture Collection (ATCC; Manassas, VA, USA). The cells were maintained in RPMI 1640 medium supplemented with 10% fetal bovine serum, 1 mM glutamine, and antibiotics (10 U/mL penicillin and 10 µg/mL streptomycin) at 37℃ in a humidified chamber, under 5% CO2.
Enzyme-linked immunoadsorbent assay
The BAL fluids were collected from the mice treated with LPS, SNAP, 1400W, SNAP combined with LPS, and 1400W combined with LPS. The concentrations of IL-1β and TNF-α in the culture medium were measured using the R&D ELISA kit (Minneapolis, MN, USA) in accordance with the manufacturer’s protocol. The assays were performed in triplicate per mouse.
RNA isolation, reverse transcription, and real-time quantative PCR
Total RNA was isolated using the Ultraspec™-II Reagent (BIOTECX Laboratories, Houston, TX, USA). The primer sequences were as follows: GAPDH (NM_001289726.1) (95 bps), forward 5′-CCTGGAGAAACCTGCCAAGTA-3′, reverse 5′-GGTCCTCAGTGTAGCCCAAGA-3′; IL-1β (NM_008361.3) (89 bps), forward 5′-GCAACTGTTCCTGAACTCAACT-3′, reverse 5′-ATCTTTTGGGGTCCGTCAACT-3′; IL-12 (NM_008352.2) (123 bps), forward 5′-TGGTTTGCCATCGTTTTGCTG-3′, reverse 5′-ACAGGTGAGGTTCACTGTTTCT-3′; IL-6 (NM_031168.1) (305 bps), forward 5′-TGTGCAATGGCAATTCTGAT-3′, reverse 5′-GGAAATTGGGGTAGGAAGGA-3′; and iNOS (NM_010927.3) (158 bps), forward 5′-CCCTTCAATGGTTGGTACATGG-3′, reverse 5′-ACATTGATCTCCGTGACAGCC-3′. The cDNA was synthesized from 1 µg of total RNA with oligo-dT primers using reverse transcriptase. The PCRs were performed using the cDNA (5 µL of diluted reverse transcription product) as the template with SYBR1 Green PCRMaster Mix (Applied Biosystems, Warrington, UK) in the presence of primer oligonucleotide specific for each gene. The relative expression was quantified by the comparative Ct (cycle threshold) method using GAPDH as internal control.
Western blot and immunoprecipitation assays
The cell lysates were prepared using PBSTD lysis buffer (1% Nonidet P-40, 50 mM Tris-HCl pH 7.4, 1 mM Na3VO4, 1 mM EDTA, 1 mM PMSF and 1% protease inhibitor cocktail). The soluble lysates (40 µg) were mixed with an equal volume of SDS-sample buffer and were resolved by 12% SDS-PAGE. The proteins were transferred onto a nitrocellulose membrane, and the membrane was incubated with antibodies as indicated. After the primary antibody incubation, the membranes were washed three times in TBS-T and were incubated with HRP-conjugated (1:1000) anti-rabbit antibody. The signals were detected using the Fujifilm LAS-4000 BioSpectrum, and the intensity of the selected bands was analyzed using Fujifilm software. For the immunoprecipitation (IP), the samples were pre-cleared using 20 µL of anti-rabbit IgG IP beads (Millipore, Bedford, MA, USA) for 1 h, and the pre-cleared samples were incubated overnight with 2 µg of anti-IKKα/β antibody at 4℃. The antigen–antibody complex was collected using 20 µL of protein A-agarose beads. The beads were washed three times with lysis buffer and were eluted using sample buffer. Subsequently, Western blotting was performed, and the blots were probed with anti-Hsp90 or IKK antibody.
Cell transfection and NF-κB reporter assay
The RAW 264.7 cells (1 × 106) were seeded in 6-well plates and transfected on the following day with 2 µg of NF-κB reporter DNA using Lipofectamine 2000 in accordance with the manufacturer’s recommendations. After 2 days, the cells were treated with LPS (1 µg/mL) SNAP (0.5 mM) or 1400W (10 µM) for 6 h. The cells were lysed, and the luciferase activity was measured using a dual luciferase kit (Promega).
Statistical analysis
All cell experiments were conducted in triplicate and repeated for at least three independent experiments. An unpaired, two-tailed t-test was performed to test the significance of the correlation. One way ANOVA (SPSS version 18.0; SPSS inc, Chicago, IL, USA) followed by post hoc test with Bonferroni correction was performed for the animal work. A P < 0.05 was considered statistically significant.
Results
Nitric oxide attenuates LPS-induced airway inflammation in allergic mice
To generate the allergic mice, BALB/c mice were sensitized using an intraperitoneal injection of OVA on experimental days 0 and 14, and the mice were challenged using the intratracheal inhalation of OVA on day 28. Thereafter, the mice were stimulated by inhaling LPS to elicit a proinflammatory response mimicking the situation during pulmonary infections. To determine whether the NO affected the LPS-stimulated response, the mice were injected intraperitoneally with SNAP (NO donor, 50 µg/mouse)21 for 1 h before the LPS treatment on day 31. The changes in the concentrations of proinflammatory cytokines, TNF-α and IL-1β, in the BAL fluid were analyzed after the LPS inhalation for 24 h. Increased TNF-α and IL-1β levels were observed after the LPS treatment, indicating the LPS-stimulated responses (Figure 1). Significantly, the SNAP treatment decreased the LPS-induced TNF-α (Figure 1a) and IL-1β (Figure 1b) in the mice. These findings suggested that NO attenuates the LPS-induced airway inflammation in the allergic mice. To demonstrate whether the removal of NO may promote the LPS-stimulated proinflammatory response, we used the iNOS inhibitor 1400W to suppress NO production. After the treatment with 1400W, increased levels of LPS-stimulated TNF-α (Figure 1c) and IL-1β (Figure 1d) were observed, suggesting that the removal of NO promotes the LPS-stimulated proinflammatory response in allergic mice.
Figure 1.

NO inhibited LPS-induced proinflammatory cytokines in BAL fluid of the OVA-allergic mouse model. BAL fluid was collected from the OVA-allergic mice (n = 5), and the cytokines were determined using ELISA. The mice were treated with SNAP (50 µg), 1400W (250 µg) or PBS (0.2 mL) 1 h before the LPS inhalation. The concentration (pg/mL) of TNF-α (a and c) and IL-1β (b and d) was analyzed in the treatments as indicated. The bar indicates the means ± SD of five mice performed in triplicate. **P < 0.01 compared with control. #P < 0.05 and ##P < 0.01 compared with LPS-treated mice
Nitric oxide inhibits the LPS-induced proinflammatory response in RAW 264.7 cells
Macrophages are the major cells secreting proinflammatory cytokines in the BAL fluid after the airway administration of LPS in the mouse allergy model. To further explore the molecular mechanism of NO-mediated attenuation of the inflammation in macrophages, we used RAW 264.7 macrophages as a model. In the presence of NO, the LPS-induced IL-1β, IL-6, IL-12, and iNOS mRNA were reduced by approximately 33%, 25%, 33%, and 15%, respectively (Figure 2a). Because the NF-κB transcription factor is involved in the proinflammatory response, we measured the transcription activity of NF-κB following stimulation of the cells with LPS for 12 h after treatment with SNAP for 3 h. The LPS-induced NF-κB activation was decreased in the NO-treated cells, showing that the inhibition of NF-κB is consistent with the expression levels of the proinflammatory response genes (Figure 2b).
Figure 2.
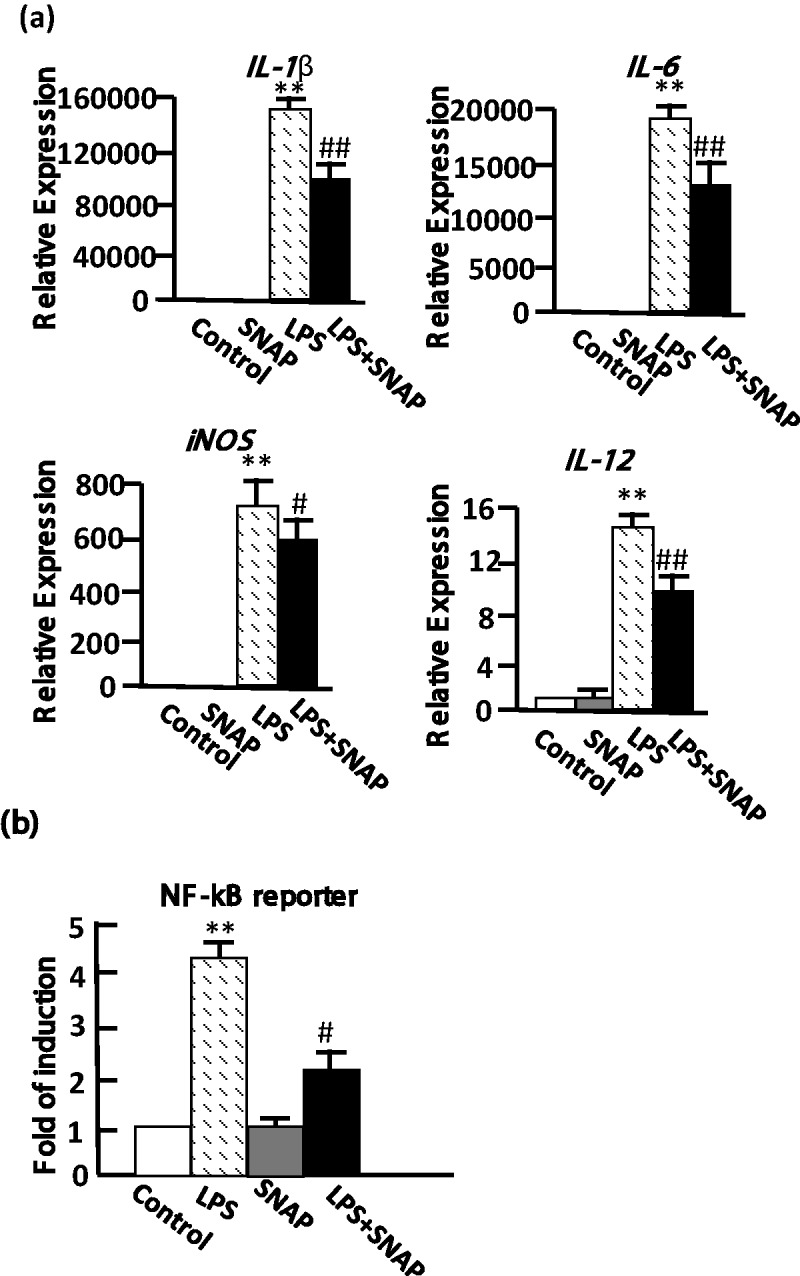
NO repressed LPS-induced proinflammatory genes and NF-κB activity in RAW 264.7 cells. (a) The gene expression was analyzed using RT-qPCR after LPS treatment of the cells. The IL-1β, IL-6, IL-12, and iNOS mRNA levels were determined after 0.5 mM SNAP for 10 h, 1 µg/mL LPS for 6 h, and 0.5 mM SNAP pretreatment for 4 h following 1 µg/mL LPS treatment for 6 h. The expression level of GAPDH was used as an expression control, and the genes were normalized to the level of GAPDH. Relative expression represents the expression level in treated versus control cells. (b) The NF-κB activity was determined using a transfected luciferase reporter plasmid containing the NF-κB binding sites. The NF-κB reporter activity was analyzed after 0.5 mM SNAP alone for 15 h, 1 µg/mL LPS alone for 12 h and pretreatment with 0.5 mM SNAP for 3 h before 1 µg/mL LPS treatment for 12 h. The data represent the means ± SD from three independent experiments in triplicate. #P < 0.05 and ##P < 0.01 compared with the value of LPS-treated cells. **P < 0.01 compared with control
Nitric oxide reduces the expression of IKK
Since IKK is an upstream kinase and plays an important role in the activation of NF-κB, we used anti-IKK and anti-phospho-IKK (pIKK) to analyze whether NO is involved in the regulation of IKK. The expression of IKK was decreased after the SNAP pretreatment (Figure 3a) compared with the untreated cells. Our findings demonstrate that the NO suppressed the LPS-stimulated proinflammatory response resulting from the down-regulation of IKK. The expression level of IKK after the SNAP treatment for 1, 4, and 6 h decreased in a time-dependent manner by 20%, 30%, and 40%, respectively (Figure 3b). The degradation was inhibited by the proteosome inhibitor MG-132, indicating that the NO-induced IKK degradation occurs via the proteosome-dependent pathway (Figure 3c).
Figure 3.
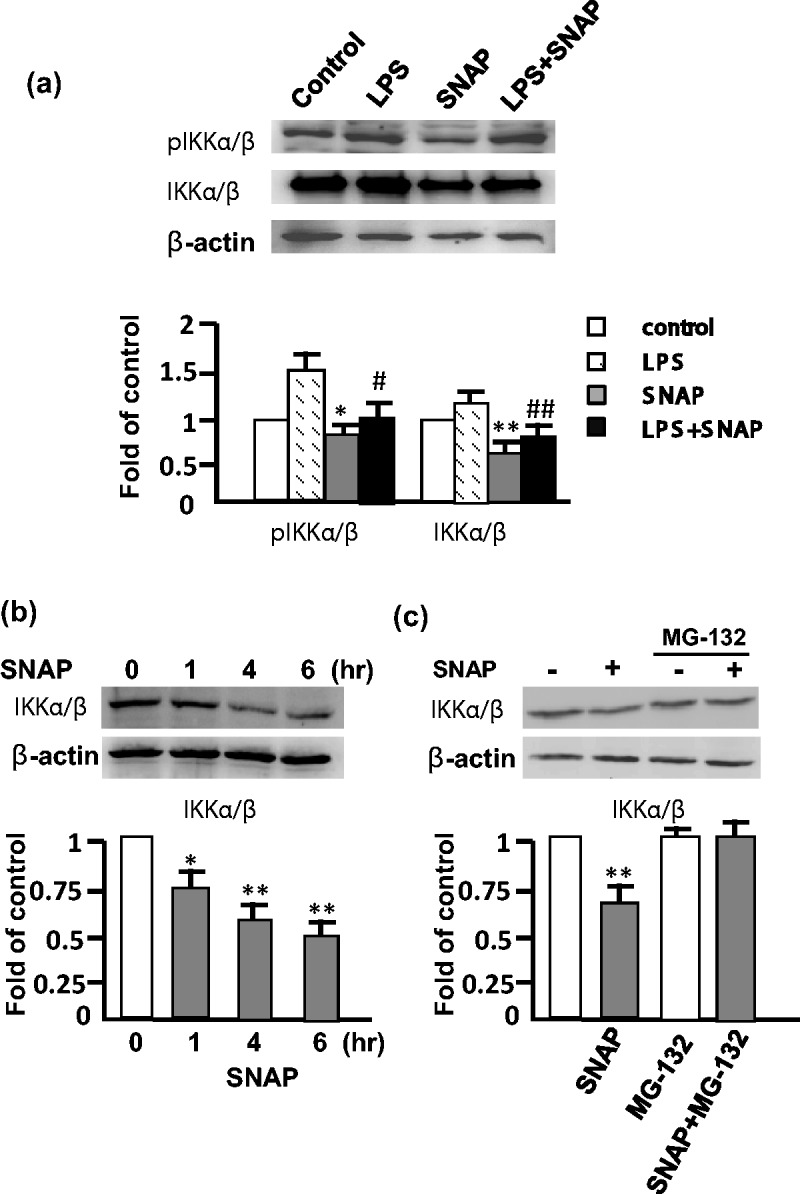
NO reduced IKK expression and activation by proteosomal degradation in RAW 264.7 cells. (a) The expression of IKK and phospho-IKK (pIKK) was analyzed by Western blot after the treatment of 0.5 mM SNAP for 4 h, and 0.5 mM SNAP for 3 h followed by 1 µg/mL LPS for 1 h. (b) The cells were treated with SNAP for 0, 1, 4 and 6 h, and IKK was analyzed by Western blot. (c) IKK expression in the presence or absence of 10 µM MG-132 for 30 min before the treatment of 0.5 mM SNAP for 3 h. Actin was used as the loading control. The level of IKK and pIKK was normalized to actin, and fold of control represents the level in treated versus control cells. The experiment was repeated three times, and a representative image is herein shown. *P < 0.05 and **P < 0.01 compared with control cells. #P < 0.05 and ##P < 0.01 compared with LPS-treated cells
Nitric oxide attenuates the interaction of Hsp90 with IKK
Hsp90 is a crucial regulator of the IKK signalosome via the interaction with Hsp90 leading to stabilization of IKK. To analyze whether NO interferes with the interaction, we performed an immunoprecipitation using the anti-IKKα/β antibody, and the presence of Hsp90 was detected by Western blots using the anti-Hsp90 antibody. Significantly, the Hsp90 interacting with IKK was decreased by 40% in the cells pretreated with SNAP for 1 h, and the pretreatment with SNAP for 6 h decreased the interaction by more than 60% (Figure 4a and b). In contrast, the amount of total Hsp90 was not changed after SNAP treatment, but the reducing interaction of Hsp90 with IKK and the decrease of IKK were observed at 1 h and at 4 h after SNAP treatment, respectively (Figure 4c). Therefore, NO is able to disrupt the interaction of Hsp90 with IKK. Our findings demonstrate that the inhibition of inflammation by NO was attributed to the decrease in the interaction of Hsp90 with IKK leading to a down-regulation of IKK.
Figure 4.
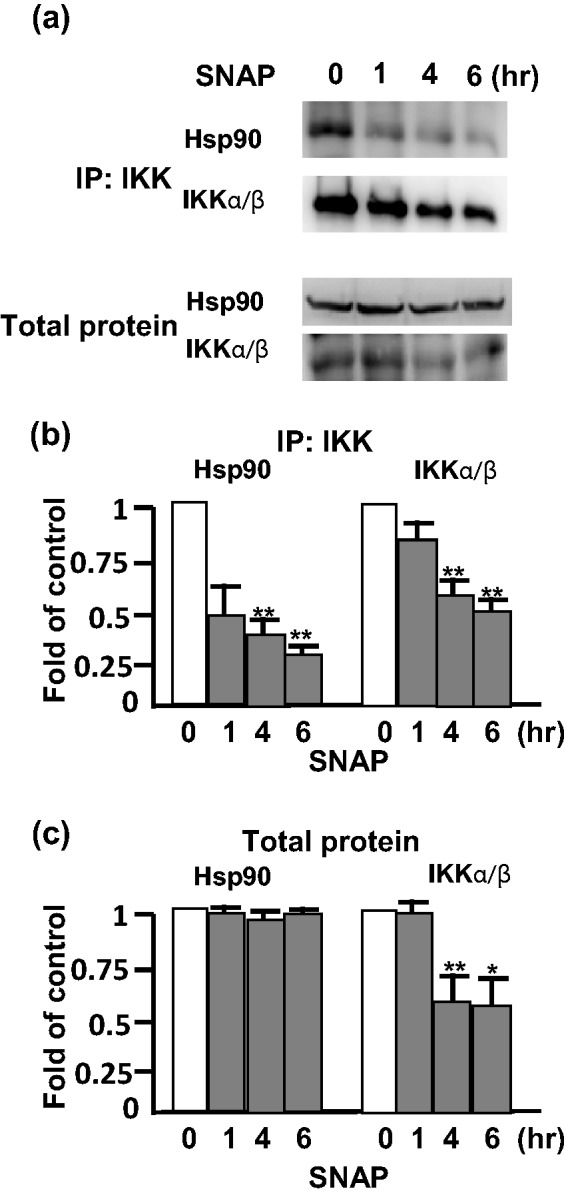
Inhibition of IKK interacting with Hsp90 by NO in RAW 264.7 cells. (a) The cells were pretreated with 0.5 mM SNAP for 0, 1, 4, and 6 h. The interaction of IKK with Hsp90 was analyzed by immunoprecipitation (IP) with anti-IKK antibody, and the presence of Hsp90 and IKK was examined by Western blot using anti-Hsp90 and anti-IKK antibody, respectively. (b) Results from IP and (c) total protein were showed as fold of control. The experiment was repeated three times, and a representative image is herein shown. *P < 0.05 and **P < 0.01 compared with untreated cells
Removal of NO enhances the expression of IKK in the infiltrated monocytes
Since NO blocked the interaction of Hsp90 with IKK, we analyzed whether the iNOS inhibitor 1400W enhances the amount of IKK by removal of NO after LPS stimulation. Our findings show that the inhibition of iNOS was enhanced 50% in the interaction of Hsp90 with IKK, as determined by the co-immunoprecipitation (Figure 5a). Moreover, IKK was increased after 1400W treatment compared with untreated cells. Consistent with the expression of IKK, the LPS-stimulated NF-κB transcription activity was also increased after removal of NO by 1400W (Figure 5b). To further validate the histopathology of NO in the lung, the allergic mice treated with LPS showed infiltrated cells containing dense monocytes around the blood vessels and bronchioles (Figure 6). The IKKα/β-positive cells were stained (brown) in the DAB chromogen assay, exhibiting the expected cytoplasmic staining pattern (Figure 6a). The scattered and dense staining of IKK in the immunostaining was observed in the inflammatory infiltrated monocytes. The numbers of monocytes expressing IKK were decreased in the infiltrated cells pretreated with SNAP but increased following the 1400W treatment (Figure 6b). Our findings demonstrate that NO is able to reduce the number of infiltrated monocytes expressing IKK in the lung of OVA-stimulated allergic mice after LPS-induced inflammation, whereas removal of NO increases the infiltrated monocytes in the infiltrated inflammatory cells.
Figure 5.
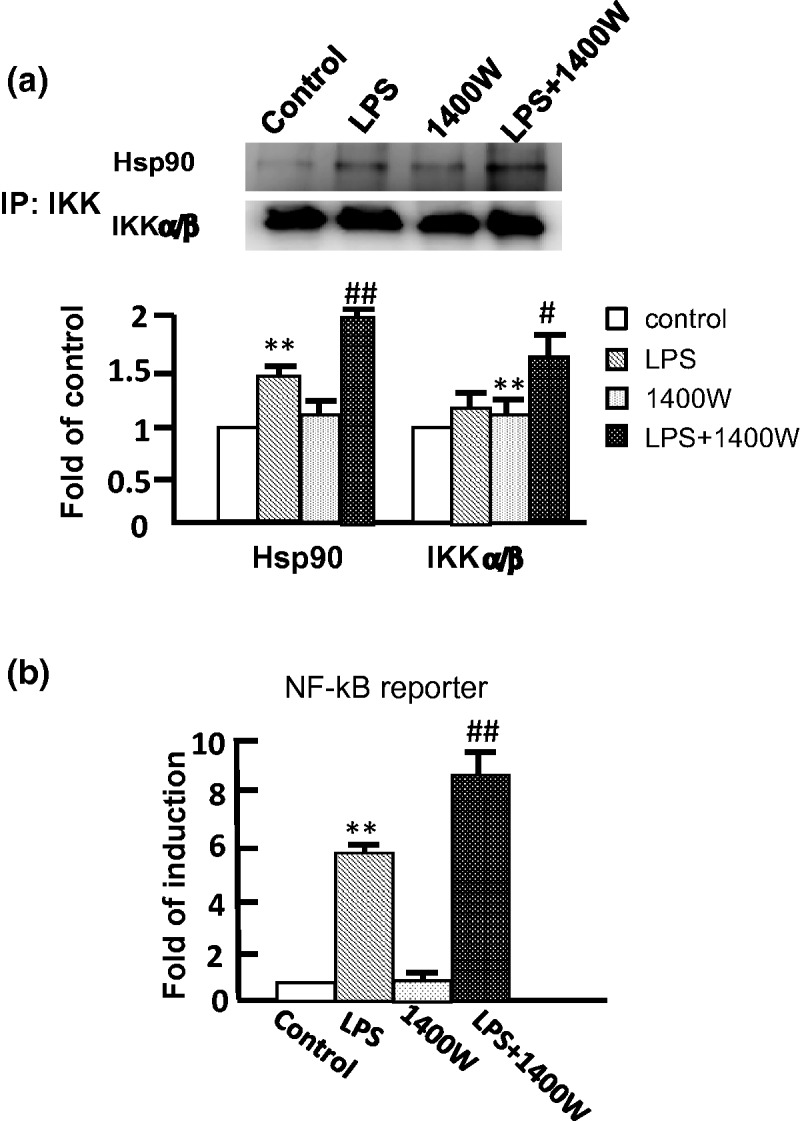
Increase in the interaction of IKK with Hsp90 by removal of NO in RAW 264.7 cells. (a) The cells were pretreated with 1400W (10 µM) and then treated LPS for 6 h. The interaction of IKK with Hsp90 was analyzed by immunoprecipitation (IP) with anti-IKK antibody, and the presence of Hsp90 and IKK was examined by Western blot using anti-Hsp90 and anti-IKK antibodies, respectively. Results from IP were shown as fold of control. The experiment was repeated three times, and a representative image is herein shown. (b) The NF-κB reporter activity was determined after the treatment of 10 µM 1400W alone for 6 h, 1 µg/mL LPS alone for 6 h, co-treatment with 10 µM 1400W, and 1 µg/mL LPS for 6 h. The data represent the means ± SD from three independent experiments. **P < 0.01 compared with control. #P < 0.05 and ##P < 0.01 compared with LPS-treated cells
Figure 6.
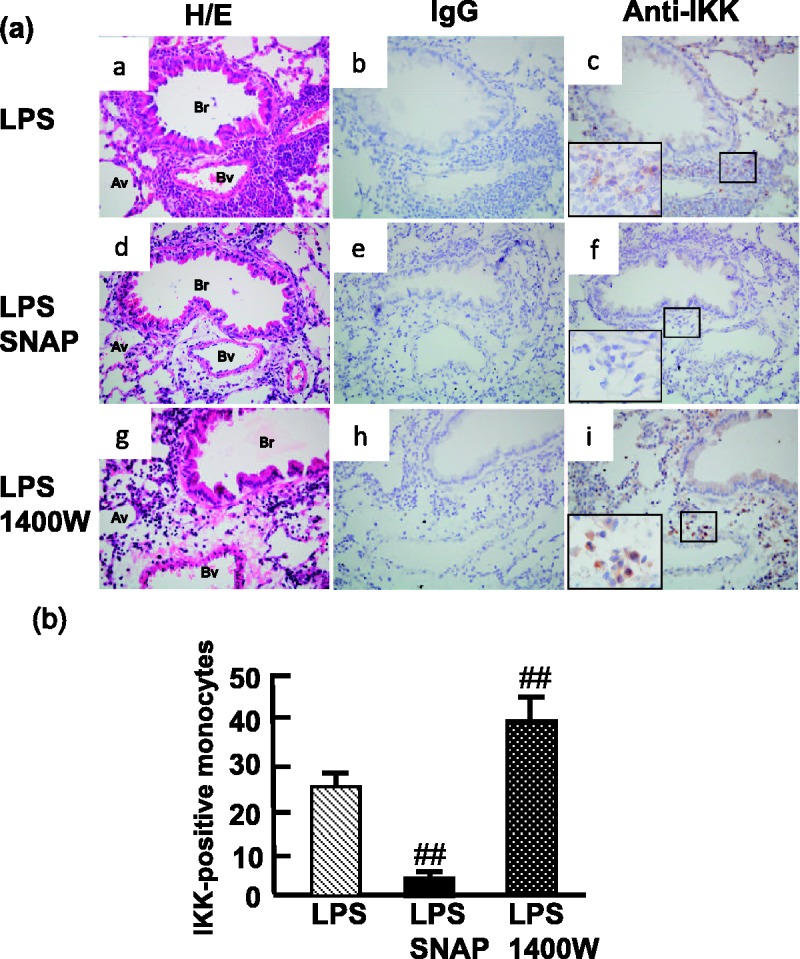
NO decreases IKK in the infiltrated monocytes in the OVA-treatment mice. (a) The expression of IKKα/β in the lung of OVA-allergic mice (n = 5) in response to LPS exposure (a, b, and c), LPS following the pretreatment of SNAP (n = 5; d, e, and f), or LPS following pretreatment with 1400W (n = 5; g, h, and i). Representative sections were stained with hematoxylin and eosin (H/E; a, d, and g), negative control (IgG; b, e, and h), and anti-IKKα/β antibody (c, f, and i; DAB staining in brown). Av: alveoli; Br: bronchiole; Bv: blood vessel. Original magnification:× 400. (b) The mean number of monocytes stained positive IKKα/β was quantitated by light microscopy from five fields selected randomly from one section and performed triplicate per mouse. ##P < 0.01 compared with LPS-treated mice
Discussion
Asthma associated with an increased risk of serious bacterial pneumonia is attributed to allergic airway inflammation, leading to a reduction in innate immunity and a decrease in pulmonary antibacterial host defense.3 The increased NO is able to deactivate the Toll-like receptor (TLR) signaling pathway and alleviate innate immunity by down-regulating IL-12 release.22 NF-κB signaling in macrophages is essential for the clearance of bacteria from the lung.23 Our study demonstrates that NO decreased the LPS-induced NF-κB activation in OVA-sensitized allergic mice and RAW 264.7 cells by reducing the interaction of Hsp90 with IKK. These results show that NO in mouse models of allergic asthma suppresses the LPS-stimulated proinflammatory response, reduces the infiltrated monocytes, and is implicated in the mechanism underlying the increased risk for invasive pneumonia infections observed among individuals with asthma.
The beneficial or protective roles of NO in subsets of asthmatic individuals, as predicted by mouse models of airway inflammation, are often contradictory.24 Recent studies have demonstrated that the protective or damaging functions of NO are dependent on the stage of infection or on the background of the mouse strain.25 However, steroid treatment alone provided only a limited degree of protection against bronchoconstriction compared with concomitant treatment using an NO donor and steroid.26 Furthermore, the absence of iNOS enhanced the LPS-induced lung inflammatory response in mice, whereas TNF-α and IL-1β levels in the BAL fluid were decreased significantly by NO in LPS-treated mice in an allergic model.27 In the OVA-stimulated lung to evaluate the expression of IKK induced by LPS following pretreatment with SNAP or 1400W, the expression of IKK in the infiltrated cells was suppressed by NO (Figure 6). Our data demonstrate a suppressive role for NO in the OVA-stimulated allergic mice, and NO produced by iNOS in allergic mouse may suppress the proinflammatory response.
In the lung, macrophages play an important role in the secretion of proinflammatory cytokines following stimulation such as exposure to LPS.28 Our data demonstrate that NO decreased the LPS-induced IL-1β, IL-6, IL-12, and iNOS mRNA levels in the RAW 264.7 cells, consistent with the observations in the BAL fluid. It has been reported that co-treatment with SNAP and LPS suppresses iNOS expression and NO production in hepatocytes, macrophages, and epithelial cells.29–31 NO is a potent inhibitor of cytokine production in stimulated human macrophages, and NF-κB activation by LPS is decreased by NO in alveolar macrophages.10 Moreover, we demonstrated that endogenous NO decreased the interaction of IKK with Hsp90 to attenuate the LPS-induced inflammation in the RAW 264.7 cells. Our findings indicate that a molecular feedback mechanism regulates LPS-induced inflammation through the production of NO.
Three NOS isozymes of iNOS, endothelial NOS (eNOS) and neuronal NOS (nNOS) are expressed in different tissues to generate NO for specific physiological roles. In the proinflammatory response, iNOS is able to generate NO in micromolar concentrations for involvement in immediate immune defense reactions. eNOS and nNOS are constitutively expressed and are involved in blood pressure regulation and neurotransmission, respectively. They generate nanomolar concentrations of NO regulated by changes of enzymatic activity upon increasing intracellular Ca2+ concentrations.32 eNOS are well known in the role of endothelium-mediated relaxation of vascular smooth muscle. Based on different model systems and different cell types/tissues examined, eNOS is both proinflammatory and anti-inflammatory and exhibits inhibition of apoptosis and promotion of angiogenesis and cell mobility.33 Moreover, nNOS knock-out mice were resistant to LPS-induced inflammation, suggesting that nNOS is involved in the initiation of inflammatory response and ultimately in lung injury, sepsis, and mortality.34 SNAP might significantly suppress the LPS-induced iNOS expression, whereas the expression of eNOS was decreased slightly by SNAP in asthma mouse model (data not shown). All isoforms of NOS are inhibited by NO. However, relatively higher concentration of NO is required to inhibit iNOS than either nNOS or eNOS.35 In addition, in RAW 264.7 macrophages, LPS increased the activity and expression of iNOS, but not eNOS.36 In this study, SNAP is NO donor that supplies NO not relying on the activity of NOSs, although SNAP might suppress the activity of iNOS. 1400W is a highly selective inhibitor of iNOS (kd = 7 nM), and Ki values of nNOS and eNOS are 2 and 50 µM, respectively. 1400W was greater than 50-fold more potent against iNOS than eNOS in a rat model.37 Since NO blocked the interaction of Hsp90 with IKK, the iNOS inhibitor 1400W enhanced the LPS-induced proinflammatory response by promoting NF-κB activity (Figure 5b).
In this study, the treatment of SNAP reduced the activation of LPS-induced NF-κB as well as the level of IKK and pIKK, whereas 1400W increased the activation of the IKK-NF-κB pathway. The activation of NF-κB regulates the production of cytokines and chemokines in inflammation, and IKK is responsible for the activation of the NF-κB pathway. The IKKβ inhibitor improves allergic airway inflammation and hyperresponsiveness in mice.13 Moreover, Hsp90 is a crucial regulator of the IKK signalosome and is associated with the severity of asthma. NO may inhibit the NF-κB DNA binding activity via S-nitrosylation of the cysteine residue in the p50 subunit.38 Hsp90 ATPase activity and its positive effect on NOS activity are both inhibited by S-nitrosylation,39 suggesting that S-nitrosylation may regulate the function of Hsp90 and provide a feedback mechanism in the NF-κB activation pathway acting as a molecular sensor of local NO concentrations and adjusting NO production accordingly. Our study is the first to demonstrate that NO attenuated the interaction between IKK and Hsp90 leading to a decrease in LPS-induced inflammation in RAW 264.7 cells. Hsp90 plays an important role in inflammation.17 Hsp90 inhibitors ameliorate the acute lung injury caused by severe sepsis, suggesting that the decrease in the interaction of Hsp90 with IKK may reduce lung injury.40 However, Hsp90 inhibitors may alleviate inflammation-induced NO leading to a decrease in the antimicrobial activity. Therefore, the use of Hsp90 inhibitors to inhibit the inflammatory response may require further investigation to evaluate the outcomes in asthma treatments.
Although asthma patients are readily infected by microbes leading to serious outcomes, the mechanism by which the inflammatory response occurs is unexplored. In this study, the LPS-induced inflammation was attenuated in the RAW 264.7 cells and allergic mice in the presence of NO, demonstrating that NO negatively regulates inflammation by interrupting the interaction of IKK with Hsp90 to reduce the NF-κB-involved inflammatory pathway.
ACKNOWLEDGEMENTS
The work was supported by grants from the National Science Council (101-2321-B-010-005), Tri-Service General Hospital and National Defense Medical Center (TSGHC101060), Taoyuan Armed Forces General Hospital (AFTYGH-10028, AFTYGH-10131, AFTYGH-10209, AFTYGH-10310), and Taipei City Hospital, Taiwan. The authors have no financial conflicts of interest.
Authors’ contributions
MYL was involved in conception and design, assembly of data, data analysis and interpretation, and manuscript writing; KHS did conception and design, and data analysis and interpretation; CPC, YCT, CFH, and GHS took part in data analysis and interpretation; and HYL and SJT were involved in conception and design, assembly of data, data analysis and interpretation, manuscript writing, financial and administrative support, and final approval of manuscript.
References
- 1.Lukacs NW, Strieter RM, Chensue SW, Kunkel SL. Activation and regulation of chemokines in allergic airway inflammation. J Leukoc Biol 1996; 59: 13–13. [DOI] [PubMed] [Google Scholar]
- 2.Kraft M. The role of bacterial infections in asthma. Clin Chest Med 2000; 21: 301–301. [DOI] [PubMed] [Google Scholar]
- 3.Beisswenger C, Kandler K, Hess C, Garn H, Felgentreff K, Wegmann M, Renz H, Vogelmeier C, Bals R. Allergic airway inflammation inhibits pulmonary antibacterial host defense. J Immunol 2006; 177: 1833–1833. [DOI] [PubMed] [Google Scholar]
- 4.Calhoun WJ, Ameredes BT, King TS, Icitovic N, Bleecker ER, Castro M, Cherniack RM, Chinchilli VM, Craig T, Denlinger L, DiMango EA, Engle LL, Fahy JV, Grant JA, Israel E, Jarjour N, Kazani SD, Kraft M, Kunselman SJ, Lazarus SC, Lemanske RF, Lugogo N, Martin RJ, Meyers DA, Moore WC, Pascual R, Peters SP, Ramsdell J, Sorkness CA, Sutherland ER, Szefler SJ, Wasserman SI, Walter MJ, Wechsler ME, Boushey HA. Comparison of physician-, biomarker-, and symptom-based strategies for adjustment of inhaled corticosteroid therapy in adults with asthma: The BASALT randomized controlled trial. JAMA 2012; 308: 987–987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lee MY, Lai YS, Yang KD, Chen CJ, Hung CH. Effects of montelukast on symptoms and eNO in children with mild to moderate asthma. Pediatr Int 2005; 47: 622–622. [DOI] [PubMed] [Google Scholar]
- 6.Mabalirajan U, Ahmad T, Leishangthem GD, Joseph DA, Dinda AK, Agrawal A, Ghosh B. Beneficial effects of high dose of L-arginine on airway hyperresponsiveness and airway inflammation in a murine model of asthma. J Allergy Clin Immunol 2010; 125: 626–626. [DOI] [PubMed] [Google Scholar]
- 7.Keller AC, Rodriguez D, Russo M. Nitric oxide paradox in asthma. Mem Inst Oswaldo Cruz 2005; 100: 19–19. [DOI] [PubMed] [Google Scholar]
- 8.Thomassen MJ, Buhrow LT, Connors MJ, Kaneko FT, Erzurum SC, Kavuru MS. Nitric oxide inhibits inflammatory cytokine production by human alveolar macrophages. Am J Respir Cell Mol Biol 1997; 17: 279–279. [DOI] [PubMed] [Google Scholar]
- 9.Kang JL, Park W, Pack IS, Lee HS, Kim MJ, Lim CM, Koh Y. Inhaled nitric oxide attenuates acute lung injury via inhibition of nuclear factor-kappaB and inflammation. J Appl Physiol 2002; 92: 795–795. [DOI] [PubMed] [Google Scholar]
- 10.Raychaudhuri B, Dweik R, Connors MJ, Buhrow L, Malur A, Drazba J, Arroliga AC, Erzurum SC, Kavuru MS, Thomassen MJ. Nitric oxide blocks nuclear factor-kappaB activation in alveolar macrophages. Am J Respir Cell Mol Biol 1999; 21: 311–311. [DOI] [PubMed] [Google Scholar]
- 11.Chang K, Lee SJ, Cheong I, Billiar TR, Chung HT, Han JA, Kwon YG, Ha KS, Kim YM. Nitric oxide suppresses inducible nitric oxide synthase expression by inhibiting post-translational modification of IkappaB. Exp Mol Med 2004; 36: 311–311. [DOI] [PubMed] [Google Scholar]
- 12.Razani B, Reichardt AD, Cheng G. Non-canonical NF-kappaB signaling activation and regulation: Principles and perspectives. Immunol Rev 2011; 244: 44–44. [DOI] [PubMed] [Google Scholar]
- 13.Sugita A, Ogawa H, Azuma M, Muto S, Honjo A, Yanagawa H, Nishioka Y, Tani K, Itai A, Sone S. Antiallergic and anti-inflammatory effects of a novel IkappaB kinase beta inhibitor, imd-0354, in a mouse model of allergic inflammation. Int Arch Allergy Immunol 2009; 148: 186–186. [DOI] [PubMed] [Google Scholar]
- 14.El-Hashim AZ, Renno WM, Abduo HT, Jaffal SM, Akhtar S, Benter IF. Effect of inhibition of the ubiquitin-proteasome-system and IkappaB kinase on airway inflammation and hyperresponsiveness in a murine model of asthma. Int J Immunopathol Pharmacol 2011; 24: 33–33. [DOI] [PubMed] [Google Scholar]
- 15.Whitesell L, Lindquist SL. HSP90 and the chaperoning of cancer. Nat Rev Cancer 2005; 5: 761–761. [DOI] [PubMed] [Google Scholar]
- 16.Perisic T, Sreckovic M, Matic G. An imbalance in antioxidant enzymes and stress proteins in childhood asthma. Clin Biochem 2007; 40: 1168–1168. [DOI] [PubMed] [Google Scholar]
- 17.Bucci M, Roviezzo F, Cicala C, Sessa WC, Cirino G. Geldanamycin, an inhibitor of heat shock protein 90 (Hsp90) mediated signal transduction has anti-inflammatory effects and interacts with glucocorticoid receptor in vivo. British J Pharmacol 2000; 131: 13–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hinz M, Broemer M, Arslan SC, Otto A, Mueller EC, Dettmer R, Scheidereit C. Signal responsiveness of IkappaB kinases is determined by Cdc37-assisted transient interaction with Hsp90. J Biol Chem 2007; 282: 32311–32311. [DOI] [PubMed] [Google Scholar]
- 19.Shimp SK, Parson CD, Regna NL, Thomas AN, Chafin CB, Reilly CM. Nichole Rylander M. HSP90 inhibition by 17-DMAG reduces inflammation in J774 macrophages through suppression of Akt and nuclear factor-kappaB pathways. Inflam Res 2012; 61: 521–521. [DOI] [PubMed] [Google Scholar]
- 20.Yu CK, Shieh CM, Lei HY. Repeated intratracheal inoculation of house dust mite (Dermatophagoides farinae) induces pulmonary eosinophilic inflammation and IgE antibody production in mice. J Allergy Clin Immunol 1999; 104: 228–228. [DOI] [PubMed] [Google Scholar]
- 21.McKay DM, Jedrzkiewicz JLS, Ho W, Sharkey KA. Nitric oxide participates in the recovery of normal jejunal epithelial ion transport following exposure to the superantigen, Staphylococcus aureus enterotoxin B. J Immunol 1999; 163: 4519–4519. [PubMed] [Google Scholar]
- 22.Xiong H, Zhu C, Li F, Hegazi R, He K, Babyatsky M, Bauer AJ, Plevy SE. Inhibition of interleukin-12 p40 transcription and NF-kappaB activation by nitric oxide in murine macrophages and dendritic cells. J Biol Chem 2004; 279: 10776–10776. [DOI] [PubMed] [Google Scholar]
- 23.Lai JF, Zindl CL, Duffy LB, Atkinson TP, Jung YW, van Rooijen N, Waites KB, Krause DC, Chaplin DD. Critical role of macrophages and their activation via MyD88-NFkappaB signaling in lung innate immunity to Mycoplasma pneumoniae. PloS One 2010; 5: e14417–e14417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mathrani VC, Kenyon NJ, Zeki A, Last JA. Mouse models of asthma: Can they give us mechanistic insights into the role of nitric oxide? Curr Med Chem 2007; 14: 2204–2204. [DOI] [PubMed] [Google Scholar]
- 25.Breitbach K, Wongprompitak P, Steinmetz I. Distinct roles for nitric oxide in resistant C57BL/6 and susceptible BALB/c mice to control Burkholderia pseudomallei infection. BMC Immunol 2011; 12: 20–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jonasson S, Hedenstierna G, Hjoberg J. Concomitant administration of nitric oxide and glucocorticoids improves protection against bronchoconstriction in a murine model of asthma. J Appl Physiol 2010; 109: 521–521. [DOI] [PubMed] [Google Scholar]
- 27.Speyer CL, Neff TA, Warner RL, Guo RF, Sarma JV, Riedemann NC, Murphy ME, Murphy HS, Ward PA. Regulatory effects of iNOS on acute lung inflammatory responses in mice. Am J Pathol 2003; 163: 2319–2319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sweet MJ, Hume DA. Endotoxin signal transduction in macrophages. J Leukoc Biol 1996; 60: 8–8. [DOI] [PubMed] [Google Scholar]
- 29.Taylor BS, Kim YM, Wang Q, Shapiro RA, Billiar TR, Geller DA. Nitric oxide down-regulates hepatocyte-inducible nitric oxide synthase gene expression. Arch Surg 1997; 132: 1177–1177. [DOI] [PubMed] [Google Scholar]
- 30.Kim YM, Son K, Hong SJ, Green A, Chen JJ, Tzeng E, Hierholzer C, Billiar TR. Inhibition of protein synthesis by nitric oxide correlates with cytostatic activity: Nitric oxide induces phosphorylation of initiation factor eIF-2 alpha. Mol Med 1998; 4: 179–179. [PMC free article] [PubMed] [Google Scholar]
- 31.Cavicchi M, Gibbs L, Whittle BJ. Inhibition of inducible nitric oxide synthase in the human intestinal epithelial cell line, DLD-1, by the inducers of heme oxygenase 1, bismuth salts, heme, and nitric oxide donors. Gut 2000; 47: 771–771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Alderton WK, Cooper CE, Knowles RG. Nitric oxide synthases: structure, function and inhibition. Biochem J 2001; 357: 593–593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ying L, Hofseth LJ. An emerging role for endothelial nitric oxide synthase in chronic inflammation and cancer. Cancer Res 2007; 67: 1407–1407. [DOI] [PubMed] [Google Scholar]
- 34.BAIG MS, Bakhshi F, Ye RD, Gao X, Malik AB, Minshall RD, Ganter B, Bonini M. Role of nNOS in the progression of systemic inflammatory response induced by lipopolysaccharide. FASEB J 2012; 26: lb546–lb546. [Google Scholar]
- 35.Griscavage JM, Hobbs AJ, Ignarro LJ. Negative Modulation on Nitric Oxide Synthase by Nitric Oxide and Nitroso Compounds. Adv Pharmacol 1995; 34: 215–215. [DOI] [PubMed] [Google Scholar]
- 36.Paul A, Pendreigh RH, Plevin R. Protein kinase C and tyrosine kinase pathways regulate lipopolysaccharide-induced nitric oxide synthase activity in RAW 264.7 murine macrophages. Br J Pharmacol 1995; 114: 482.–482.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Garvey EP, Oplinger JA, Furfine ES, Kiff RJ, Laszlo F, Whittle BJ, Knowles RG. 1400W is a slow, tight binding, and highly selective inhibitor of inducible nitric-oxide synthase in vitro and in vivo. J Biol Chem 1997; 272: 4959.–4959.. [DOI] [PubMed] [Google Scholar]
- 38.Colasanti M, Persichini T. Nitric oxide: An inhibitor of NF-kappaB/Rel system in glial cells. Brain Res Bull 2000; 52: 155–155. [DOI] [PubMed] [Google Scholar]
- 39.Martínez-Ruiz A, Villanueva L, González de Orduña C, López-Ferrer D, Higueras MA, Tarín C, Rodríguez-Crespo I, Vázquez J, Lamas S. S-nitrosylation of Hsp90 promotes the inhibition of its ATPase and endothelial nitric oxide synthase regulatory activities. Proc Natl Acad Sci USA 2005; 102: 8525–8525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chatterjee A, Dimitropoulou C, Drakopanayiotakis F, Antonova G, Snead C, Cannon J, Venema RC, Catravas JD. Heat shock protein 90 inhibitors prolong survival, attenuate inflammation, and reduce lung injury in murine sepsis. Am J Respir Crit Care Med 2007; 176: 667–667. [DOI] [PMC free article] [PubMed] [Google Scholar]