

Abstract
Turoctocog alfa (NovoEight) is a third-generation recombinant factor VIII (rFVIII) with a truncated B-domain that is manufactured in Chinese hamster ovary cells. No human or animal-derived materials are used in the process. The aim of this study is to describe the molecular design and purification process for turoctocog alfa. A five-step purification process is applied to turoctocog alfa: protein capture on mixed-mode resin; immunoaffinity chromatography using a unique, recombinantly produced anti-FVIII mAb; anion exchange chromatography; nanofiltration and size exclusion chromatography. This process enabled reduction of impurities such as host cell proteins (HCPs) and high molecular weight proteins (HMWPs) to a very low level. The immunoaffinity step is very important for the removal of FVIII-related degradation products. Manufacturing scale data shown in this article confirmed the robustness of the purification process and a reliable and consistent reduction of the impurities. The contribution of each step to the final product purity is described and shown for three manufacturing batches. Turoctocog alfa, a third-generation B-domain truncated rFVIII product is manufactured in Chinese hamster ovary cells without the use of animal or human-derived proteins. The five-step purification process results in a homogenous, highly purified rFVIII product.
Keywords: B-domain, development, factor VIII, haemophilia, purification, turoctocog alfa
Introduction
During the past three decades, the treatment of patients with haemophilia A has progressed from cryoprecipitate to partly purified plasma-derived factor VIII (pd-FVIII) concentrates and, more recently, to highly purified recombinant FVIII (rFVIII) proteins. Recombinantly produced FVIII molecules, with a minimized risk of viral contamination, are a major step forward [1]; in addition to having a good safety profile, rFVIII products provide efficacious replacement therapy for prophylaxis and treatment of bleeding episodes in patients with haemophilia A. However, access to therapy remains limited in some parts of the world, especially in developing countries. The introduction of new rFVIII products into the market may secure the supply of these recombinant products. Novo Nordisk has developed turoctocog alfa (NovoEight, Novo Nordisk A/S, Bagsværd, Denmark), a third-generation rFVIII replacement therapy, to offer the haemophilia A community an additional treatment option.
The human FVIII gene encodes a mature protein consisting of a single chain of 2332 amino acid residues [1,2] with six distinct domains: A1, A2, B, A3, C1, C2 (Fig. 1a). During secretion, the single chain form is cleaved by furin (a transmembrane protease) into a heavy chain and a light chain held together by metal ions [3–5]. FVIII is converted into FVIIIa by thrombin cleavage at three sites, resulting in a three-chain molecule without the B-domain (Fig. 1a).
Fig. 1.
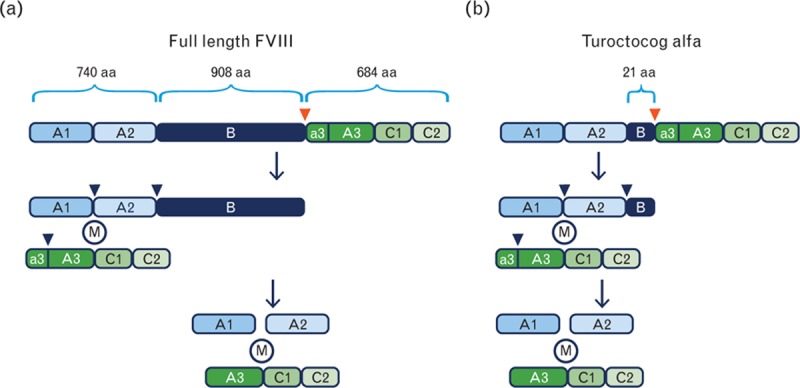
Molecular structure of FVIII and turoctocog alfa. (a) Changes to the molecular structure of FVIII from its full length (upper section) to its thrombin-activated form (lowest section). The red and blue arrowheads show the site of cleavage by the transmembrane protease furin and the three sites of thrombin cleavage, which converts FVIII to FVIIIa, respectively. M represents metal ions keeping the heavy chain and light chain together. (b) Structure of turoctocog alfa and thrombin activation. aa, amino acids; FVIII, factor VIII.
Designing the optimal pharmaceutical product on the basis of a rFVIII protein entails a number of considerations, including choice of cell line for protein expression; cell culturing conditions; construct (e.g. full-length FVIII or B-domain truncated); design of the purification process; product formulation (e.g. temperature stability); and how the product can be administered in a well tolerated, convenient and efficient manner.
Intensive investigations have shown that the B-domain may not be needed for FVIII biological function [6,7]. Expressing full-length FVIII molecules with an intact B-domain in mammalian cells has been considerably more difficult than expressing B-domain truncated variants [8], presumably because of the 19 potential N-glycosylation sites in the B-domain. In manufacture, B-domain truncated rFVIII products are expected to be more homogenous, making it easier to design the downstream purification process.
Consequently, when developing turoctocog alfa, we chose an FVIII-encoding sequence consisting of an intact human FVIII heavy chain sequence of 740 amino acid residues followed by a 21-amino acid residue truncated B-domain. The light chain sequence of 684 amino acid residues was kept intact. The 21-amino acid residue truncated B-domain comprises 10 amino acids from the N-terminal of the naturally occurring B-domain linked to 11 amino acids from the C-terminal of the B-domain. This truncated B-domain is removed following thrombin cleavage of turoctocog alfa, generating a rFVIIIa molecule identical to endogenous FVIIIa (Fig. 1b).
Turoctocog alfa is produced in Chinese hamster ovary (CHO) cells under serum-free conditions. A five-step purification process is applied as outlined in Fig. 2, including the following steps: capture on a mixed-model resin and detergent treatment; anti-rFVIII immunoaffinity chromatography (using a unique recombinant mAb F25); anion exchange (AIEX) chromatography; nanofiltration (20-nm filter) and size exclusion chromatography (SEC) [9]. This purification process produces turoctocog alfa to a high degree of purity and with no major host cell impurities that may activate the immune system. We have previously described the purification and characterization of this rFVIII molecule [7], and its in-vitro functional characteristics [10]. Here, we describe the considerations behind the process design that ensure robustness of the large-scale manufacturing process and a high-quality product. The overall aim was to develop a rFVIII product that is well tolerated, effective and convenient using the latest scientific and technological knowledge.
Fig. 2.
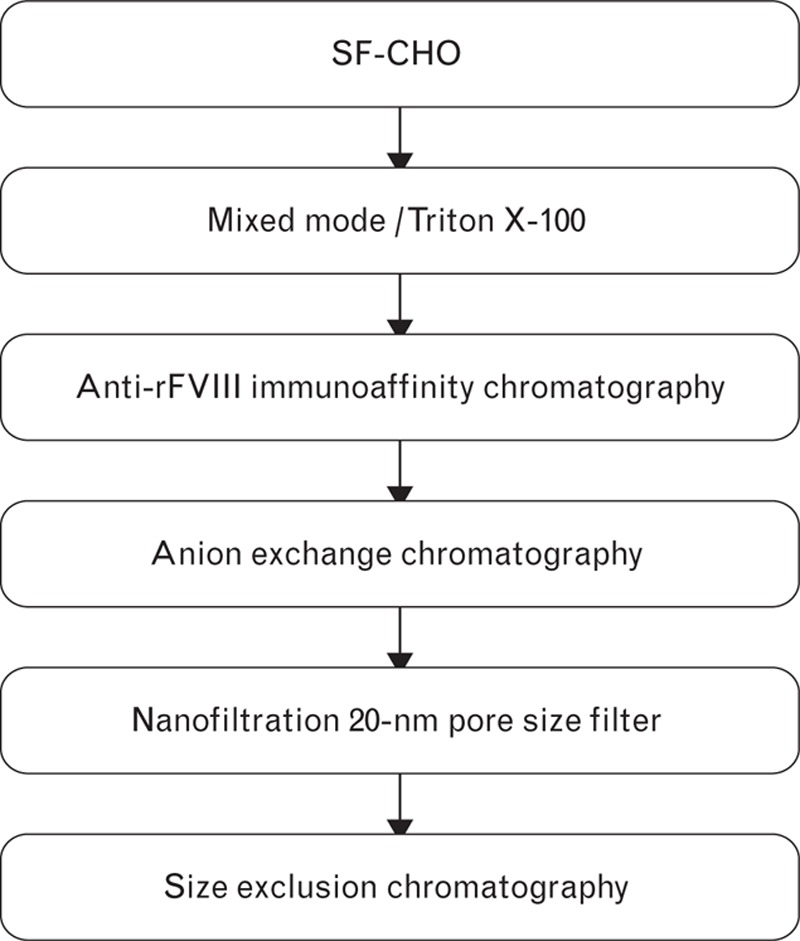
The five-step purification process developed for turoctocog alfa production following its secretion from CHO cells grown in serum-free conditions. CHO, Chinese hamster ovary; rFVIII, recombinant factor VIII.
Materials and methods
Production cell line
A turoctocog alfa coding DNA sequence was established from human kidney FVIII cDNA [6,11] and inserted into a mammalian expression plasmid. CHO cells transfected with the expression construct were selected with the dihydrofolate reductase gene as a selection marker and cloned by limiting dilution. A high-producing clone was selected as the turoctocog alfa production cell line and an animal component-free production process was developed.
Purification
Equipment, columns and filters
Chromatographic isolation of turoctocog alfa from the cell culture media used an Äkta system from GE Healthcare (Uppsala, Sweden). Capto MMC and cyanogen bromide (CNBr)-activated Sepharose were from GE Healthcare.
Anti-rFVIII immunoaffinity chromatography with immobilized F25 mAb
For production of the F25 mAb, immunoglobulin G (IgG) light and heavy chain cDNA were amplified by reverse transcription PCR from total RNA extracted from F25 hybridoma cells. The cDNA was inserted into expression vectors and clones were sequenced, leading to the identification of two different IgG light chain cDNA sequences and two different IgG heavy chain cDNA sequences. Upon coexpression of the four different light and heavy chain combinations, only one of these (HC2, LC1) exhibited anti-FVIII reactivity (Fig. 3). Thus, the cDNA sequences encoding the recombinant F25 (rF25) mAb were identified. The cDNA encoding light and heavy chain rF25 mAb was inserted into an expression vector containing a glutamine synthetase selection marker and transfected into CHO cells. Clones of transfected cells were selected with 37.5 mmol/l of methionine sulphoximine, a glutamine synthetase inhibitor. A number of CHO cell clones were screened and a suitable cell line for production of rF25 mAb was selected. The production was carried out under serum and animal component-free conditions. The rF25 mAb was purified using a modified generic platform for antibody purification and was coupled to CNBr-activated Sepharose Fast Flow (GE Healthcare) according to the manufacturer's instructions.
Fig. 3.
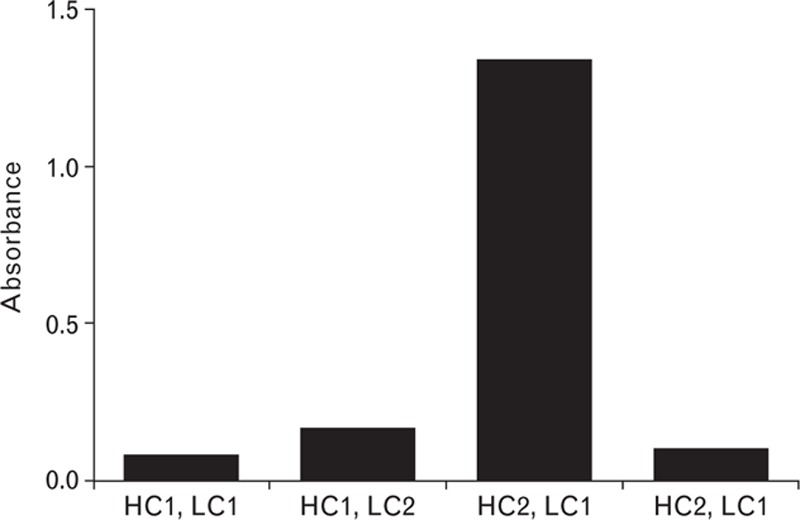
Identification of the cDNA encoding F25 mAb. Reactivity of combinations of IgG heavy chain and light chain cloned from F25 hybridoma cells, coexpressed in HEK293 cells, and utilized as the detection antibody in an ELISA. IgG, immunoglobulin G.
Hydrogen-deuterium exchange mass spectrometry
HDX mass spectrometry identified the epitope of F25 on FVIII. The procedure was performed as described previously, with minor adjustment [12,13]. In brief, turoctocog alfa and F25 were dialysed to a buffer comprising 20 mmol/l imidazole (pH 7.3), 10 mmol/l CaCl2 and 150 mmol/l NaCl, and the proteins were kept at 2°C until the start of the experiment. HDX was initiated by incubating 3 μmol/l FVIII in the absence or presence of 4.5 μmol/l F25 in the same buffer in 98% deuterium oxide (D2O) instead of H2O, at nine time intervals (10, 20, 30, 40, 60, 120, 240, 480 and 960 s). The HDX experiments were performed using the Waters SYNAPT G2 HDMS in combination with the nanoACQUITY UPLC with HDX technology.
Analytical methods
SDS-gel electrophoresis
Samples were denatured and reduced at 70°C for 10 min in SDS sample buffer (Life Technologies, Naerum, Denmark) containing 50 mmol/l DTT. Electrophoresis was performed using 7% TA Pre-Cast Novex gels and TA buffer (Life Technologies). Silver staining was carried out using SilverQuest (Life Technologies) and Coomassie staining using GelCode (Pierce; Thermo Fisher Scientific Inc., Rockford, Illinois, USA) according to the manufacturer's instructions.
Size exclusion HPLC determination of high molecular weight protein content
High molecular weight protein (HMWP) content (turoctocog alfa polymers) was determined using size exclusion HPLC, with the column BioSep SEC 3000, 290 Å, (Phenomenex, Værl⊘se, Denmark), 300 × 7.8 mm, 5 μm and 10 mmol/l Tris + 10 mmol/l CaCl2 + 300 mmol/l NaCl + 5% isopropanol, pH 7.0, as mobile phase. The analysis was run at 0.4 ml/min, with column temperature at 30°C.
Results and discussion
Cell lines
The CHO cell line was selected as the production cell for expressing turoctocog alfa. CHO cells are a reliable, well established cell line for the production of recombinant proteins for medicinal purposes. Biopharmaceuticals produced in CHO cells have been administered to a large number of patients for more than two decades and have proven to be compatible and bioactive, and to have good safety profiles in humans [14]. Production in a nonhuman CHO cell line minimizes the risk of cell culture infection with viruses infectious to humans [14]. Furthermore, CHO cells are robust and can be cultivated at a large scale in the absence of serum or other human or animal-derived materials in a highly reproducible manner, securing consistent production of a homogenous, high-quality protein.
Step 1: capture on Capto MMC
In the first step of the purification process, turoctocog alfa is selectively captured from the cell culture media. One advantage of using a mixed-mode resin for capture of the basic protein is that it enables direct loading of the harvest to the column without the need for further conditioning (e.g. adjustment of pH and conductivity). Virus inactivation was achieved by incubating the resin with adsorbed turoctocog alfa, with a Triton X-100 buffer. The highest yields of FVIII were achieved by combining high concentrations of both NaCl and ethylene glycol in the elution buffer (Fig. 4) [15]. As proper FVIII elution was not possible using either salt or ethylene glycol alone, a wash step with a high salt concentration was included to remove impurities. The application volume was 500 column volume or less and the eluate pool was 2 column volume, giving a 250-fold reduction in volume. SDS-PAGE showed enrichment of FVIII in the capture pool (Fig. 5), confirmed by the chromogenic activity assay, which showed a 100-fold increase in FVIII-specific activity within the pool versus the harvest. Using a combination of salt and ethylene glycol for elution also generated a pool that remained stable throughout freezing and thawing, without loss of activity. This is an advantage in manufacturing, as the purification process can be stopped at the capture stage if necessary.
Fig. 4.
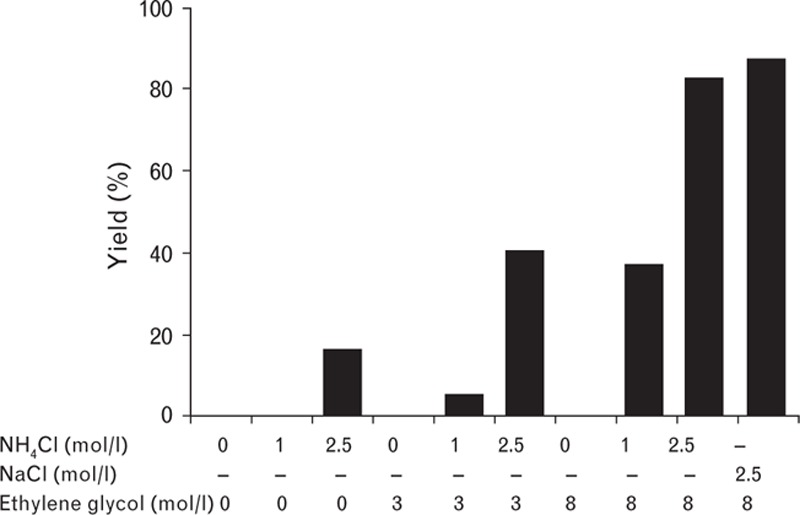
Elution of turoctocog alfa from Capto MMC.
Fig. 5.
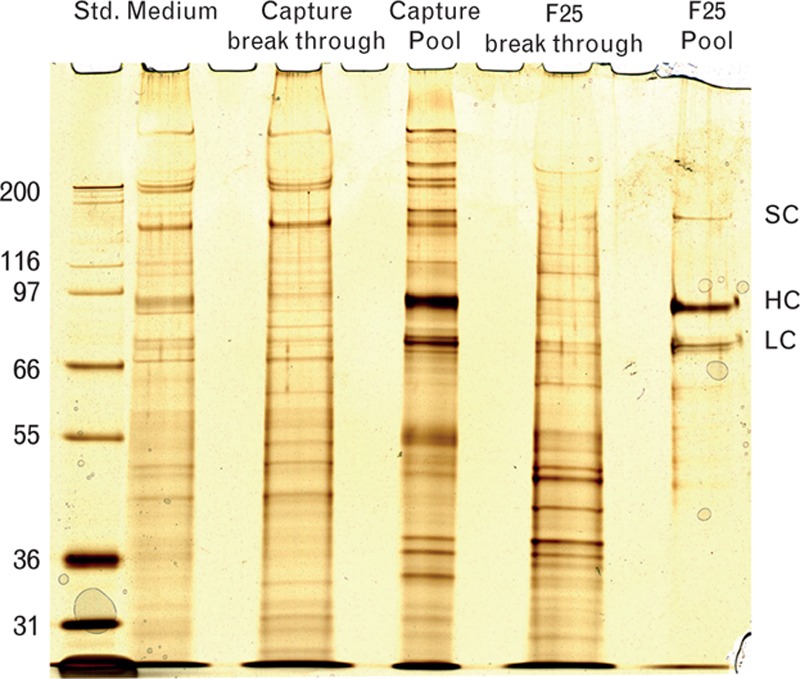
Silver-stained SDS-PAGE of samples taken during purification. Molecular weights are given in kDa for the standard marker on the left side of the gel. HC, heavy chain; LC, light chain; SC, single chain; Std., standard.
Step 2: immunoaffinity with immobilized F25 antibody
In the immunoaffinity chromatography step, the immunoaffinity resin was immobilized with F25, a recombinant IgG mAb specific for FVIII molecules with an intact A2 domain. This second step in the purification process is crucial for the homogeneity of the final turoctocog alfa product, as the cell culture supernatant from the production cell line contains FVIII-related degradation products with heavy chains shortened at the C-terminal in the A2 domain. The F25 mAb, which recognizes the turoctocog alfa molecule at the far C-terminal of the heavy chain (amino acids 735–739), does not bind to FVIII molecules with incomplete heavy chains. Consequently, heavy chain degradation products processed at amino acid 720 are not copurified with turoctocog alfa [16]. SDS-PAGE analysis clearly demonstrated the high purity of the turoctocog alfa molecule (Fig. 5).
FVIII regions that contribute to F25 antibody binding
HDX mass spectrometry, used to identify the epitope of F25 on FVIII, exploits the characteristic that HDX in a protein can be followed by mass spectrometry upon replacement of H2O by D2O in the aqueous solvent [17]. The peptic peptides undergoing HDX mass spectrometry analysis covered 91% of the turoctocog alfa primary sequence. The obtained peptic peptides are mapped onto a model on the basis of the X-ray crystallographic structure of NovoEight [5]. Loops missing in the structure (residues 17–45, 211–228, 1716–1726) have been modelled using the homology modelling tool, Modeller [18] and a1 (residues 336–372) and a2–a3 (residues 711–741, 1648–1690) domains have been folded using the ab initio folding tool, Rosetta [19]. After folding, the domains were docked to the X-ray structure so that their termini fitted the corresponding termini in the structure. Of these peptides, 96% displayed an exchange pattern that was unaffected by the binding of F25, suggesting that amino acid residues within these regions do not contribute to F25 binding. In contrast, a limited number of peptides were protected from HDX in the presence of F25.
The peptides displaying protection could be divided into two groups: 721–734 displayed protection 1.4 times the standard deviation of data from deuterium incorporation in the presence of antibody F25 (Fig. 6) [18,19] compared with the other group, which showed a level of protection more than twice the standard deviation. The higher level of protection indicates a higher affinity for F25 binding. The FVIII region that displayed this prolonged protection included 11 partially overlapping peptides clustered on the C-terminal of the A2 domain. The analysis of partially overlapping peptides defined the residues that are protected by F25 binding, that is amino acid residues 735–739, which are protected from HDX in the presence of F25 by more than twice the standard deviation of the data.
Fig. 6.
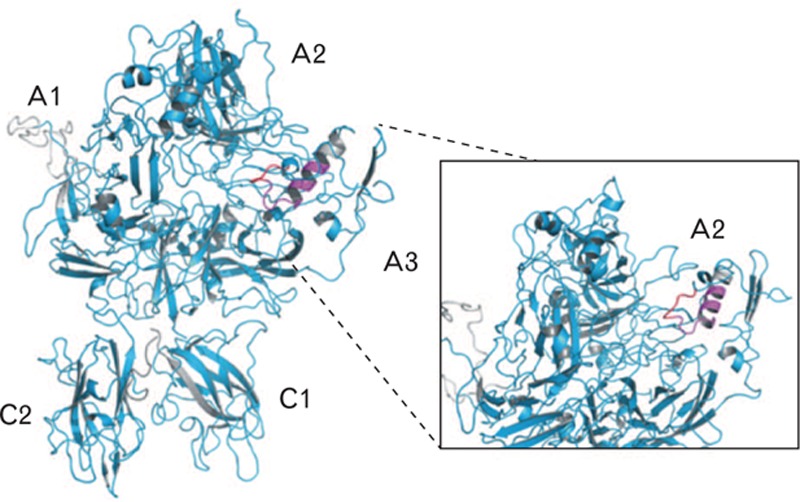
FVIII peptides 721–734 and 729–738, but not 738–750, exhibit reduced hydrogen-deuterium exchange in the presence of antibody F25. The deuterium incorporation level upon F25 binding was identified to be unchanged for cyan areas, reduced more than twice the standard deviation of data for red areas and below twice the standard deviation of data for magenta areas. The inset zooms in on the FVIII A2 domain. Grey areas are uncovered by the peptide map. The obtained peptic peptides are mapped onto a model on the basis of the X-ray crystallographic structure of NovoEight [5]. FVIII, factor VIII.
In summary, amino acid residues 735–739 were protected by F25 from efficient HDX; in comparison, a lower HDX protection was observed for residues 721–734. This demonstrates that residues within these regions contribute to F25 binding.
Step 3: anion exchange
The presence of residual host cell protein (HCP), that is CHO and leaked mAb, in the final product may activate the immune system and it is therefore important to maintain a low level of HCP and mAb. The AIEX chromatography step reduces HCP and mAb leakage from Step 2 and further purifies and concentrates the intermediate product to obtain the target drug substance concentration after Step 5. The final drug substance concentration is a critical quality attribute in product formulation. Ensuring a sufficient drug substance concentration is made more challenging by dilution of the protein solution during virus filtration and SEC. Dilution during virus filtration is primarily due to washing of the filter to promote protein displacement; during SEC, dilution occurs by dispersion as the loaded peak moves down the column. The amount loaded on the AIEX step varies to a certain extent. As gradient volume in chromatographic processes is normally fixed to a certain value, any variation in protein load produces variations in pool concentration (Table 1). This variation, together with the dilution from the virus filter, causes variation in the feed concentration of the SEC step and subsequently the drug substance concentration (pool from SEC step).
Table 1.
Pool concentration after Step 3 according to the amount of protein loaded onto the column when using the traditional approach with fixed gradient length
| Load (g/l) | Gradient (CV) | Pool concentration (g/l) |
| 5 | 10 | 1.8 |
| 10 | 10 | 3.0 |
| 15 | 10 | 3.9 |
| 25 | 10 | 5.0 |
Pool concentration varies depending on the load. CV, column volume.
To circumvent variations in pool concentration of the AIEX chromatography step, we derived a simple algorithm in which the volume of the elution gradient (measured in column volume) is related to the load (g/l). The elution gradient volume can be set equal to the load (in g/l resin) in the range of 5–25 g/l and maintained at 5 column volume for loads less than 5 g/l. So, for a protein load of 10 g/l, a gradient volume of 10 column volume was used. The higher the load, the shallower the gradient must be to obtain the same pool concentration. Using this approach, only small variations in pool concentration were observed despite large variations in protein loaded onto the column (Table 2).
Table 2.
Relationship of elution gradient volume to load and subsequent pool concentration in Step 3
| Load (g/l) | Gradient (CV) | Pool concentration (g/l) |
| 5 | 5 | 2.6 |
| 10 | 10 | 3.0 |
| 15 | 15 | 3.1 |
| 25 | 25 | 3.1 |
By adjusting the gradient length to the load, a more consistent pool concentration is obtained. CV, column volume.
A simulation was set up to relate protein load to gradient length. The pool concentrations obtained during laboratory validation and full-scale production were compared with simulated values (Fig. 7). Pool concentration remained stable when gradient volume was changed according to load. In general, the data were slightly above the simulated line, as the conservative model parameters ensure that the pool concentration, if unknown, is underestimated.
Fig. 7.
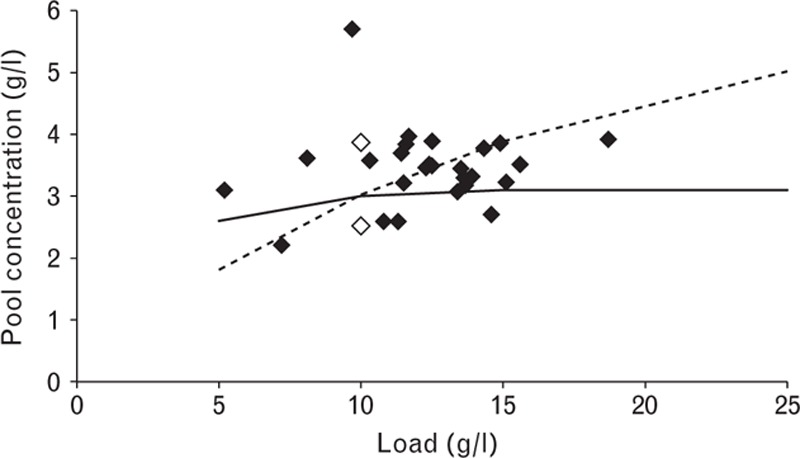
The pool concentration for Step 3 as a function of the load when applying a variable gradient. Filled diamonds are from production scale, empty diamonds are laboratory validation runs, the solid line is the design obtained using simulation and the dashed line represents the simulated data for a fixed gradient length.
Process evaluation used a mechanistic model calibrated with experimental data. Computer simulations assessed process robustness within parameter limits by evaluating important output parameters (turoctocog alfa and HCP content; anti-FVIII mAb leakage). The process was found to be robust within the specified limits. During process performance qualification, the parameters were kept as constant as possible, while in the laboratory-scale experiments, variation was introduced deliberately. Laboratory data and manufacturing-scale data were similar, with the same variation, confirming the robustness of the process step.
Step 4: virus filtration
After the three chromatography steps, the turoctocog alfa protein product underwent nano-filtration using a 20-nm filter to ensure a high level of viral inactivation. Detailed data on virus clearance will be published separately and are not discussed in this paper.
Step 5: size exclusion chromatography
SEC allows separation of substances of different size and is a final polishing step in the manufacturing process. In this step, the content of HMWP is reduced, the elution buffer is exchanged for the final drug substance buffer and HCP content and mAb leakage are further reduced. The HCP level was higher on the ascending side of the peak and there was a direct correlation between reduction of HMWP and HCP (Fig. 8). At this step, anti-FVIII mAb leakage was reduced to around the limit of quantification. Specific activity was maintained at the same level. As this was the last purification step, it is important to obtain a consistent concentration of turoctocog alfa at every run.
Fig. 8.
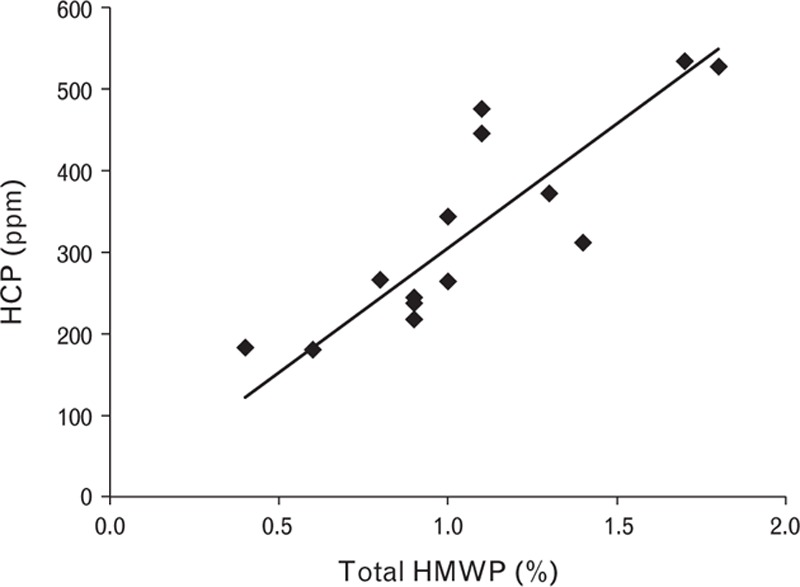
Correlation between high molecular weight protein and host cell protein reduction. HCP, host cell protein; HMWP, high molecular weight protein.
Initially, the set point for the SEC step was to load 0.05 column volume of feed solution; however, as variations in the feed concentration directly impact pool concentration, an alternative control strategy was developed to ensure a sufficient and consistent pool concentration in preparation for subsequent drug substance formulation. Simulations investigated pool concentration as a function of feed concentration and feed volume. The procedure for column loading was changed from volume-based (fixed column volume) to mass-based (load in g/l), so that a higher volume was loaded for dilute feed stocks. This significantly reduced variation in pool concentration.
In the new control strategy, load was expressed in g/l column instead of column volume. This means that for dilute feed solutions, a higher volume is loaded, while for concentrated solutions, a smaller volume is loaded. Experimental verification data showed that the model can predict pool concentration (Fig. 9).
Fig. 9.
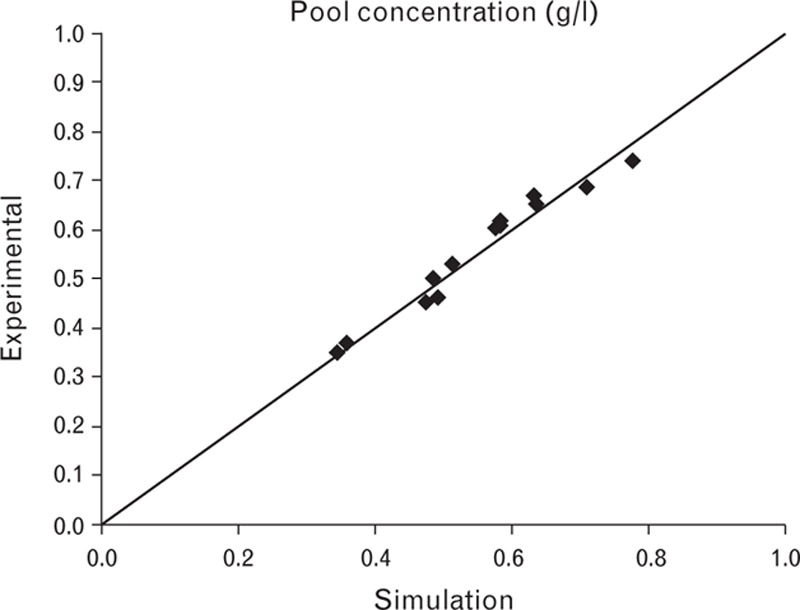
Experimental data versus simulated values for the pool concentration of Step 5.
It is important to obtain a sufficient pool concentration while avoiding excessively high concentrations, as this could result in the inclusion of HMWP impurities. Furthermore, the loading volume was limited to 0.10 column volume to obtain the required separation.
As in Step 3, process evaluation used a model-based approach in which simulations assessed process robustness regarding content and purity within the desired ranges. Robustness was confirmed by experimental verification.
Process-related impurities
Manufacturing-scale data confirmed the robustness of the purification process and its ability to reduce impurities to a very low level (Tables 3 and 4). HCP (CHO) content was reduced throughout the process (reduction factor 17 292–22 980), with the major contribution to the reduction from immunoaffinity chromatography (reduction factor 266–383). The leakage from the immunoaffinity chromatography step was efficiently reduced by the subsequent AIEX (reduction factor 5–6) and SEC (reduction factor 10–15) steps. The reduction factor by AIEX and SEC for mAb leakage from the immunoaffinity chromatography step was 160 (batch 1), whereas in batches 2 and 3, it was around 60–75. The pool from immunoaffinity chromatography in batch 1 had a higher content of mAb, but the final product from all three batches had the same very low mAb content (0.04–0.05 ng/10 000 IU) (Table 4), demonstrating that even though the level of mAb leakage varies from batch to batch, the process efficiently reduces leakage. HMWP was reduced mainly by SEC (reduction factor 5–18) with almost no contribution from AIEX. As for mAb, the reduction factor for HMWP varied by content from the previous step but resulted in the same final low level (0.3–0.8%) (Table 4).
Table 3.
Reduction of three important impurities during the purification process
| RF for HCP | RF for mAb | RF for HMWP | |||||||
| Batch 1 | Batch 2 | Batch 3 | Batch 1 | Batch 2 | Batch 3 | Batch 1 | Batch 2 | Batch 3 | |
| Affinity chromatography | 270 | 266 | 383 | – | – | – | ND | ND | ND |
| AIEX | 5 | 5 | 6 | 32 | 15 | 24 | 1 | 1 | 1 |
| SEC | 15 | 13 | 10 | 5 | 5 | 2.5 | 18 | 6 | 5 |
| Total RF | 20 250 | 17 290 | 22 960 | 160 | 75 | 60 | 18 | 6 | 5 |
Results are from three different manufacturing batches. AIEX, anion exchange chromatography; HCP, host cell protein; HMWP, high molecular weight protein; ND, not determined; RF, reduction factor (calculated as amount in the load/amount in the pool): total RF is calculated by multiplication of RF values in each column; SEC, size exclusion chromatography.
Table 4.
Content of host cell protein, mAb and high molecular weight protein in manufactured batches of purified turoctocog alfa
| Batch 1 | Batch 2 | Batch 3 | |
| Specific activity (IU/mg) | 7480 | 10 324 | 8146 |
| HCP (ng 1000 IU−1) | 40 | 33 | 38 |
| mAb (ng 1000 IU−1) | 0.05 | 0.04 | 0.05 |
| HMWP (%) | 0.3 | 0.8 | 0.4 |
HCP, host cell protein; HMWP, high molecular weight protein; IgG, immunoglobulin G.
Conclusion
Turoctocog alfa, a third-generation B-domain truncated rFVIII product, offers an alternative treatment option for haemophilia A. The molecule is manufactured in CHO cells without the use of human or animal-derived proteins. The five-step purification process results in a homogenous, highly purified rFVIII product. The most important step in the purification process is the immunoaffinity chromatography, wherein an anti-FVIII mAb is used. This mAb, which recognizes the turoctocog alfa molecule at the far C-terminal of the heavy chain (amino acids 735–739), does not bind to FVIII molecules with incomplete heavy chains. Consequently, heavy chain degradation products processed at amino acid 720 are not copurified with turoctocog alfa. The robustness of the AIEX and SEC steps was further ensured by development of mechanistic mathematical models and use of mathematical algorithm. Manufacturing data confirmed the robustness of the process.
Acknowledgements
This study was funded by Novo Nordisk A/S, Denmark.
H.A. analysed the data and contributed to writing the article. E.B.H., J.H.F., L.S., J.K., G.B., J.J.H., L.T. performed the research, analysed the data and contributed to writing the article.
Conflicts of interest
All authors are employed by Novo Nordisk A/S and hold shares in the company. Medical writing support was provided by Katherine Ayling-Rouse (PAREXEL) and financially supported by Novo Nordisk Healthcare AG. This study was funded by Novo Nordisk A/S, Denmark.
References
- 1.Gitschier J, Wood WI, Goralka TM, Wion KL, Chen EY, Eaton DH, et al. Characterization of the human factor VIII gene. Nature 1984; 312:326–330. [DOI] [PubMed] [Google Scholar]
- 2.Vehar GA, Keyt B, Eaton D, Rodriguez H, O’Brien DP, Rotblat F. Structure of human factor VIII. Nature 1984; 312:337–342. [DOI] [PubMed] [Google Scholar]
- 3.Andersson LO, Forsman N, Huang K, Larsen K, Lundin A, Pavlu B, et al. Isolation and characterization of human factor VIII: molecular forms in commercial factor VIII concentrate, cryoprecipitate, and plasma. Proc Natl Acad Sci U S A 1986; 83:2979–2983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kaufman RJ, Wasley LC, Dorner AJ. Synthesis, processing, and secretion of recombinant human factor VIII expressed in mammalian cells. J Biol Chem 1988; 263:6352–6362. [PubMed] [Google Scholar]
- 5.Svensson LA, Thim L, Olsen OH, Nicolaisen EM. Evaluation of the metal binding sites in a recombinant coagulation factor VIII identifies two sites with unique metal binding properties. Biol Chem 2013; 394:761–765. [DOI] [PubMed] [Google Scholar]
- 6.Burke RL, Pachl C, Quiroga M, Rosenberg S, Hagiwood N, Nordfang O, et al. The functional domains of coagulation factor VIII:C. J Biol Chem 1986; 261:12574–12578. [PubMed] [Google Scholar]
- 7.Pittman DD, Alderman EM, Tomkinson KN, Wang JH, Giles AR, Kaufman RJ. Biochemical, immunological, and in vivo functional characterization of B-domain-deleted factor VIII. Blood 1993; 81:2925–2935. [PubMed] [Google Scholar]
- 8.Toole JJ, Pittman DD, Orr EC, Murtha P, Wasley LC, Kaufman RJ. A large region (approximately equal to 95 kDa) of human factor VIII is dispensable for in vitro procoagulant activity. Proc Natl Acad Sci U S A 1986; 83:5939–5942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ezban M, Vad K, Kjalke M. Turoctocog alfa (NovoEight®) – from design to clinical proof of concept. Eur J Haematol 2014; 93:369–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Christiansen ML, Balling KW, Persson E, Hilden I, Bagger-S⊘rensen A, S⊘rensen BB, et al. Functional characteristics of N8, a new recombinant FVIII. Haemophilia 2010; 16:878–887. [DOI] [PubMed] [Google Scholar]
- 11.Truett MA, Blacher R, Burke RL, Caput D, Chu C, Dina D, et al. Characterization of the polypeptide composition of human factor VIII:C and the nucleotide sequence and expression of the human kidney cDNA. DNA 1985; 4:339–349. [DOI] [PubMed] [Google Scholar]
- 12.Andersen MD, Faber JH. Structural characterization of both the nonproteolytic and proteolytic activation pathways of coagulation Factor XIII studied by hydrogen–deuterium exchange mass spectrometry. Int J Mass Spectrom 2011; 302:139–148. [Google Scholar]
- 13.Bloem E, van den Biggelaar M, Wroblewska A, Voorberg J, Faber JH, Kjalke M, et al. Factor VIII C1 domain spikes 2092-2093 and 2158-2159 comprise regions that modulate cofactor function and cellular uptake. J Biol Chem 2013; 288:29670–29679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kim JY, Kim YG, Lee GM. CHO cells in biotechnology for production of recombinant proteins: current state and further potential. Appl Microbiol Biotechnol 2012; 93:917–930. [DOI] [PubMed] [Google Scholar]
- 15.Bang S, Thim L, Karlsson J. Purification of factor VIII using a mixed-mode or multimodal resin. Patent No. WO 2009/007451 A1. http://www.google.com/patents/WO2009007451A1?cl=en [Accessed 5 March 2015]. [Google Scholar]
- 16.Thim L, Vandahl B, Karlsson J, Klausen NK, Pedersen J, Krogh TN, et al. Purification and characterization of a new recombinant factor VIII (N8). Haemophilia 2010; 16:349–359. [DOI] [PubMed] [Google Scholar]
- 17.Wales TE, Engen JR. Hydrogen exchange mass spectrometry for the analysis of protein dynamics. Mass Spectrom Rev 2006; 25:158–170. [DOI] [PubMed] [Google Scholar]
- 18.Eswar N, Webb B, Marti-Renom MA, Madhusudhan MS, Eramian D, Shen MY, et al. Comparative protein structure modeling using Modeller. Curr Protoc Bioinformatics 2006; Chapter 5:Unit 5.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bonneau R, Ruczinski I, Tsai J, Baker D. Contact order and ab initio protein structure prediction. Protein Sci 2002; 11:1937–1944. [DOI] [PMC free article] [PubMed] [Google Scholar]