

Abstract
Vγ9Vδ2 T effectors lyse cells in response to phosphorus-containing small molecules, providing primates a unique route to remove infected or malignant cells. Yet, the triggering mechanisms remain ill-defined. We examined lysis mediated by human Vγ9Vδ2 T effectors in response to the naturally-occurring (E)-4-hydroxy-3-methyl-but-2-enyl diphosphate (HMBPP) or a synthetic cell-permeable prodrug, bis (pivaloyloxymethyl) (E)-4-hydroxy-3-methyl-but-2-enyl phosphonate (POM2-C-HMBP). CD27+/CD45RA− Th1-like effector cells killed K562 target cells though a mechanism that could be enhanced by either compound or TCR antibody and blocked by Src inhibition or BTN3A1 disruption. Pre-treatment at 4°C decreased HMBPP-induced lysis but did not reduce lysis induced by POM2-C-HMBP. Together, our results show that internalization of HMBPP into target cells is required for BTN3A1-dependent lysis by Vγ9Vδ2 T effectors. The enhanced activity of the prodrug analog is due to its ability to bypass the pathways required for entry of HMBPP. These findings support an inside-out model of T cell triggering driven by small-molecule induction of BTN3A1.
Keywords: phosphoantigen, CD277, gamma delta T cell, butyrophilin
Introduction
It is essential for organisms to recognize and destroy foreign invaders, and the immune system has evolved a variety of means to do so. In adaptive immunity, T cells can employ the TCR to recognize a tremendous diversity of peptide antigens bound to the MHC. In contrast, innate cells can utilize pattern recognition receptors such as the TLRs to detect foreign small molecules known as pathogen-associated molecular patterns. The human Vγ9Vδ2 T cells are unique in that they respond to pathogen-associated small molecules in a way that appears dependent on TCR signaling [1]. The mechanisms controlling this non-traditional activation process are a matter of current interest [2, 3].
Vγ9Vδ2 T cells detect the small molecule (phosphoantigen) HMBPP [4]. As HMBPP is not produced by mammals, this sensitivity is indicative of a role for Vγ9Vδ2 T cells in pathogen elimination [5]. However, a lower-affinity molecule, IPP, is produced by mammals and can be elevated in malignant cells, suggesting a role for Vγ9Vδ2 T cells in immunosurveillance [6, 7]. Likewise, treatment with bisphosphonate drugs which activate Vγ9Vδ2 T cells can reduce risk of certain cancers [8]. If cells contain high levels of IPP, as in some cancerous cells, or HMBPP, as in infected cells, they will activate Vγ9Vδ2 T cells to cause a multifaceted immune response [3, 9, 10] including direct target cell lysis. HMBPP or its analogs may find clinical use as a way to direct Vγ9Vδ2 T cells to attack the malignant cells [11].
A B7 family protein, butyrophilin 3 isoform A1 (BTN3A1), is required for HMBPP to activate Vγ9Vδ2 T cells [12–15]. There are conflicting x-ray crystallography reports of how this occurs [16, 17]. One suggests a model of extracellular presentation similar to the traditional MHC-peptide-TCR complex [16]. However, another model clearly demonstrates that ligands bind to the intracellular domain of BTN3A1 [17], and our own NMR studies support the latter [18]. However, much remains unknown with respect to the mechanisms by which these small molecules work [19]. To address this question through alternative means, we recently described a cell-permeable prodrug analog of HMBPP, POM2-C-HMBP [18, 20]. Here, we use HMBPP and POM2-C-HMBP to reveal new information about the mechanisms governing their cellular uptake leading to lysis by Vγ9Vδ2 effectors and the excellent potency of the novel prodrug.
Materials and Methods
Reagents and supplies
RPMI-1640, FBS, non-essential amino acids, pyruvate, penicillin streptomycin, HEPES, 2-ME, HMBPP and FITC-γδ TCR antibody (clone 5A6.E91) were obtained through Fisher Scientific. IL-2 and the MACS γδ T cell negative selection kit were obtained through Miltenyi. PE-CD3 (UCHT1), APC-TNFα (MAb11), PE-IFNγ (4S.B3), FITC-IL-2 (MQ1-17H12), APC-CD45RA (HI-100), PerCP-eFluor710-CD27 (LG.7F9), FITC-Annexin V and PerCP-eFluor710-Annexin V were from eBioscience. K562 cells were from ATCC while Research Blood Components (Boston, MA) supplied blood. DiD, calcein AM, pcDNA3.1 and DNA primers were obtained through Life Technologies, while CRISPR/Cas9 repair templates were obtained through IDT. Restriction enzymes including XhoI, EcoRV and NheI were obtained through New England Biolabs. PE-BTN3A1 (CD277) antibody (BT3.1), FITC-CD183 (G025H7), functional grade anti-human TCR γ/δ Antibody (B1), and human IFNγ ELISA MAX deluxe kit were obtained from Biolegend while G418, PP2 and phytohaemagglutinin P were obtained from Sigma. Anti-myc (9E10) antibody was purified from hybridoma cells obtained from the Developmental Studies Hybridoma Bank at The University of Iowa. POM2-C-HMBP was a kind gift of Dr. David Wiemer at the University of Iowa.
Vγ9Vδ2 T cell expansion and purification
Human PBMCs from healthy anonymous donors were isolated from heparinized blood using Lymphoprep (Axis-Shield) within 24 hours of collection. Cells were counted by hemocytometer in the presence of trypan blue then aliquoted in freezing media (10% DMSO, 20% FBS, 70% media) and stored in liquid nitrogen. PBMC were resuspended at 1 × 106 cells/mL in fresh T cell media (RPMI-1640, 10% heat-inactivated FBS, 1x HEPES, pyruvate, NEAA, and BME). Cells were stimulated with 0.01 μM HMBPP or 0.01 μM POM2-C-HMBP for 3 days and cultured for another 4–18 days after compound removal. Fresh IL-2 (5 ng/mL) was provided every 3 days. After 7–21 days, Vγ9Vδ2 T cells were purified by negative selection. Experiments were performed at least three times using at least two donors.
K562 cell culture
K562 cells were cultured in RPMI-1640, 10% Fetal Clone III Serum (Hyclone) and 1% penicillin streptomycin and maintained between 0.2 and 1 × 106 cells per mL.
Surface staining
For surface staining experiments (γδ TCR/CD3, CD27/CD45RA, validation of BTN3A1 disruption), cells were washed twice in FACS buffer (2% BSA/PBS), suspended in FACS buffer and incubated at 4 °C for 30 minutes with antibodies (2–5 μL in 100 μL). Cells were washed twice in FACS buffer and analyzed using a BD FACSCalibur. In dual color experiments, gates were determined using single stained controls. Data was analyzed with FlowJo 9 and X software at the University of Connecticut Flow Cytometry & Confocal Microscopy Facility.
Intracellular cytokine staining
Purified expanded Vγ9Vδ2 T cells (4 × 106 cells in 1 mL in a 12 well plate) were incubated with PMA (10 ng/mL), ionomycin (1 μM), and brefeldin A solution (1x) according to manufacturer’s protocol (BD). Cells were incubated 4 hours at 37 °C, washed in FACS buffer, and fixed by addition of 4% paraformaldehyde for 10 minutes. Cells were washed in FACS buffer, permeabilized by addition of 0.1% saponin, and blocked with anti-CD16/CD32 (Fc Block) for 10 minutes. Cells were stained for 30 minutes with fluorescent antibodies, washed twice, and analyzed. In four color experiments, gates were drawn using controls deficient in each antibody.
IFNγ ELISA
K562 cells were suspended in T cell media and were treated with various concentrations of test compounds or PHA as described in the text for 2 hours. K562 cells were washed twice and mixed with T cells at an effector:target ratio of 3:1 in 200 μL in duplicate. Mixtures were incubated for 24 hours, following which IFNγ was assessed by ELISA according to manufacturer’s protocol (Biolegend).
Lysis assay
K562 cells were stained with DiD (2 minutes in 4 μM DiD in BSA/PBS), quenched by addition of an equal volume of FBS, and washed twice in T cell media. K562 cells were treated as described in text. K562 cells were mixed with T cells at an effector:target ratio of 3:1 in 200 μL. Mixtures were incubated for 4 hours at 37 °C then placed on ice for 5 minutes. FITC- or PerCP-eFluor710-Annexin V (3 μL) was added for 15 minutes on ice, then cells were diluted by addition of 200–300 μL binding buffer (BD) and analyzed by flow cytometry. In the multicolor experiments, K562 cells were stained with DiD or calcein-AM (15 minutes in 1 μM in media) and washed. DiD+ cells were exposed to test compounds while calcein+ cells were untreated. Cells were washed twice in media. Cells were mixed at a 1:1 DiD:Calcein ratio, and mixed with Vγ9Vδ2 T cells at an effector:target ratio of 3:1 in 200 μL.
Live imaging
ICAM-1 was obtained by protein G sepharose purification of CHO cell spent media as described [21, 22]. 384-well clear bottom plates were coated with 5 μg/μL (ICAM-1) for 60 min. Plates were washed with PBS, blocked with 2% BSA at 37 °C for 30 minutes. K562 cells pre-treated with HMBPP or POM2-C-HMBP were added and incubated at 37 °C for 30 minutes. T cells were added to the wells in a 3:1 ratio (effector:target). Cells were visualized using an Andor confocal microscope set up for multi-dimension acquisition and live imaging. Images were taken every 30–60 seconds for 30–240 minutes at 37 °C.
Disruption and re-expression of BTN3A1
CRISPR/Cas9 was used to disrupt BTN3A1 according to the method of Ran with slight modification [23]. sgRNAs of the following sequences designed to remove a 45 bp region of genomic DNA containing the BTN3A1 start codon were cloned into pX335-U6-Chimeric_BB-CBh-hSpCas9n (D10A) (Addgene): 5′-AAT GAA AAT GGC AAG TTT CC-3′ (sense) and 5′-TAT CAG GAG ATA CTG GAA GG-3′ (antisense). To facilitate homology directed repair, cells were co-transfected with a 192-bp single stranded repair template (IDT) (5′-GGT CAC CAT ACT TGA GTT AGC TCT AGG GAA GTG GAG GTT TCC ATT TGG AAT TCT ATA GCT TCT TCC AGG TCA TAG TGT CTG CCC CCC ACC CTC GAG GAT ATC CCT GGC CTT CCT TCT GCT CAA CTT TCG TGT CTG CCT CCT TTT GCT TCA GCT GCT CAT GCC TCA CTC AGG TAG GGA ACA ATT CCA CGC TTG-3′). The repair template contained central restriction sites for XhoI and EcoRV to facilitate genotyping. Transfection was performed by electroporation with a gene pulser XCELL (Biorad) using 316 volts, 500 μF capacitance and 4 mm conditions. The transfection was performed using 1 × 107 cells, 10 μg of each sgDNA plasmid, and 200 pg ssODN in 400 μL volume. At 24 hours post-transfection, single cells were separated by limiting dilution and allowed to grow for 3–4 weeks. Genomic DNA from individual clones was isolated by re-suspension of the cells from 1 mL culture in buffer (8 mM Tris, 5 mM EDTA, 200 mM NaCl, 0.2% SDS pH 8.0) containing 200 μg/mL proteinase K (Fisher) followed by heating at 55 °C for 1 hour, isopropanol precipitation, 70% ethanol wash, and re-suspension in water. Genotyping was performed by PCR of genomic DNA with the following primers: 5′-GTG AAG ACA CTG AAG GAC AGA A-3′ and 5′-CAG GTT TGG GAA GGA GTC AA-3′. The amplicon was 336 bp for the WT BTN3A1 and 303 bp for the expected mutant. The mutant DNA was further assessed by test digest with XhoI and EcoRV. Sanger sequencing was performed.
In BTN3A1 deficient cells, BTN3A1 was re-introduced by transfection and selection. BTN3A1 was cloned into pcDNA3.1 (Life Technologies) using the following primers from Life Technologies: forward primer into NheI site of pcDNA3.1 5′-GCT ATC GCT AGC ATG AAA ATG GCA AGT TTC-3′, reverse primer into XhoI site of pcDNA3.1 including myc tag and stop codon 5′-GCT ATC CTC GAG TCA CAG ATC CTC TTC TGA GAT GAG TTT TTG TTC CGC TGG ACA AAT AGT CAG and validated by Sanger sequencing. The plasmid (10 μg) was electroporated using the above conditions and selected with G418.
Statistical Analysis
One-way ANOVA was used to calculate significance in bar graphs. Comparisons were done relative to the control or between pairs of conditions. Columns in bar graphs represent the mean ± SEM. An α level of 0.05 was used. Dose response curves were analyzed using non-linear regression (log [agonist] versus response) in GraphPad Prism to generate EC50 values and confidence intervals.
Results
Stimulation with HMBPP or POM2-C-HMBP generates effector Vγ9Vδ2 T cells with a Th1 phenotype
Human blood contains Vγ9Vδ2 T cells that express sub-populations reminiscent of naïve and memory T cells [24]. We first assessed the phenotypes of Vγ9Vδ2 T cells following exposure to HMBPP or POM2-C-HMBP (Figure 1A). HMBPP was used in this study because it is the most potent naturally-occurring Vγ9Vδ2 T cell agonist, with activity that is 4 to 5 orders of magnitude greater than any other known natural molecule [25]. POM2-C-HMBP was used because we predicted it would rapidly deliver the BTN3A1 B30.2 ligand C-HMBP to the cytoplasmic binding site by passive transmembrane diffusion (Figure 1A) [18]. PBMCs were treated with either HMBPP or POM2-C-HMBP, cultured, and assessed for proliferation, Th1/Th2 polarization, and naïve/memory phenotype. Treatment with the compounds and IL-2 caused an increase in the proportion of γδ TCR+/CD3+ cells, which were isolated to >97% purity (shown for HMBPP in Figure 1B/C).
Figure 1. HMBPP and POM2-C-HMBP stimulate expansion of Th1 polarized effector Vγ9Vδ2 T cells.

A) Chemical structures of compounds used in this study. The pKa of each acidic hydrogen atom (as determined by Marvin software) is given in italics. B) Timeline showing HMBPP (10 nM) and IL-2 stimulation as well as experimental window used to expand effector T cells from PBMC. C) Vγ9Vδ2 T cell expansion analysis by flow cytometry of cell populations before and after purification by negative selection. D) Intracellular cytokine staining of IL-2, IFNγ, and TNFα in effector T cells as assessed by flow cytometry. E) Quantification of cytokine production by effector cell populations. Values represent the mean of three independent experiments, no significant differences were observed. F) Effector cell memory phenotype as assessed by flow cytometric staining for CD45RA and CD27. Flow plots in all panels are representative of greater than three independent experiments.
We assessed the polarization by intracellular cytokine staining (Figure 1D). Cells were labeled with antibodies to TNFα, IFNγ, IL-2, and IL-10. Few IL-10 positive cells were observed (not shown). In contrast, the majority of the expanded cells were positive for both TNFα and IFNγ. A subset of these cells expressed IL-2, while another subset was positive only for IFNγ (Figure 1E). No differences in cytokine production were observed in cells that were expanded by HMBPP versus POM2-C-HMBP. The pattern of cytokine production is consistent with a Th1 phenotype, and the cells were of sufficient quality to produce multiple cytokines regardless of the stimulatory compound used for initial expansion.
We assessed both compounds for their ability to generate effector cells by staining for CD27 and CD45RA. Unstimulated Vγ9Vδ2 TCR+/CD3+ cells were largely CD27+ with a range of CD45RA expression (Figure 1F). Following stimulation a strong majority of the cells became CD45RA−. Therefore, most of the cells used in this study were Vγ9Vδ2 TCR+/CD3+/CD27+/CD45RA−, consistent with an antigen experienced effector memory phenotype. Again, no differences were observed after exposure to HMBPP versus POM2-C-HMBP. Thus, regardless of whether the initial stimulation was performed with HMBPP or POM2-C-HMBP, the resultant Vγ9Vδ2 effector cells are of consistent phenotype.
Effector Vγ9Vδ2 T cells kill target cells in a Src-dependent manner
To measure the lytic capabilities of the effector T cells, we used a flow cytometric method [26] based on annexin V staining of the target cells that allows tracking both the killing of the target cells and the effectors (Figure 2). We mixed DiD+ K562 cells with the effectors for 4 hours in the continuous presence of HMBPP with or without the Src kinase inhibitor PP2. As expected, treatment with HMBPP induced T cell mediated killing of the K562 cells (Figure 2A/B). This effect was blocked by PP2. Therefore, inducible Vγ9Vδ2 T cell mediated killing is dependent on Src kinase activation.
Figure 2. Target cell lysis by Vγ9Vδ2 T effectors requires activity of Src kinases and is enhanced by POM2-C-HMBP during short-term pre-treatment.

A) Target and effectors cells were mixed and simultaneously exposed to 100 nM HMBPP with or without 10 μM PP2 for four hours. B) Quantification of panel A. C) Effector T cells demonstrate autolysis in the continued presence of HMBPP. D) K562 cell population gating scheme for 2 hour pre-treatment of K562 cells with phosphorus compounds (100 nM), followed by 4 hour incubation with effector cells. E) Dose response quantification of 2 hour pre-treatment lysis assays for both HMBPP and POM2-C-HMBP with EC50 values. F) Dose response quantification of IFNγ secretion following a 2 hour phosphorus compound pre-treatment and 24 hour incubation with effector cells. Flow plots are representative of greater than three independent experiments. *, significantly different from control; # significantly different from treatment.
Short-term preloading of target cells enhances specificity while reducing background Vγ9Vδ2 effector autolysis
During these studies, we observed that a number of effector T cells became annexin V positive. We directly tested for T cell autolysis under these conditions (Figure 2C). In the absence of K562 cells, the continuous presence of HMBPP significantly increased the number of annexin V positive T cells. To avoid this unwanted autolysis, we pre-loaded the K562 target cells alone for 2 hours, washed, and introduced them to the effectors for 4 hours. This pre-loading strategy resulted in lower baseline levels of target cell lysis in the absence of stimulation, typically to the 5–20% range, and reduced the amount of T cell autolysis, allowing for a more clear measurement of inducible call lysis. Figure 2D shows the gating scheme of the lysis assays used throughout this paper, where DiD+ K562 target cells were analyzed for annexin V binding.
Activity of the effector cells for lysis of the target cells could be increased by pre-treatment of the target cells with HMBPP or POM2-C-HMBP in a dose-dependent manner (Figure 2D, 2E). Pre-treatment of the target cells with the compounds resulted in a more specific response by the effector cells than did co-treatment for target cell lysis because it reduced the amount of T cell autolysis. Furthermore, preloading with POM2-C-HMBP (EC50 = 1.2 nM) was more effective at promoting lysis than was preloading with HMBPP (EC50 = 19 nM) (Figure 2E). Preloading the target cells with POM2-C-HMBP was also more effective at promoting IFNγ secretion than was preloading with HMBPP, where EC50 values of 22 nM for POM2-C-HMBP and 590 nM for HMBPP were observed (Figure 2F). To directly monitor the efficacy of the prodrug strategy, we also measured IFNγ production in response to the active metabolite of POM2-C-HMBP, C-HMBP, which showed a much larger EC50 value of 32 μM (1500x difference) (Figure S1). The superior activity of the neutral compound versus the charged compound during short incubations supports a model in which target cell internalization is critical for inducible T cell mediated killing and cytokine production by effector Vγ9Vδ2 T cells.
BTN3A1 disruption in target cells
BTN3A1 is necessary for prenyl diphosphates to induce proliferation of Vγ9Vδ2 T cells [27] and HMBPP and its analogs interact with the intracellular domain of BTN3A1 [17, 18], but it is not yet clear whether BTN3A1 functions in a similar capacity in experienced cytotoxic effector cells during cytolysis. To clarify the role of BTN3A1 in target cells, we created cell lines deficient of functional BTN3A1 using CRISPR/Cas9 (Figure S2A). All cell lines were analyzed by PCR to determine if the desired BTN3A1 mutation was produced. Of 35 clones, a range of indel mutations was observed, and clones 17, 18 and 26 were identified as “knockouts” (Figure S2B) as judged by the successful deletion of the start codon and insertion of a restriction site as intended by the single stranded DNA repair template. To validate loss of BTN3A1, we assessed surface levels of BTN3A1 in WT, clone 4 (WT clone), and KO clones 17, 18 and 26 by flow cytometry. A significant decrease in the fluorescence intensity of BTN3A1 staining was seen in KO cell lines, when compared to WT K562 cells and clone 4 (Figure S2C). Surface levels of CD183 were also analyzed by flow cytometry and collectively show no reduction in surface expression levels (Figure S2D).
BTN3A1 in target cells is required for lysis induced by HMBPP or POM2-C-HMBP
To evaluate whether or not BTN3A1 is required for inducible target cell lysis by Vγ9Vδ2 T cells, we performed lysis assays with the three clones of BTN3A1-disrupted cells. One WT clone (clone 4) and parental WT K562 cells were both used as controls. Cells were pre-treated for 2 hours with HMBPP or POM2-C-HMBP, then exposed to Vγ9Vδ2 effector cells. In the absence of ligand, all three BTN3A1-deficient clones showed no change in lysis when compared to WT K562 cells and the WT clone 4 (Figure 3A). However, when the BTN3A1-deficient cells were treated with HMBPP or POM2-C-HMBP, inducible lysis was significantly decreased relative to WT cells (Figure 3A–C).
Figure 3. Disruption of BTN3A1 expression in target cells reduces their lysis by Vγ9Vδ2 T effectors in response to direct-acing ligands.

A) Lysis assay results of WT, BTN3A1 WT clone 4, and BTN3A1 deficient clones 17, 18 and 26. K562 cells were pre-loaded for 2 hours with 100 nM HMBPP or 100 nM POM2-C-HMBP. B/C) Quantification of lysis assays shown in panel A. D) Lysis assay results of WT, clone 26 and clone 26+BTN3A1 K562 cells pre-treated for 2 hours with 100 nM test compound at 37°C followed by a 4 hour co-incubation with effector cells. E) Quantification of lysis assay described in D, where there is a significant rescue of lysis in clone 26 when expressing myc-tagged BTN3A1. F) Flow diagrams of K562 cell lysis induced by pre-treatment of T effector cells with TCR antibody. G) Quantification of panel F. Flow plots are representative of greater than three independent experiments. *, significance between treatments or groups identified.
To show that the change in cell lysis induced by the small molecules was due to lack of BTN3A1, we generated a BTN3A1-myc construct and electroporated it in to the CRISPR/Cas9 clone 26, producing a stable rescue cell line by selection in G418. Clone 26 cells expressing exogenous BTN3A1 showed a significant increase in lysis compared to clone 26 (Figure 3D/E). Together, loss of target cell BTN3A1 expression reduces the ability of both HMBPP and POM2-C-HMBP to trigger activation of Vγ9Vδ2 effector cells.
As further validation of the role of BTN3A1 in activation of Vγ9Vδ2 effector cells and the functionality of our CRISPR mutants, we loaded WT K562 cells or BTN3A1 deficient clones 17, 18, and 26 with various concentrations of C-HMBP and assessed their ability to stimulate production of IFNγ (Figure S3). As expected, preloaded WT K562 cells stimulated IFNγ in a dose-dependent manner (Figure S3A). All three of the BTN3A1 deficient clones were unable to trigger IFNγ production. WT K562 cells that had been pre-exposed to phytohemagglutinin (PHA) for 2 hours also triggered a dose-dependent increase in IFNγ production (Figure S3B). However, the effect of PHA on IFN production was only mildly reduced by loss of BTN3A1.
BTN3A1 in target cells is dispensable for lysis induced by TCR antibody
To determine whether these BTN3A1-deficient cells were still susceptible to killing by Vγ9Vδ2 effector cells, we then examined the ability of a γδ TCR antibody to induce their lysis (Figure 3F/G). T cells were pre-incubated with the antibody then mixed with target cells. In WT K562 cells, the TCR antibody was sufficient to induce the T cells to kill the K562 cells. In contrast to the inducible lysis stimulated by HMBPP, the loss of BTN3A1 function had no effect on inducible lysis stimulated by the TCR antibody. Thus, BTN3A1 is required for lysis induced by phosphorous compounds but loss of BTN3A1 does not affect the ability of the target cells to be lysed when stimulated by TCR crosslinking.
HMBPP, but not POM2-C-HMBP, utilizes an energy-dependent uptake pathway to enter target cells, which is required for inducible Vγ9Vδ2 T cell mediated lysis
Based on the more potent activity of the charge-neutral POM2-C-HMBP compared to the negatively charged HMBPP (Figure 2E, F), we hypothesized that these target cells utilize an active uptake process to internalize HMBPP. To further investigate this possibility we pre-treated K562 cells with HMBPP or POM2-C-HMBP at either 37 °C or 4 °C for 2 hours, as incubation of cells at low temperatures inhibits energy dependent uptake mechanisms [28]. After pre-incubation, we mixed the target cells with the effectors and assessed their lysis (Figure 4A). HMBPP-treated cells exhibited a significant decrease in inducible lysis when pre-loaded at 4 °C in comparison to those treated at 37 °C, while POM2-C-HMBP treated cells did not lose activity when loaded at the lower temperature (Figure 4B). Thus, energy-dependent cellular entry is required for the activity of HMBPP.
Figure 4. HMBPP, but not POM2-C-HMBP, utilizes energy dependent uptake to enter target cells.

A) Flow cytometry plots of lysis assays where K562 cells were pre-treated with test compound (100 nM) for 2 hours at either 37 °C or 4 °C, followed by 4 hour incubation with effector cells. B) Quantification of lysis assays described in panel A. Percent lysis was found by the percentage of Annexin V/K562 target cells for each of the treatments, 0 hour control, 4 hour control, 100 nM HMBPP and 100 nM POM2-C-HMBP at both 37°C and 4°C. C) Live imaging of K562 cells untreated or pre-treated with 100 nM POM2-C-HMBP at 37°C. White arrow heads indicate effector Vγ9Vδ2 T cells; black arrow heads indicate target K562 cells. D) Quantification of imaging analysis, percent lysis was determined by the number of cells lysed within 1 hour, divided by the total number of K562 cells in the visual field multiplied by 100. E) HMBPP dose response lysis assay at 37°C or 4°C. F) POM2-C-HMBP dose response lysis assay at 37°C or 4°C. Flow plots are representative of greater than three independent experiments. *, significance between treatments or groups identified.
These findings are also evident from live cell imaging experiments (Figure 4C/D). The K562 cells were exposed to compounds as described above, plated onto ICAM-1 to allow for traction [21, 22, 29] and exposed to the effectors. Cells that had been loaded with POM2-C-HMBP were lysed by the T cells, while untreated cells were scanned but not killed (Figure 4C). Again, pre-treatment at 4 °C blocked the ability of HMBPP but not POM2-C-HMBP to trigger T cell mediated lysis (Figure 4D), while POM2-C-HMBP demonstrated enhanced potency relative to HMBPP.
To further investigate these effects, we used flow cytometry to generate dose response curves for both compounds at both temperatures. Lysis induced by HMBPP was significantly reduced by loading at 4 °C relative to 37 °C, while the activity of POM2-C-HMBP was unchanged (Figure 4E/F) (Table I). The charged molecule C-HMBP was also analyzed in the same manner and showed a similar pattern to that of HMBPP (Figure S4A). These data show that the activity of both HMBPP and C-HMBP are decreased at 4 °C, indicating an energy-dependent uptake mechanism is required for their activity, but not that of POM2-C-HMBP. Together, these data oppose the model of extracellular ligand binding.
Table I.
Phosphorus compound EC50 values at 37°C and 4°C. 1
| EC50 37°C (95% CI) | EC50 4°C (95% CI) | Fold change | |
|---|---|---|---|
| HMBPP | 0.042 (0.017–0.10) | 1.1 (0.57–2.3) | 26 |
| POM2-C-HMBP | 0.0041 (0.0017–0.010) | 0.0077 (0.0017–0.034) | 1.9 |
| C-HMBP | 2.1 (0.55–8.0) | 48 (41–57) | 23 |
EC50 values determined by dose response lysis assays at both 37°C and 4°C. 95% confidence intervals are shown in parenthesis. All values are in μM. n=3.
Inducible lysis by Vγ9Vδ2 T cells is selective for loaded target cells
It was not clear whether the effectors were sensitive enough to kill only cells that contain HMBPP and not HMBPP-deficient neighboring cells. Therefore we assessed the ability of Vγ9Vδ2 T cells to lyse a mixture of K562 cells in which only some cells were loaded (Figure 5). We labeled K562 cells with one of two fluorescent probes (DiD and calcein). DiD+ cells were then loaded with either HMBPP or POM2-C-HMBP for 2 hours, washed, and exposed to effectors. In this mixed population, some constitutive lysis was observed, which was consistent regardless of the label. Treatment with HMBPP or POM2-C-HMBP increased the fraction of lysed cells. Vγ9Vδ2 T cells effectively lysed the targets that contained either HMBPP or POM2-C-HMBP while ignoring the non-loaded cells in the mixture (Figure 5A/B).
Figure 5. Vγ9Vδ2 T cells specifically target cells containing phosphorus compounds.
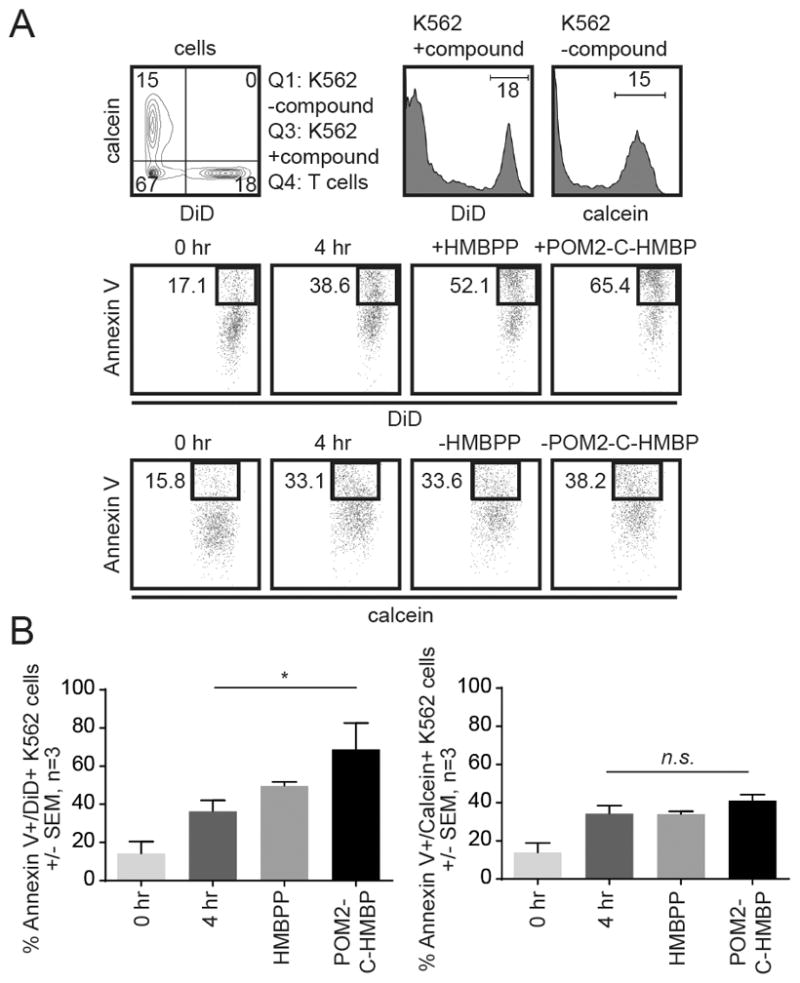
A) Cells were stained with either DiD or calcein. DiD+ cells were pre-treated for 2 hours with HMBPP (100 nM) or POM2-C-HMBP (100 nM), then mixed with effector cells for 4 hours. B) Quantification of panel A. Flow plots are representative of three independent experiments. *, significance of treatments or groups identified compared to control.
POM2-C-HMBP remains active within target cells while HMBPP is quickly metabolized
Once HMBPP is internalized, the target cells are susceptible to lysis by the Vγ9Vδ2 T effectors. However, it is not clear how long target cells retain this susceptibility. We tested this by pre-loading target cells with 100 nM HMBPP or 100 nM POM2-C-HMBP for 2 hours and then resting the target cells for 0 (no rest), 12 or 24 hours. After this time, the target cells were mixed with Vγ9Vδ2 T effectors for 4 hours and analyzed by flow cytometry as described above. Results shown in Figure 6A/B reveal that the effect of HMBPP is lost within 12 hours, while POM2-C-HMBP remains fully active at 12 hours and 24 hours post-loading.
Figure 6. HMBPP, but not POM2-C-HMBP, is rapidly metabolized.

A) Flow cytometry data of lysis assays where K562 cells were pre-treated with compound (100 nM) for 2 hours at 37 °C, followed by a 0 hour (no rest), 12 hour or 24 hour resting period. Once rested, cells were incubated with effector cells for 4 hours. B) Quantification of lysis assay described in panel A. *, significance between treatments or groups identified. C) Time course lysis assay of K562 cells pre-treated for 2 hours with 100 nM HMBPP. K562 cells were rested for 0 hours (no rest), 6 hours, 12 hours, 24 hours, 48 hours or 72 hours. Once rested, cells were incubated with effector cells for 4 hours. *, significant compared to no rest. D) Time course lysis assay of K562 cells pre-treated for 2 hours with 100 nM POM2-C-HMBP, as described in C. *, significant compared to no rest. E) Time course lysis assay of K562 cells pre-treated for 2 hours with 10 nM POM2-C-HMBP, as described in C. *, significant compared to no rest. Flow plots are representative of greater than three independent experiments.
To further investigate these findings, we extended our experiment to include additional time points. The activity gained by pre-treatment with 100 nM HMBPP was again lost between 6 and 12 hours (Figure 6C), while the activity of 100 nM POM2-C-HMBP is lost only after 48 hours (Figure 6D). Even at 10 nM (Figure 6E), POM2-C-HMBP remains active for longer than 100 nM HMBPP. This analysis was also done using C-HMBP at 10 μM (Figure S4B), which lost its activity between 12 and 24 hours, similar to that of 10 nM POM2-C-HMBP. Thus, target cells are capable of HMBPP destruction or removal in the absence of T cell mediated cytolysis. POM2-C-HMBP and C-HMBP, appear to delay this metabolic fate, presumably due in part to the metabolic stability of the phosphonate bond relative to the analogous phosphate linkage of HMBPP.
Discussion
In this study, we use HMBPP and a cell-cleavable prodrug analog, POM2-C-HMBP, to clarify the mechanisms of Vγ9Vδ2 TCR triggering during target cell lysis and the role of cellular internalization in this process. We demonstrate that an energy-dependent uptake mechanism is required for charged phosphorus compounds to use BTN3A1 to trigger Vγ9Vδ2 mediated cell lysis. This uptake mechanism can be readily bypassed by the charge-neutral POM2-C-HMBP, leading to a dramatic increase in BTN3A1-mediated killing and cytokine production relative to the charged compound HMBPP.
Exposure of PBMCs to either compound generates effector cells that produce IFNγ, TNFα, and IL-2. This is similar to the indirect-acting zoledronate [30]. These effectors can kill K562 target cells in a way that is enhanced by addition of either phosphorus compound. The inducible lysis is dependent upon Src kinase activity, as it can be blocked by co-treatment with PP2, which presumably inhibits the T cell specific Src isoforms Lck and/or Fyn in this model. Given that K562 cells do not express MHC [31–33], the process is MHC-independent. Inducible lysis is dependent upon the presence of BTN3A1, as disruption of this gene by CRISPR/Cas9 blocks the ligand-induced lysis while re-expression of BTN3A1 rescues the effect. Likewise, disruption of BTN3A1 fully abrogates the ability of phosphoantigens, but not PHA, to stimulate cytokine production by Vγ9Vδ2 effectors. However, BTN3A1 disruption does not affect lysis induced by TCR antibody, so it is likely that the function of BTN3A1 is specific to HMBPP and its analogs. The sensitivity to lysis induced by HMBPP is selective enough that it can kill only the loaded cells within a mixed population, but it is transient, disappearing in 6–12 hours after exposure.
We also determined that entry of charged ligands but not the neutral prodrug decreases at low temperatures, which supports a role for a type of energy dependent uptake, likely endocytosis and internalization in the activity of HMBPP. This finding is consistent with a model in that, given the pKa values of HMBPP, diffusion into the cytoplasm is strongly enhanced as the molecule becomes protonated during acidification of endocytic vesicles, similar as had been previously described with respect to cellular entry of bisphosphonate drugs [34]. Vγ9Vδ2 T cells recognize cells containing these molecules bound to the BTN3A1 B30.2 domain and form an MHC-independent immunological synapse with the target cell that leads to the activation of the Vγ9Vδ2 T cell. This active uptake mechanism would not be necessary for Vγ9Vδ2 T cells to sense metabolites produced by intracellular pathogens which can readily access the binding site. However, internalization is likely critical when Vγ9Vδ2 effector cells respond to distantly affected cells in the absence of contact, such as has been reported in Plasmodium infection [35] and response to tumor-derived IPP [36].
There are only two naturally-occurring compounds that are currently known to activate BTN3A1 directly- HMBPP and IPP, the former of which clearly has the stronger binding [18]. Therefore, BTN3A1 can be described as a receptor for HMBPP, with IPP and the synthetic compounds being analogs of HMBPP that also bind the receptor. Use of HMBPP and its analogs provides clarity over use of zoledronate and the other bisphosphonates, which although they are clinically-used agents, act indirectly and display a multitude of cellular effects resulting from depletion of isoprenoids and sterols, including cell toxicity. The direct acting BTN3A1 ligands HMBPP and POM2-C-HMBP display markedly increased therapeutic indices relative to zoledronate [18]. Furthermore, as we show in the present study, POM2-C-HMBP offers several differences over HMBPP- it retains the ability to generate high quality Th1 effector cells, it shows faster cell uptake that is independent of endocytosis, and it sensitizes cells for a longer time. It is not yet clear whether these differences would result in therapeutic benefit.
Although it has been recognized that HMBPP and its analogs activate T cells that express the Vγ9Vδ2 TCR [33], the mechanisms have remained enigmatic [2, 3]. Investigators have tried to fit these small molecules into traditional models of TCR stimulation triggered by peptide antigens. In part, this is because the TCR extracellular domain [37] and downstream signaling is required for HMBPP activity [38], which our findings with PP2 and the TCR antibody support. At the same time, HMBPP activity has been reported to be independent of MHC [16], which is consistent with our findings because K562 cells do not express MHC. The apparent discordance of these results has led others to search for an antigen presenting protein that is functionally analogous to MHC and could serve to present HMBPP directly to the Vγ9Vδ2 TCR on the cell surface, which is exemplified by the models put forth initially by Morita and Brenner [27, 33] and subsequently by De Libero [16].
However, now that the importance of the internal B30.2 domain of BTN3A1 in HMBPP activity has been identified [3, 17, 18, 27, 39], it has become apparent that Vγ9Vδ2 T cell activation does not fit the traditional models. The earlier studies that suggested external presentation [33] were likely confounded by use of high ligand concentrations which allowed for transmembrane diffusion and BTN3A1 activation even in lightly fixed cells. Because BTN3A1 is a B7 family protein, it is possible that HMBPP functions through inducible BTN3A1-mediated co-stimulation (Figure 7). Unfortunately, this implies that the term phospho‘antigen’ may be a confusing misnomer as HMBPP and its analogs are not actually stimulatory MHC:TCR ligands (i.e. antigens). Instead, they are ligands which control the function of a B7 family member. Thus, these non-peptide “antigens” which activate Vγ9Vδ2 T cells may be better classified as pathogen associated molecular-patterns that bind to a type of pattern recognition receptor-the B30.2 domain of BTN3A1.
Figure 7. Hypothetical models of BTN3A1 mediated T cell activation.

Phosphoantigens such as HMBPP are internalized by an energy dependent process, which can be bypassed by POM2-C-HMBP. Both BTN3A1 and the Vγ9Vδ2 TCR are required for T cell activation in response to these phosphoantigens. It is unknown how BTN3A1 activation may trigger a T cell response, but this could occur either through direct TCR engagement or through a co-stimulatory process.
The outstanding unknown is the identity of the extracellular binding partners of BTN3A1 and the Vγ9Vδ2 TCR. Specifically, models in which zero, one or two extracellular ligands are unknown can be envisioned. Although the TCRγ J region has been reported to be required for detection of prenyl diphosphates [37], direct extracellular binding between the TCR and BTN3A1 has not been observed [17]. However, these studies did not exclude the possibility of multimeric complex of BTN3A1 directly binding to the TCR (i.e. the direct triggering model, Figure 7). On the other hand, if the traditional two-signal aggregation/conformational change mechanisms are true [27], then both proteins would require an extracellular ligand on the opposing cell. This model of TCR binding with BTN3A1 co-stimulation is supported by studies that show additional factors on chromosome 6 influence phosphoantigen detection [40]. However, existing data does not appear to exclude a kinetic segregation model as described in other T cell types [41]. If this model were true, the Vγ9Vδ2 TCR would be required only as a signaling platform, with the TCRγ J region required but not used for binding the opposing cell. T cell activation would occur not through extracellular ligand binding to an MHC-like complex, but rather through inducible co-stimulation leading to synapse formation and steric exclusion of inhibitory proteins. In fact, CD45 exclusion from the Vγ9Vδ2 synapse already has been observed [42, 43]. Identification of the extracellular binding partner(s), if any exist, will further elucidate the mechanisms underlying this unique cross-membrane signaling event.
Supplementary Material
Acknowledgments
The assistance of Carol Norris at the University of Connecticut Flow Cytometry and Confocal Microscopy Facility is greatly appreciated.
Source of support: National Institutes of Health under Award Number R01CA186935 (A.J.W., P.I.)
Abbreviations
- POM2-C-HMBP
bis (pivaloyloxymethyl) (E)-4-hydroxy-3-methyl-but-2-enyl phosphonate
- HMBPP
(E)-4-hydroxy-3-methyl-but-2-enyl diphosphate
- IPP
isopentenyl diphosphate
- BTN3A1
butyrophilin 3 isoform A1
References
- 1.Wrobel P, Shojaei H, Schittek B, Gieseler F, Wollenberg B, Kalthoff H, Kabelitz D, Wesch D. Lysis of a broad range of epithelial tumour cells by human gamma delta T cells: involvement of NKG2D ligands and T-cell receptor- versus NKG2D-dependent recognition. Scand J Immunol. 2007;66:320–328. doi: 10.1111/j.1365-3083.2007.01963.x. [DOI] [PubMed] [Google Scholar]
- 2.Adams EJ, Gu S, Luoma AM. Human gamma delta T cells: Evolution and ligand recognition. Cell Immunol. 2015;296:31–40. doi: 10.1016/j.cellimm.2015.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Harly C, Peigne CM, Scotet E. Molecules and Mechanisms Implicated in the Peculiar Antigenic Activation Process of Human Vgamma9Vdelta2 T Cells. Front Immunol. 2014;5:657. doi: 10.3389/fimmu.2014.00657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wiemer DF, Wiemer AJ. Opportunities and challenges in development of phosphoantigens as Vgamma9Vdelta2 T cell agonists. Biochem Pharmacol. 2014;89:301–12. doi: 10.1016/j.bcp.2014.03.009. [DOI] [PubMed] [Google Scholar]
- 5.Wiemer AJ, Hsiao CHC, Wiemer DF. Isoprenoid Metabolism as a Therapeutic Target in Gram-Negative Pathogens. Curr Top Med Chem. 2010;10:1858–1871. doi: 10.2174/156802610793176602. [DOI] [PubMed] [Google Scholar]
- 6.Harwood HJ, Jr, Alvarez IM, Noyes WD, Stacpoole PW. In vivo regulation of human leukocyte 3-hydroxy-3-methylglutaryl coenzyme A reductase: increased enzyme protein concentration and catalytic efficiency in human leukemia and lymphoma. J Lipid Res. 1991;32:1237–52. [PubMed] [Google Scholar]
- 7.Gober HJ, Kistowska M, Angman L, Jeno P, Mori L, De Libero G. Human T Cell Receptor Cells Recognize Endogenous Mevalonate Metabolites in Tumor Cells. J Exp Med. 2003;197:163–168. doi: 10.1084/jem.20021500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Santolaria T, Robard M, Leger A, Catros V, Bonneville M, Scotet E. Repeated systemic administrations of both aminobisphosphonates and human Vgamma9Vdelta2 T cells efficiently control tumor development in vivo. J Immunol. 2013;191:1993–2000. doi: 10.4049/jimmunol.1300255. [DOI] [PubMed] [Google Scholar]
- 9.Ryan-Payseur B, Frencher J, Shen L, Chen CY, Huang D, Chen ZW. Multieffector-functional immune responses of HMBPP-specific Vgamma2Vdelta2 T cells in nonhuman primates inoculated with Listeria monocytogenes DeltaactA prfA*. J Immunol. 2012;189:1285–93. doi: 10.4049/jimmunol.1200641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vantourout P, Hayday A. Six-of-the-best: unique contributions of gammadelta T cells to immunology. Nat Rev Immunol. 2013;13:88–100. doi: 10.1038/nri3384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fournie JJ, Sicard H, Poupot M, Bezombes C, Blanc A, Romagne F, Ysebaert L, Laurent G. What lessons can be learned from gammadelta T cell-based cancer immunotherapy trials? Cell Mol Immunol. 2013;10:35–41. doi: 10.1038/cmi.2012.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Messal N, Mamessier E, Sylvain A, Celis-Gutierrez J, Thibult ML, Chetaille B, Firaguay G, Pastor S, Guillaume Y, Wang Q, Hirsch I, Nunes JA, Olive D. Differential role for CD277 as a co-regulator of the immune signal in T and NK cells. Eur J Immunol. 2011;41:3443–54. doi: 10.1002/eji.201141404. [DOI] [PubMed] [Google Scholar]
- 13.Palakodeti A, Sandstrom A, Sundaresan L, Harly C, Nedellec S, Olive D, Scotet E, Bonneville M, Adams EJ. The molecular basis for modulation of human Vgamma9Vdelta2 T cell responses by CD277/butyrophilin-3 (BTN3A)-specific antibodies. J Biol Chem. 2012;287:32780–90. doi: 10.1074/jbc.M112.384354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Harly C, Guillaume Y, Nedellec S, Peigne CM, Monkkonen H, Monkkonen J, Li J, Kuball J, Adams EJ, Netzer S, Dechanet-Merville J, Leger A, Herrmann T, Breathnach R, Olive D, Bonneville M, Scotet E. Key implication of CD277/butyrophilin-3 (BTN3A) in cellular stress sensing by a major human gammadelta T-cell subset. Blood. 2012;120:2269–2279. doi: 10.1182/blood-2012-05-430470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gu S, Nawrocka W, Adams EJ. Sensing of Pyrophosphate Metabolites by Vgamma9Vdelta2 T Cells. Front Immunol. 2014;5:688. doi: 10.3389/fimmu.2014.00688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vavassori S, Kumar A, Wan GS, Ramanjaneyulu GS, Cavallari M, El Daker S, Beddoe T, Theodossis A, Williams NK, Gostick E, Price DA, Soudamini DU, Voon KK, Olivo M, Rossjohn J, Mori L, De Libero G. Butyrophilin 3A1 binds phosphorylated antigens and stimulates human gammadelta T cells. Nat Immunol. 2013;14:908–16. doi: 10.1038/ni.2665. [DOI] [PubMed] [Google Scholar]
- 17.Sandstrom A, Peigne CM, Leger A, Crooks JE, Konczak F, Gesnel MC, Breathnach R, Bonneville M, Scotet E, Adams EJ. The Intracellular B30.2 Domain of Butyrophilin 3A1 Binds Phosphoantigens to Mediate Activation of Human Vgamma9Vdelta2 T Cells. Immunity. 2014;40:490–500. doi: 10.1016/j.immuni.2014.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hsiao CH, Lin X, Barney RJ, Shippy RR, Li J, Vinogradova O, Wiemer DF, Wiemer AJ. Synthesis of a phosphoantigen prodrug that potently activates Vgamma9Vdelta2 T-lymphocytes. Chem Biol. 2014;21:945–54. doi: 10.1016/j.chembiol.2014.06.006. [DOI] [PubMed] [Google Scholar]
- 19.Kabelitz D. CD277 takes the lead in human gammadelta T-cell activation. Blood. 2012;120:2159–2161. doi: 10.1182/blood-2012-07-442731. [DOI] [PubMed] [Google Scholar]
- 20.Wiemer AJ, Wiemer DF. Prodrugs of phosphonates and phosphates: crossing the membrane barrier. Top Curr Chem. 2015;360:115–60. doi: 10.1007/128_2014_561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wernimont SA, Wiemer AJ, Bennin DA, Monkley SJ, Ludwig T, Critchley DR, Huttenlocher A. Contact-Dependent T Cell Activation and T Cell Stopping Require Talin1. J Immunol. 2011;187:6256–6267. doi: 10.4049/jimmunol.1102028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wiemer AJ, Wernimont S, Huttenlocher A. Live imaging of LFA-1-dependent T-cell motility and stop signals. Methods Mol Biol. 2012;757:191–204. doi: 10.1007/978-1-61779-166-6_13. [DOI] [PubMed] [Google Scholar]
- 23.Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, Zhang F. Genome engineering using the CRISPR-Cas9 system. Nat Protoc. 2013;8:2281–308. doi: 10.1038/nprot.2013.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Holtmeier W, Kabelitz D. gammadelta T cells link innate and adaptive immune responses. Chem Immunol Allergy. 2005;86:151–83. doi: 10.1159/000086659. [DOI] [PubMed] [Google Scholar]
- 25.Morita CT, Jin C, Sarikonda G, Wang H. Nonpeptide antigens, presentation mechanisms, and immunological memory of human Vgamma2Vdelta2 T cells: discriminating friend from foe through the recognition of prenyl pyrophosphate antigens. Immunol Rev. 2007;215:59–76. doi: 10.1111/j.1600-065X.2006.00479.x. [DOI] [PubMed] [Google Scholar]
- 26.Goldberg JE, Sherwood SW, Clayberger C. A novel method for measuring CTL and NK cell-mediated cytotoxicity using annexin V and two-color flow cytometry. J Immunol Methods. 1999;224:1–9. doi: 10.1016/s0022-1759(98)00038-6. [DOI] [PubMed] [Google Scholar]
- 27.Wang H, Henry O, Distefano MD, Wang YC, Raikkonen J, Monkkonen J, Tanaka Y, Morita CT. Butyrophilin 3A1 Plays an Essential Role in Prenyl Pyrophosphate Stimulation of Human Vgamma2Vdelta2 T Cells. J Immunol. 2013;191:1029–1042. doi: 10.4049/jimmunol.1300658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Batzri S, Korn ED. Interaction of phospholipid vesicles with cells. Endocytosis and fusion as alternate mechanisms for the uptake of lipid-soluble and water-soluble molecules. J Cell Biol. 1975;66:621–34. doi: 10.1083/jcb.66.3.621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wernimont SA, Legate KR, Simonson WT, Fassler R, Huttenlocher A. PIPKI gamma 90 negatively regulates LFA-1-mediated adhesion and activation in antigen-induced CD4+ T cells. J Immunol. 2010;185:4714–4723. doi: 10.4049/jimmunol.1001445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gruenbacher G, Gander H, Rahm A, Nussbaumer W, Romani N, Thurnher M. CD56+ human blood dendritic cells effectively promote TH1-type gammadelta T-cell responses. Blood. 2009;114:4422–4431. doi: 10.1182/blood-2009-06-227256. [DOI] [PubMed] [Google Scholar]
- 31.Holoshitz J, Vila LM, Keroack BJ, McKinley DR, Bayne NK. Dual antigenic recognition by cloned human gamma delta T cells. J Clin Invest. 1992;89:308–14. doi: 10.1172/JCI115577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kozbor D, Trinchieri G, Monos DS, Isobe M, Russo G, Haney JA, Zmijewski C, Croce CM. Human TCR-gamma+/delta+, CD8+ T lymphocytes recognize tetanus toxoid in an MHC-restricted fashion. J Exp Med. 1989;169:1847–51. doi: 10.1084/jem.169.5.1847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Morita CT, Beckman EM, Bukowski JF, Tanaka Y, Band H, Bloom BR, Golan DE, Brenner MB. Direct presentation of nonpeptide prenyl pyrophosphate antigens to human gamma delta T cells. Immunity. 1995;3:495–507. doi: 10.1016/1074-7613(95)90178-7. [DOI] [PubMed] [Google Scholar]
- 34.Thompson K, Rogers MJ, Coxon FP, Crockett JC. Cytosolic entry of bisphosphonate drugs requires acidification of vesicles after fluid-phase endocytosis. Mol Pharmacol. 2006;69:1624–1632. doi: 10.1124/mol.105.020776. [DOI] [PubMed] [Google Scholar]
- 35.Guenot M, Loizon S, Howard J, Costa G, Baker DA, Mohabeer SY, Troye-Blomberg M, Moreau JF, Dechanet-Merville J, Mercereau-Puijalon O, Mamani-Matsuda M, Behr C. Phosphoantigen Burst Upon Plasmodium Falciparum Schizont Rupture Can Distantly Activate Vgamma9-Vdelta2 T-Cells. Infect Immun. 2015 doi: 10.1128/IAI.00446-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Benzaid I, Monkkonen H, Stresing V, Bonnelye E, Green J, Monkkonen J, Touraine JL, Clezardin P. High phosphoantigen levels in bisphosphonate-treated human breast tumors promote Vgamma9Vdelta2 T-cell chemotaxis and cytotoxicity in vivo. Cancer Res. 2011;71:4562–72. doi: 10.1158/0008-5472.CAN-10-3862. [DOI] [PubMed] [Google Scholar]
- 37.Bukowski JF, Morita CT, Band H, Brenner MB. Crucial role of TCR gamma chain junctional region in prenyl pyrophosphate antigen recognition by gamma delta T cells. J Immunol. 1998;161:286–293. [PubMed] [Google Scholar]
- 38.Gong G, Shao L, Wang Y, Chen CY, Huang D, Yao S, Zhan X, Sicard H, Wang R, Chen ZW. Phosphoantigen-activated V gamma 2V delta 2 T cells antagonize IL-2-induced CD4+CD25+Foxp3+ T regulatory cells in mycobacterial infection. Blood. 2009;113:837–845. doi: 10.1182/blood-2008-06-162792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang H, Morita CT. Sensor Function for Butyrophilin 3A1 in Prenyl Pyrophosphate Stimulation of Human Vgamma2Vdelta2 T Cells. J Immunol. 2015;195:4583–94. doi: 10.4049/jimmunol.1500314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Riano F, Karunakaran MM, Starick L, Li J, Scholz CJ, Kunzmann V, Olive D, Amslinger S, Herrmann T. Vgamma9Vdelta2 TCR-activation by phosphorylated antigens requires butyrophilin 3 A1 (BTN3A1) and additional genes on human chromosome 6. Eur J Immunol. 2014;44:2571–6. doi: 10.1002/eji.201444712. [DOI] [PubMed] [Google Scholar]
- 41.Davis SJ, van der Merwe PA. The kinetic-segregation model: TCR triggering and beyond. Nat Immunol. 2006;7:803–9. doi: 10.1038/ni1369. [DOI] [PubMed] [Google Scholar]
- 42.Espinosa E, Tabiasco J, Hudrisier D, Fournie JJ. Synaptic transfer by human gamma delta T cells stimulated with soluble or cellular antigens. J Immunol. 2002;168:6336–43. doi: 10.4049/jimmunol.168.12.6336. [DOI] [PubMed] [Google Scholar]
- 43.Favier B, Espinosa E, Tabiasco J, Dos Santos C, Bonneville M, Valitutti S, Fournie JJ. Uncoupling between immunological synapse formation and functional outcome in human gamma delta T lymphocytes. Journal of immunology (Baltimore, Md: 1950) 2003;171:5027–5033. doi: 10.4049/jimmunol.171.10.5027. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.