

Abstract
Pulmonary alveolar proteinosis (PAP) is a rare lung syndrome caused by the accumulation of surfactants in the alveoli. The most prevalent clinical form of PAP is autoimmune (aPAP) whereby IgG autoantibodies neutralize granulocyte-macrophage colony-stimulating factor (GM-CSF). GM-CSF is a pleiotropic cytokine that promotes the differentiation, survival, and activation of alveolar macrophages, the cells responsible for surfactant degradation. IgG-mediated neutralization of GM-CSF thereby inhibits alveolar macrophage homeostasis and function, leading to surfactant accumulation and innate immunodeficiency. Importantly, there are no rodent models for this disease, and therefore underlying immune mechanisms regulating GM-CSF-specific IgG in aPAP are not well understood. Herein, we identify that autoimmune-prone Rasgrp1-deficient mice develop aPAP: 1) Rasgrp1-deficient mice exhibit reduced pulmonary compliance as well as lung histopathology characteristic of PAP; 2) alveolar macrophages from Rasgrp1-deficient mice are enlarged and exhibit reduced surfactant degradation; 3) the concentration of GM-CSF-specific IgG is elevated in both serum and bronchial-alveolar lavage fluid (BALF) from Rasgrp1-deficient mice; 4) GM-CSF-specific IgG is capable of neutralizing GM-CSF bioactivity; and 5) Rasgrp1-deficient mice also lacking CD275/ICOSL, a molecule necessary for conventional T cell-dependent antibody production, have reduced GM-CSF-specific autoantibody and do not develop PAP. Collectively these studies reveal that Rasgrp1-deficient mice represent the first rodent model for aPAP.
Keywords: pulmonary alveolar proteinosis, granulocyte-macrophage colony-stimulating factor, autoantibodies, alveolar macrophages, pulmonary surfactants, mouse model
Introduction
Pulmonary alveolar proteinosis (PAP) is a rare pulmonary syndrome defined by the accumulation of pulmonary surfactant in the alveoli leading to respiratory insufficiency. Pulmonary surfactant levels are maintained in part by granulocyte-macrophage colony-stimulating factor (GM-CSF, also called colony stimulating factor 2)1-5. GM-CSF is a pleotropic cytokine that is required for the differentiation, maintenance, and function of alveolar macrophages6, 7. In the absence of GM-CSF, degradation of pulmonary surfactants by alveolar macrophages is impaired leading to an accumulation in the alveoli. GM-CSF-deprived alveolar macrophages also are less adherent, and they exhibit impaired phagocytosis and pathogen killing6. Defective phagocytosis and pathogen killing by alveolar macrophages likely contributes to increased susceptibility to infections with opportunistic pathogens such as Nocardia species (spp.), Cryptococcus spp. and Aspergillus spp. in PAP patients8.
PAP is classified in three clinical forms including congenital, secondary, and autoimmune, with the latter representing the predominant form comprising 90% of patients9. In autoimmune PAP (aPAP) GM-CSF-specific autoantibodies are elevated in the serum and bronchial-alveolar lavage fluid (BALF)10, 11. Moreover, transfer of GM-CSF-specific autoantibodies from human aPAP patients to healthy non-human primates recapitulates the disease, establishing a causal relationship12. aPAP patients treated with rituximab, a B cell depleting anti-CD20 monoclonal antibody, exhibit improvement in lipid homeostasis and overall outcome, illustrating that B cells and antibody are necessary for the pathology13-15. GM-CSF-specific autoantibodies in aPAP patients are polyclonal and somatically hypermutated IgG antibodies16, 17. Interestingly, GM-CSF-specific IgG autoantibodies are also detectable in healthy individuals, though titers are significantly lower compared to aPAP patients18. GM-CSF-specific autoantibodies from both healthy individuals and aPAP patients are comparable in regards to IgG subclass distribution18 raising a key question of how immunological tolerance is regulated in healthy and pathologic settings. However, identifying underlying immune mechanism(s) resulting in the production of pathological GM-CSF-specific autoantibodies is problematic due to a lack of appropriate animal models.
RasGRP1 is a guanine exchange factor expressed by B and T cells that activates Ras downstream of B cell and T cell antigen receptor stimulation19. Mice deficient in RasGRP1 are lymphopenic early in life with a block at the double positive stage in T cell development20. Later in life, Rasgrp1-deficient mice develop a lymphoproliferative disorder with multiple autoimmune features including enlarged secondary lymphoid tissues, spontaneous formation of germinal centers, and autoantibody production including anti-nuclear autoantibodies21. The autoimmune features in Rasgrp1-deficient mice are T cell-dependent since Rasgrp1-deficient mice crossed to athymic nude mice have attenuated splenomegaly and lack anti-nuclear antibodies22. In addition, studies utilizing Rasgrp1-deficient mice harboring an autoreactive B cell receptor transgene revealed that B cells in Rasgrp1-deficient mice also breach tolerance at multiple checkpoints, implicating both T cell-independent and –dependent mechanisms23.
In the current study, we show that autoimmune prone Rasgrp1-deficient mice develop PAP. The disease in Rasgrp1-deficient mice features characteristic accumulation of pulmonary surfactants in alveolar spaces, reduced pulmonary compliance, impaired alveolar macrophage function, and age-dependent increases in GM-CSF neutralizing autoantibodies in serum and BALF that correspond with disease onset. Lastly, aged mice lacking both RasGRP1 and CD275/ICOSL, a molecule necessary for conventional T follicular helper cell development and the production of T cell-dependent antibody, do not develop PAP. Therefore, from these data, we suggest that Rasgrp1-deficient mice represent the first rodent model for aPAP.
Materials and Methods
Mice
Mice were housed in an American Association for the Accreditation of Laboratory Animal Care (AAALAC)-certified pathogen-free facility at the University of South Alabama College of Medicine. Mice were more than 10 generations backcrossed on a C57BL/6 background. Both C57BL/6 mice and mice heterozygous for Rasgrp1 were used as controls as indicated. Importantly, PAP was not observed in either control group. Mice referred to as ‘young’ herein ranged from 2-3 months of age, while those referred to as ‘aged’ ranged from 7-12 months of age. All procedures involving mice were performed according to approved protocols by the University of South Alabama Institutional Animal Care and Use Committee.
Pulmonary Compliance
Pulmonary compliance was determined by removing the chest wall and injecting 2-20 ml/Kg of air at 50μl increments and recording pressure at each increment after a 2-3 second pause for the pressure to plateau. Compliance was calculated from the slope of the linear portion of the inflation curve.
Histology and Immunohistochemistry
Lungs of mice were inflation-fixed in situ with 10% phosphate buffered formalin at a pressure of 15 cmH2O, embedded in paraffin, sectioned and stained with hematoxylin and eosin (H&E) or periodic acid-Schiff (PAS). For quantification of proteinaceous material, non-overlapping lung sections imaged at low power (40X total) encompassing the entire lung section were analyzed using ImageJ software. Percent proteinaceous accumulation was defined as binary area of proteinaceous material divided by binary area of the total lung section analyzed. Total percent proteinaceous accumulation for each mouse was calculated using the mean of three lung sections including an upper, middle, and lower portion of the left lung lobe for each mouse.
To identify surfactant proteins (SP), sections were deparaffinized and immunostained with rabbit anti-mouse SP-A, SP-B (EMD Millipore Corporation, Temecula, CA), SP-C (Santa Cruz Biotechnology, Dallas, Texas) SP-D (Bioss, Inc., Woburn, MA), or an rabbit IgG isotope control (Cell Signaling Technology, Danvers, MA). Biotinylated horse anti-rabbit IgG (Vector Laboratories, Burlingame, CA) was used as a secondary antibody which was detected using HRP conjugated streptavidin (BD Biosciences, San Jose, CA) and using 3-amino-9-ethylcarbazole (Thermo-Fisher Scientific Waltham, MA) as substrate.
Bronchial-alveolar lavage (BAL), alveolar macrophage diameter, and surfactant degradation assay
BAL was collected in situ by washing 3 times with 0.8 mL of phosphate buffered saline (PBS, pH 7.4) i.t. at a pressure of 15 cmH2O. To determine alveolar macrophage diameter, BAL cells were cytocentrifuged and stained with PAS. Alveolar macrophage diameter was determined using ImageJ. To determine light scatter of alveolar macrophages, BAL cells were stained for flow cytometry using the following antibodies: BV510-conjugated anti-CD11c (N418, Biolegend, San Diego, CA), PE-conjugated anti-Siglec F (E50-2440, BD Biosciences, Franklin Lakes, NJ). For the surfactant degradation assay, BAL cells were evenly divided into 3 samples and incubated at 37°C 5% CO2 for 30 minutes and washed to remove non-adherent cells. A 1:1 mixture of Rhodamine-labeled 1,2-dipalmitoyl-sn-glycero-3-phosphoethanolamine (R-DPPE) (Avanti Polar Lipids, Inc., Alabaster, AL) and bovine surfactant (Infasurf, Forest Pharmaceuticals, St. Louis, MO), or surfactant mixed with a vehicle control was added to the cells and incubated for 30 mins at 37°C 5% CO2. Surfactant uptake was determined after 30 minutes by washing cells three times to remove excess surfactant, trypsinizing, and measuring rhodamine fluorescence by flow cytometry. After an additional 4 hours, a separate sample was measured to detect degradation. The degradation of labeled surfactant was quantified using the following equation: (30 min MFI - 4 hr MFI) / (30 min MFI - control MFI).
Real Time Quantitative PCR (RT-qPCR)
mRNA was prepared from lungs or alveolar macrophages using the RNeasy mini kit (Qiagen, Valencia, CA) and reverse transcribed to cDNA using an iScript cDNA preparation kit (Bio-Rad Laboratories, Hercules, CA). Quantitative PCR was performed by SYBR Green incorporation using a CFX96 (Bio-Rad) thermocycler with the following primers: β2-microglobulin (5’-CCCGCCTCACATTGAAATCC/ GCGTATGTATCAGTCTCAGTGG-3’) SP-A (5’- GTGCACCTGGAGAACATGGA/ TGGATCCTTGCAAGCTGAGG-3’), SP-B (5’- CCTCACAAAGATGACCAAGGA/ CTGGCTCTGGAAGTAGTCAATAA-3’), SP-C (5’- AGAGGTCCTGATGGAGAGTCC/ CATGAGCAGAGCCCCTACAA-3’), SP-D (5’- GGGCCAACAAGGAAGCAATC/ GGCCATTCTCTGTTGGGCTA-3’), PU.1 (5’-CCATCGGATGACTTGGTTACTT/ GTTCTCAAACTCGTTGTTGTGG-3’), PPARγ (5’-GCCCTTTACCACAGTTGATTTC/ GTGGAGATGCAGGTTCTACTT-3’), ABCG1 (5’CAGACGAGAGATGGTCAAAGAG/ CTGGTGGGCTCATCAAAGAA-3’).
GM-CSF-specific autoantibody ELISA
ELISA plates were coated with 1 µg/ml mouse recombinant GM-CSF (Shenandoah Biotechnology, Inc., Warwick, PA) overnight, and then blocked with StabilGuard (SurModics, Eden Prairie, MN) for 1 hour at room temperature. Biotinylated goat anti-mouse IgG Fc specific (Southern Biotechnology, Birmingham, AL) was used to detect GM-CSF autoantibodies in serial dilutions of serum followed by HRP conjugated streptavidin (BD Biosciences, San Jose, CA). Absorbance of serial dilutions was compared to a standard curve generated from non-specific IgG captured with a goat anti-mouse Ig antibody (Southern Biotechnology).
GM-CSF neutralization assay
Neutralization of GM-CSF was determined using the GM-CSF-dependent FDC-P1 cell line (American Type Culture Collection, Manassas, VA). IgG was isolated from pooled serum from 15 mice using protein G-coupled magnetic beads following manufacturer’s instructions (Promega, Madison, WI). Serially diluted serum IgG from mice was added to FDC-P1 cell cultures grown in the presence of 0.01 ng/ml GM-CSF and proliferation was measured after 3 days by addition of 10 µL of 5 mg/mL 3-(4, 5-dimethylthiazol-2-YL)-2, 5-diphenyltetrazolium bromide (MTT, Amresco, Solon, OH). Rat GM-CSF-specific IgG monoclonal antibody (MP1-31G6, Biolegend, San Diego, CA) was used a positive control. Absorbance was measured at 540 nm following addition of DMSO.
Statistics
Statistics were determined using GraphPad Prism 6 software. Kaplan-Meier survival curves were compared using a Mantel-Cox log rank test. Unpaired Student’s t test was used to compare two means with equal variances and Welch’s correction was applied when variances were unequal as determined by an F test. One-way ANOVA was used to compare more than two means with the exception of body mass data that was compared using a two-way ANOVA.
Results
Morbidity and mortality of aged Rasgrp1-deficient mice
Mice deficient in RasGRP1 are autoimmune-prone, with breaks in lymphocyte tolerance in both B and T cell compartments23, 24. To understand the effects of autoimmunity, mice were aged. As controls, Rasgrp1-sufficient littermate mice achieved an average body mass of 35.5 and 42.0 grams for females and males, respectively, and over 90% of the mice survived past one year of age (Figure 1a, b). By comparison, aged Rasgrp1-deficient mice exhibited significantly reduced body mass (25.1 and 30.5 grams for females and males, respectively), and survival decreased beginning at 21 weeks of age with only 38% of the mice surviving beyond one year of age. Aged Rasgrp1-deficient mice also exhibited labored breathing, prompting us to evaluate lung function. Pulmonary compliance of aged control and Rasgrp1-deficient mice was assessed by generating pressure volume curves. While aged control mice displayed pressure volume curves with typical hysteresis within the normal volume range for mice, aged Rasgrp1-deficient mice had comparatively right shifted pressure-volume curves characteristic of restrictive lung disease (Figure 1c). Calculation of pulmonary compliance showed that, relative to aged control mice, aged Rasgrp1-deficient mice had reduced compliance (Figure 1d; 0.032 ± 0.011, control vs.0.025 ± 0.004, Rasgrp1-deficient). Therefore, lung function was impaired in aged Rasgrp1-deficient mice.
Figure 1. Morbidity and mortality of aged Rasgrp1-deficient mice.

(a) Kaplan-Meier survival curves of control (n=12) and Rasgrp1-deficient (n=18) mice (p=0.005, Mantel-Cox log rank test). (b) Body mass of aged (7-12 months) female and male control and Rasgrp1-deficient mice. Each symbol represents the value of an individual mouse (***p=0.0002, two-way ANOVA with Sidak's multiple comparisons test). (c) Mean pulmonary pressure-volume curve from aged control (n=12) and Rasgrp1-deficient (n=11) mice. (d) Pulmonary compliance of aged control and Rasgrp1-deficient mice. Pulmonary compliance was calculated from linear portion of the inflation pressure-volume curve. Error bars represent SD (*p=0.031, unpaired Student’s t test with Welch’s correction).
Rasgrp1-deficient mice exhibit PAP-like pathology
To identify the nature of restrictive lung disease in aged Rasgrp1-deficient mice, histological examination of lungs was performed. Formalin-fixed, paraffin-embedded sections of lungs from young (2-3 months) and aged (7-12 months) control mice (both wild-type and Rasgrp1 heterozygous) demonstrated normal histology, with open alveolar spaces and thin septal walls (Figure 2a). Lungs from young Rasgrp1-deficient mice also appeared normal. In contrast, analysis of lung sections from aged Rasgrp1-deficient mice indicated an eosinophilic proteinaceous accumulation in the alveolar space characteristic of PAP (Figure 2a). Perivascular and peribronchial mononuclear cell infiltration was also apparent in the lungs of aged Rasgrp1-deficient mice (data not shown). To assess the extent of pathology, the amount of proteinaceous accumulation in the lungs was quantified (described in Materials and Methods). While lung sections from control and young Rasgrp1-deficient mice had no measurable accumulation, analysis of lung sections from aged Rasgrp1-deficient mice with pathology showed variable accumulation ranging from 5.6 to 38.5% of the lung section (Figure 2b). Notably, 8 out of 11 aged Rasgrp1-deficient mice showed measureable protein accumulation by histology (Fig. 2b), suggesting that approximately 70 percent of aged Rasgrp1-deficient mice develop PAP-like disease. Moreover, because no pathology was observed in young Rasgrp1-deficient mice, development of PAP-like disease is age-dependent.
Figure 2. Rasgrp1-deficient mice exhibit PAP-like pathology.

(a) Formalin-fixed paraffin-embedded lung sections of control and Rasgrp1-deficient lungs stained with H&E. Upper panels represent young (2-3 months) mice and lower panels represent aged (7-12 months) mice (scale bars, 50µm). (b) Quantification of the accumulating proteinaceous material as a percent of total lung section area. Each symbol represents the mean percent accumulation of an individual mouse calculated from three lung sections including the upper, middle, and lower portion of the left lung (*p=0.0006, One-way ANOVA with Tukey’s multiple comparison test).
Periodic Acid Schiff (PAS)-positive staining of the accumulated material in the alveoli is a key feature in PAP. Consistent with PAP, the accumulated material in lung sections from aged Rasgrp1-deficient mice were PAS-positive (Figure 3a). Further, because impaired surfactant homeostasis in PAP patients leads to build-up of surfactants in the alveoli, lung sections were stained for surfactant protein SP-A, -B, -C, and –D by immunohistochemistry. Consistent with impaired surfactant homeostasis, the accumulated material in lung sections from aged Rasgrp1-deficient mice stained positively for all SPs (Figure 3b).
Figure 3. Impaired surfactant homeostasis in lungs of mice with PAP.

(a) Formalin-fixed paraffin-embedded lung sections of aged control and Rasgrp1-deficient mice stained with PAS. (b) Immunohistochemical examination of lung sections from aged control and Rasgrp1-deficient mice using antibodies to SP-A, B, C, and D and counterstained with hematoxylin (scale bars, 50µm). (c) mRNA expression of SP genes from aged control and Rasgrp1-deficient mouse lungs was measured by quantitative RT-PCR. Values represent mean of duplicate wells expressed relative to β2-microglobulin [n = 9 (control) and 8 (Rasgrp1-deficient); error bars represent standard deviation, ns= not significant, unpaired Student’s t test].
Accumulation of pulmonary surfactants in PAP is reportedly caused by impaired degradation rather than increased SP expression5, 25. To investigate whether increased SP expression contributes to accumulation of surfactants in aged Rasgrp1-deficient lungs, SP gene expression was measured using quantitative RT-PCR. No measurable differences in control and Rasgrp1-deficient mice in SP-A, B C or D mRNA were observed, suggesting that the accumulation of pulmonary surfactants is not due to increased gene expression (Figure 3c). Taken together with histological analysis of lung sections, these data support that Rasgrp1-deficient mice develop PAP in an age-dependent manner.
Surfactant degradation by alveolar macrophages in Rasgrp1-deficient mice is impaired
An important function of alveolar macrophages is to maintain surfactant homeostasis. During PAP, alveolar macrophage function is impaired, rendering them unable to degrade pulmonary surfactants6. Consistent with impaired function, histological analysis of lung sections from aged Rasgrp1-deficient mice revealed enlarged PAS-positive alveolar macrophages in the alveoli of Rasgrp1-deficient mice in comparison with control mice (Figure 3a). To quantify alveolar macrophage size, BAL cells from aged control and Rasgrp1-deficient mice were isolated and cell diameters were measured. While the mean diameter of alveolar macrophages from littermate control mice was approximately 14 µm, those from Rasgrp1-deficient mice had a significantly larger mean diameter (Figure 4a, b; 13.9 ± 0.94 μm, control vs. 17.2 ± 2.25 µm, Rasgrp1-deficient). Moreover, alveolar macrophages from Rasgrp1-deficient mice were larger and more granular than those from control mice using light scatter by flow cytometry (Figure 4c). Therefore, alveolar macrophages from aged Rasgrp1-deficient mice are enlarged, consistent with an inability to degrade surfactants.
Figure 4. Impaired surfactant degradation by alveolar macrophages from Rasgrp1-deficient mice.

(a) Representative image of cytocentrifuged BAL cells stained with H&E from aged control and Rasgrp1-deficient mice (scale bars, 50µm). (b) Diameter of alveolar macrophages quantified from aged control and Rasgrp1-deficient mice. Each symbol represents the mean diameter for alveolar macrophages from an individual mouse (**p=0.004, unpaired Student’s t test with Welch’s correction). (c) Representative FSC vs SSC flow cytometry plots of alveolar macrophages from aged control (top panel) and Rasgrp1-deficient (bottom panel) mice. Alveolar macrophages were identified as CD11c and SiglecF double-positive cells. (d) Representative flow cytometry histograms of surfactant degradation by alveolar macrophages from control and Rasgrp1-deficient mice. Open histogram (dashed line) indicates unstained control; open histogram (solid line) indicates labeled surfactant uptake at 30 minutes; filled histogram represents labeled surfactant at 4 hours post uptake. (e) R-DPPE labeled surfactant degradation for individual mice was quantified as an index of degradation where 1 is complete degradation of labeled surfactant and 0 is no degradation of labeled surfactant (ns = not significant, ****p<0.0001, One way ANOVA with Tukey’s multiple comparison test).
To measure directly whether alveolar macrophages from mice with PAP have impaired surfactant degradation, BAL cells were incubated with rhodamine-labeled surfactant. At indicated times, the amount of rhodamine label in alveolar macrophages was measured by flow cytometry (Figure 4d). Surfactant degradation was quantified as the percent reduction in rhodamine signal intensity after normalizing to unstained cells (Figure 4e). As expected, alveolar macrophages from all mice showed maximal uptake of surfactant after incubating for 30 minutes, consistent with previous reports26. At 4 hours incubation, alveolar macrophages from all control mice had reduced rhodamine fluorescence, indicating that they had degraded the labeled surfactants. By comparison, alveolar macrophages from aged Rasgrp1-deficient mice, while showing surfactant uptake comparable to control cells, were less able to degrade surfactant. Importantly, alveolar macrophages from young Rasgrp1-deficient mice were able to both take up and degrade surfactants similar to alveolar macrophages from control mice. These data demonstrate that alveolar macrophage function is impaired in Rasgrp1-deficient mice with PAP. Moreover, surfactant degradation by alveolar macrophages from young Rasgrp1-deficient mice is not impaired, indicating alveolar macrophage dysfunction is progressive in Rasgrp1-deficient mice.
Rasgrp1-deficient mice have an increase in GM-CSF-neutralizing IgG autoantibodies
Rasgrp1-deficient mice develop germinal centers that contain autoreactive B cells, and have increased levels of serum anti-nuclear autoantibodies beginning at 2-3 months of age23. In aPAP patients, GM-CSF-specific IgG autoantibodies are elevated in serum and in BALF, and these antibodies neutralize GM-CSF10. To determine whether GM-CSF-specific autoantibodies were linked to development of PAP in Rasgrp1-deficient mice, the concentration of GM-CSF-specific IgG in serum and bronchial alveolar lavage fluid (BALF) was measured using ELISA. Aged control mice had a mean serum GM-CSF-specific IgG concentration of approximately 10 ng/ml (Figure 5a, 10.4 ± 4.9). In contrast, aged-matched Rasgrp1-deficient mice had a mean serum GM-CSF-specific IgG concentration of 32.5 ng/ml, representing a 3-fold increase relative to control mice. Consistent with the lack of disease in younger Rasgrp1-deficient mice, 2-3 month old Rasgrp1-deficient mice exhibited GM-CSF-specific IgG concentration comparable to control mice (Figure 5c, 3.5 ± 2.6). The concentration of GM-CSF-specific IgG in BALF from aged Rasgrp1-deficient mice was also increased in comparison to levels measured from control mice (Figure 5b). Therefore, GM-CSF-specific IgG autoantibodies are elevated in Rasgrp1-deficient mice in an age-dependent manner.
Figure 5. Elevated GM-CSF-neutralizing autoantibodies in aged Rasgrp1-deficient mice.

Concentration of GM-CSF-specific IgG in the serum (a) and BALF (b) of aged control and Rasgrp1-deficient mice measured by ELISA. Symbols represent the mean concentration for individual mice (*p=0.02,***p=0.001, unpaired Student’s t test with Welch’s correction). (c) Concentration of GM-CSF-specific IgG in serum from young Rasgrp1-deficient mice compared to the concentration from aged Rasgrp1-deficient mice in (a) (****p<0.0001, unpaired Student’s t test with Welch’s correction). (d) Inhibition of FDC-P1 cell growth by pooled IgG from aged Rasgrp1-deficient mice. Lines represent the OD450 of FDC-P1 cells grown in the absence of Csf-2 (dashed) or IgG (dotted) in the cell media. Symbols represent the mean values from duplicate wells. (representative of three independent experiments; error bars represent SEM).
To test whether GM-CSF-specific autoantibodies from Rasgrp1-deficient mice have the capacity to neutralize GM-CSF, isolated serum IgG was tested for the ability to block proliferation of GM-CSF-dependent FDC-P1 cells (Figure 5d). As a positive control, a monoclonal rat anti-mouse GM-CSF IgG prevented growth of FDC-P1 cells. Importantly, IgG from wild-type mice failed to inhibit GM-CSF-dependent growth. By comparison serum IgG isolated from aged Rasgrp1-deficient mice completely inhibited GM-CSF-dependent growth. Therefore, aged Rasgrp1-deficient mice have elevated GM-CSF-neutralizing serum IgG, suggesting that progressive development of PAP in Rasgrp1-deficient mice is autoimmune.
Alveolar macrophages from Rasgrp1-deficient mice show characteristic GM-CSF depletion
In the absence of GM-CSF, alveolar macrophages have reduced expression of transcription factors PU.16, 27 and PPARγ28, as well as the ABCG1 transporter29. Accordingly, if GM-CSF is neutralized by autoantibody in Rasgrp1-deficient mice, than PU.1, PPARγ, and ABCG1 mRNA levels would be reduced. To test the bioactivity of GM-CSF in Rasgrp1-deficient mice, the expression of PU.1, PPARγ, and ABCG1 in alveolar macrophages (GR1−, CD11c+ SiglecF+) sorted from the lungs of aged control and Rasgrp1-deficient mice were measured using quantitative RT-PCR (Figure 6). Consistent with aPAP patients, the relative expression of PU.1, PPARγ, and ABCG1 was reduced in alveolar macrophages from aged Rasgrp1-deficient mice compared to aged control mice. Collectively, these data suggest that bioactivity of GM-CSF is neutralized in Rasgrp1-deficient mice.
Figure 6. Decreased levels of PU.1, PPARγ, and ABCG1 mRNA in AM from Rasgrp1-deficient mice.
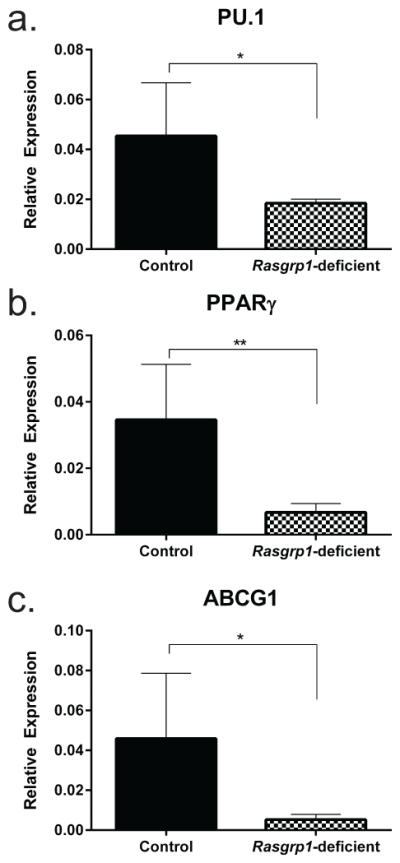
mRNA expression of (a) PU.1, (b) PPARγ, and (c) ABCG1 in alveolar macrophages from aged control and Rasgrp1-deficient mice was measured by quantitative RT-PCR. Values represent mean of duplicate wells expressed relative to β2-microglobulin [n=6 (control) and 4 (Rasgrp1-deficient); error bars represent standard deviation (*p=0.01, **p=0.004, unpaired Student’s t test with Welch’s correction)].
Rasgrp1-deficient mice also deficient in CD275 have reduced GM-CSF-specific autoantibody and do not develop PAP
CD278/ICOS-CD275/ICOSL interactions are necessary for conventional T cell-dependent humoral responses30-32. GM-CSF autoantibodies are IgG, and therefore likely to be dependent on T cell help. To test whether disrupting B cell-T cell interactions via CD278-CD275 is required for the development of aPAP and GM-CSF autoantibodies, Rasgrp1-deficient mice were crossed to mice deficient in CD275. As before, histological lung sections from aged Rasgrp1-deficient mice (n=4) revealed lesions characteristic of PAP while those from aged control mice (n=3) showed normal airways. Interestingly, 9 out of 11 aged mice lacking both RasGRP1 and CD275 (Rasgrp1−/− CD275−/−) had normal lung histology and therefore showed no signs of PAP (Figure 7a). Though lung sections from two aged Rasgrp1−/− CD275−/− mice exhibited enlarged alveolar macrophages, there was little evidence of surfactant accumulation compared to the pathology observed in aged Rasgrp1−/− mice (data not shown). To determine whether the absence of PAP in Rasgrp1−/− CD275−/− mice was due to a reduced autoimmunity, the concentration of serum GM-CSF-specific autoantibodies were measured. Similar to earlier data (Figure 5), the concentration of serum GM-CSF-specific autoantibodies from Rasgrp1-deficient mice was approximately 40 ng/ml. In contrast, the concentration of serum GM-CSF-specific autoantibodies from Rasgrp1−/− CD275−/− mice were reduced and comparable to those observed in control mice (Figure 7b). Therefore, disruption of CD278-CD275 interactions abrogated the development of aPAP.
Figure 7. Rasgrp1-deficient mice also lacking CD275/ICOSL do not develop PAP and have normal GM-CSF-specific autoantibody levels.

(a) Representative formalin-fixed paraffin-embedded lung sections from control (upper right, n=3), Rasgrp1-deficient (upper left, n=4), CD275-deficient (lower left, n=3), and Rasgrp1−/− CD275−/− (lower right, n=11) mice stained with H&E (scale bars, 50µm). (b) Concentrations of GM-CSF-specific IgG in the serum of aged control, Rasgrp1-deficient, CD275-deficient, and Rasgrp1−/− CD275−/− mice measured by ELISA. Symbols represent the mean concentration for individual mice (**p≤0.005, One way ANOVA with Tukey’s multiple comparison test).
Discussion
aPAP is a rare autoimmune disorder caused by neutralizing autoantibodies to GM-CSF. Currently, there are no identified animal models for aPAP that spontaneously produce GM-CSF-specific autoantibodies. Herein, we show that aged Rasgrp1-deficient mice have an increase in morbidity and mortality associated with reduced pulmonary compliance. The histopathology in lungs of aged Rasgrp1-deficient mice is indistinguishable from human PAP with eosinophilic, PAS- and SP-positive material within the alveoli of the lungs. Rasgrp1-deficient mice also have enlarged, foam-like alveolar macrophages that are impaired at degrading surfactants. Lastly, Rasgrp1-deficient mice exhibit an age- and CD275-dependent increase in GM-CSF-specific autoantibodies that are capable of neutralizing GM-CSF bioactivity. Collectively, the current studies strongly support that Rasgpr1-deficient mice represent the first rodent model for aPAP.
GM-CSF-specific autoantibody is elevated concomitant with disease onset, and IgG from aged Rasgrp1-deficient mice neutralizes GM-CSF suggesting that antibody-mediated neutralization of GM-CSF is the mechanism of PAP in Rasgrp1-deficient mice. Moreover, the onset of pathology in Rasgrp1-deficient mice presents at 6-8 months of age which compares to aPAP patients that typically present from ages 30-46 years of age8. This contrasts with GM-CSF- and GM-CSF receptor (GM-CSFR)-deficient mice1, 2, 4 that develop disease as early as 2 months of age. Similarly, congenital disease in humans resulting from GM-CSFR mutations, also presents early in life33-35. Importantly, Rasgrp1-deficient mice do not show signs of disease early in life and therefore we consider it highly unlikely that PAP in aged Rasgrp1-deficient mice is caused by congenital deficiency. Thus, the kinetics of disease in Rasgrp1-deficient mice is most consistent with aPAP rather than congenital disease.
It should be noted that the concentration of GM-CSF-specific autoantibodies observed in Rasgrp1-deficient mice is lower than those established in human aPAP patients18, 36, 37. This difference may be due to assay conditions used to quantify GM-CSF-specific antibodies in mouse. In our study, the concentration of GM-CSF-specific antibodies was determined by comparison to a standard curve generated using total immunoglobulin capture antibody (i.e. not GM-CSF-specific capture). Therefore, the concentrations reported herein are relative to detection of total, non-specific IgG rather than GM-CSF-specific IgG.
Reduced mRNA levels of GM-CSF-dependent genes is consistent with GM-CSF-neutralization as the cause of PAP in Rasgrp1-deficient mice. Alveolar macrophages from aPAP patients and GM-CSF-deficient mice have markedly reduced levels of ABC transporter ABCG129, a molecule involved in lipid transport38-40. The reduction of ABCG1 is attributed to lower levels of PPARγ as a consequence of GM-CSF deficiency28, 41. Interestingly, mice deficient in ABCG1 display increased surfactants in the alveoli later in life, however these mice also have elevated SP-D and reduced SP-B and –C mRNA expression consistent with disorders of surfactant production42, 43. Disorders of surfactant production are considered distinct from PAP, which is defined by an inability to degrade surfactants44. Notably, while alveolar macrophages from aged Rasgrp1-deficient mice have a reduction in ABCG1 expression, they also have a reduction in PPARγ mRNA. Further, expression of SPs in the lung is normal. Therefore, gene expression in alveolar macrophages from aged Rasgrp1-deficient mice are most consistent with GM-CSF neutralization and aPAP rather than with disorders of surfactant production. Neutralization of GM-CSF in aPAP also causes reduced levels of PU.1, a transcription factor regulated by GM-CSF6, 12, 27. Enforced expression of PU.1 in alveolar macrophages from GM-CSF-deficient mice restores cell size, adherence, and surfactant degradation6, demonstrating that PU.1 is sufficient to recoup alveolar macrophage function. Notably, alveolar macrophages from aged Rasgrp1-deficient mice have reduced PU.1 mRNA, again implicating GM-CSF neutralization as a cause of pathology in aged Rasgrp1-deficient mice.
GM-CSF deficiency or neutralization results in an increased susceptibility to pulmonary pathogens8 including many fungal pathogens such as Cryptococcus neoformans45, Pneumocystis carinii46, and Histoplasma capsulatum47. Consistent with autoantibody-mediated neutralization of GM-CSF, lung sections from aged Rasgrp1-deficient mice display positive staining for Grocott’s methenamine silver (GMS) (data not shown). While PAP can develop secondary to infection, particularly Pneumocystis spp. infection, secondary PAP remains unlikely because 1) aged Rasgrp1-deficient mice with PAP and littermate controls tested negative in a serological test for Pneumocystis spp. (data not shown); 2) aged Rasgrp1-deficient mice that do not develop PAP pathology are frequently caged with aged Rasgrp1-deficient mice that develop PAP pathology; and 3) SP-B levels are decreased in a mouse model of secondary PAP48 whereas SP-B expression is unchanged in aged Rasgrp1-deficient mice in comparison to control mice. It is more likely that autoantibody neutralization of GM-CSF contributes to impaired clearance by alveolar macrophages6 and/or inhibition of pulmonary macrophage and dendritic cell maturation7, 49. Future studies can elucidate whether fungal infections in aged Rasgrp1-deficient mice contribute to mortality since GM-CSF-deficient mice exhibit a normal lifespan1.
CD278/ICOS-CD275/ICOSL interactions are required for humoral responses to T cell-dependent antigens30. Our current study demonstrates that aged Rasgrp1-deficient mice also lacking CD275 produce normal amounts of GM-CSF-specific IgG antibodies and do not develop PAP. Thus, these data support that PAP in Rasgrp1-deficient mice is autoantibody-dependent. In MRL/lpr50, collagen-induced arthritis51, and diabetes/NOD mouse models52 disruption of CD278-CD275 interactions mitigates severity of disease. CD278-CD275 interactions are considered key for the generation of T follicular helper cells (Tfh)53, 54. In recent work, we demonstrated that Rasgrp1-deficient mice have increased frequency and number of IL-17A-producing Tfh cells55. Interestingly, Rasgrp1-deficient mice also lacking CD275 still produced Tfh17 cells, formed germinal centers, and produced similar titers of anti-nuclear autoantibodies compared with mice lacking only RasGRP1. However, while Tfh17 cells from Rasgrp1-deficient mice also produced IL-21, Tfh17 cells from Rasgrp1−/− CD275−/− mice did not. Because GM-CSF-specific autoantibodies in mice lacking both RasGRP1 and CD275 are reduced to levels comparable to control mice, we speculate that IL-21 production by Tfh17 cells is critical for GM-CSF-specific but not anti-nuclear autoantibodies. Formal testing of this hypothesis may be significant for human disease as well. To-date, no studies have examined the importance of Tfh cells, CD275-CD278 interactions, and/or IL-21 in human disease, though GM-CSF-specific autoantibodies from aPAP patients are heavily somatically hypermutated16 suggesting a role for Tfh cells.
Mutations in BCR and TCR signaling pathways can facilitate breaks in B and T cell tolerance. For example, Rasgrp1-deficiency in mice leads to a break in tolerance in B cells early in development, at the transitional stage, and in the germinal center23. These studies utilized an immunoglobulin knock-in transgene with broad specificity to ssRNA and nuclear antigens. It is unknown whether GM-CSF-specific B cells are regulated similarly. Notably, anti-nuclear and transgene-derived autoantibodies are elevated at 2-3 months of age in Rasgrp1-deficient mice, an age at which the concentration of GM-CSF-specific autoantibodies is similar to those observed in control mice. As discussed above, elevation of GM-CSF-specific autoantibodies is not evident until 6-8 months of age. Whether these age-dependent changes in anti-GM-CSF antibody levels is due to escape from discrete tolerance checkpoints is unknown. To our knowledge, there are no studies examining whether BCR and TCR signaling mutations or whether specific tolerance checkpoints associate with aPAP in humans.
In conclusion, our data strongly suggest that Rasgrp1-deficient mice are the first rodent model for aPAP. Thus, Rasgrp1-deficient mice develop age-dependent PAP-like pathology in the lung that corresponds with an age-dependent increase in GM-CSF-specific neutralizing autoantibodies. This novel aPAP model may reveal important underlying immune mechanisms and identify new therapeutic targets for human aPAP.
Acknowledgements
We thank Kristin O’Donnell for assistance with measuring pulmonary compliance, and also Audrey Vasauskas for assistance with surfactant staining.
This work was supported by the American Lung Society Biomedical Research Grant (R.A.B.) as well as start-up funds provided to R.A.B. by the University of South Alabama College of Medicine. A.F. is supported by National Institutes of Health (NIH) training grant T32-HL076125.
References
- 1.Dranoff G, Crawford AD, Sadelain M, Ream B, Rashid A, Bronson RT, Dickersin GR, Bachurski CJ, Mark EL, Whitsett JA, Mulligan RC. Involvement of granulocyte-macrophage colony-stimulating factor in pulmonary homeostasis. Science. 1994;264:713–716. doi: 10.1126/science.8171324. [DOI] [PubMed] [Google Scholar]
- 2.Stanley E, Lieschke GJ, Grail D, Metcalf D, Hodgson G, Gall JA, Maher DW, Cebon J, Sinickas V, Dunn AR. Granulocyte/macrophage colony-stimulating factor-deficient mice show no major perturbation of hematopoiesis but develop a characteristic pulmonary pathology. Proc Natl Acad Sci U S A. 1994;91:5592–5596. doi: 10.1073/pnas.91.12.5592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Robb L, Drinkwater CC, Metcalf D, Li R, Kontgen F, Nicola NA, Begley CG. Hematopoietic and lung abnormalities in mice with a null mutation of the common beta subunit of the receptors for granulocyte-macrophage colony-stimulating factor and interleukins 3 and 5. Proc Natl Acad Sci U S A. 1995;92:9565–9569. doi: 10.1073/pnas.92.21.9565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nishinakamura R, Nakayama N, Hirabayashi Y, Inoue T, Aud D, McNeil T, Azuma S, Yoshida S, Toyoda Y, Arai K, Murray R, Miyajima A. Mice deficient for the IL-3/GM-CSF/IL-5 beta c receptor exhibit lung pathology and impaired immune response, while beta IL3 receptor-deficient mice are normal. Immunity. 1995;2:211–222. doi: 10.1016/1074-7613(95)90046-2. [DOI] [PubMed] [Google Scholar]
- 5.Huffman JA, Hull WM, Dranoff G, Mulligan RC, Whitsett JA. Pulmonary epithelial cell expression of GM-CSF corrects the alveolar proteinosis in GM-CSF-deficient mice. J Clin Invest. 1996;97:649–655. doi: 10.1172/JCI118461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shibata Y, Berclaz PY, Chroneos ZC, Yoshida M, Whitsett JA, Trapnell BC. GM-CSF regulates alveolar macrophage differentiation and innate immunity in the lung through PU.1. Immunity. 2001;15:557–567. doi: 10.1016/s1074-7613(01)00218-7. [DOI] [PubMed] [Google Scholar]
- 7.Hashimoto D, Chow A, Noizat C, Teo P, Beasley MB, Leboeuf M, Becker CD, See P, Price J, Lucas D, Greter M, Mortha A, Boyer SW, Forsberg EC, Tanaka M, van Rooijen N, Garcia-Sastre A, Stanley ER, Ginhoux F, Frenette PS, Merad M. Tissue-resident macrophages self-maintain locally throughout adult life with minimal contribution from circulating monocytes. Immunity. 2013;38:792–804. doi: 10.1016/j.immuni.2013.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Seymour JF, Presneill JJ. Pulmonary alveolar proteinosis: progress in the first 44 years. Am J Respir Crit Care Med. 2002;166:215–235. doi: 10.1164/rccm.2109105. [DOI] [PubMed] [Google Scholar]
- 9.Trapnell BC, Whitsett JA, Nakata K. Pulmonary alveolar proteinosis. N Engl J Med. 2003;349:2527–2539. doi: 10.1056/NEJMra023226. [DOI] [PubMed] [Google Scholar]
- 10.Kitamura T, Tanaka N, Watanabe J, Uchida, Kanegasaki S, Yamada Y, Nakata K. Idiopathic pulmonary alveolar proteinosis as an autoimmune disease with neutralizing antibody against granulocyte/macrophage colony-stimulating factor. J Exp Med. 1999;190:875–880. doi: 10.1084/jem.190.6.875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Uchida K, Nakata K, Trapnell BC, Terakawa T, Hamano E, Mikami A, Matsushita I, Seymour JF, Oh-Eda M, Ishige I, Eishi Y, Kitamura T, Yamada Y, Hanaoka K, Keicho N. High-affinity autoantibodies specifically eliminate granulocyte-macrophage colony-stimulating factor activity in the lungs of patients with idiopathic pulmonary alveolar proteinosis. Blood. 2004;103:1089–1098. doi: 10.1182/blood-2003-05-1565. [DOI] [PubMed] [Google Scholar]
- 12.Sakagami T, Beck D, Uchida K, Suzuki T, Carey BC, Nakata K, Keller G, Wood RE, Wert SE, Ikegami M, Whitsett JA, Luisetti M, Davies S, Krischer JP, Brody A, Ryckman F, Trapnell BC. Patient-derived granulocyte/macrophage colony-stimulating factor autoantibodies reproduce pulmonary alveolar proteinosis in nonhuman primates. Am J Respir Crit Care Med. 2010;182:49–61. doi: 10.1164/rccm.201001-0008OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Malur A, Kavuru MS, Marshall I, Barna BP, Huizar I, Karnekar R, Thomassen MJ. Rituximab therapy in pulmonary alveolar proteinosis improves alveolar macrophage lipid homeostasis. Respir Res. 2012;13:46. doi: 10.1186/1465-9921-13-46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kavuru MS, Malur A, Marshall I, Barna BP, Meziane M, Huizar I, Dalrymple H, Karnekar R, Thomassen MJ. An open-label trial of rituximab therapy in pulmonary alveolar proteinosis. Eur Respir J. 2011;38:1361–1367. doi: 10.1183/09031936.00197710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Amital A, Dux S, Shitrit D, Shpilberg O, Kramer MR. Therapeutic effectiveness of rituximab in a patient with unresponsive autoimmune pulmonary alveolar proteinosis. Thorax. 2010;65:1025–1026. doi: 10.1136/thx.2010.140673. [DOI] [PubMed] [Google Scholar]
- 16.Wang Y, Thomson CA, Allan LL, Jackson LM, Olson M, Hercus TR, Nero TL, Turner A, Parker MW, Lopez AL, Waddell TK, Anderson GP, Hamilton JA, Schrader JW. Characterization of pathogenic human monoclonal autoantibodies against GM-CSF. Proc Natl Acad Sci U S A. 2013;110:7832–7837. doi: 10.1073/pnas.1216011110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Piccoli L, Campo I, Fregni CS, Rodriguez BM, Minola A, Sallusto F, Luisetti M, Corti D, Lanzavecchia A. Neutralization and clearance of GM-CSF by autoantibodies in pulmonary alveolar proteinosis. Nature communications. 2015;6:7375. doi: 10.1038/ncomms8375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Uchida K, Nakata K, Suzuki T, Luisetti M, Watanabe M, Koch DE, Stevens CA, Beck DC, Denson LA, Carey BC, Keicho N, Krischer JP, Yamada Y, Trapnell BC. Granulocyte/macrophage-colony-stimulating factor autoantibodies and myeloid cell immune functions in healthy subjects. Blood. 2009;113:2547–2556. doi: 10.1182/blood-2009-05-155689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ebinu JO, Stang SL, Teixeira C, Bottorff DA, Hooton J, Blumberg PM, Barry M, Bleakley RC, Ostergaard HL, Stone JC. RasGRP links T-cell receptor signaling to Ras. Blood. 2000;95:3199–3203. [PubMed] [Google Scholar]
- 20.Dower NA, Stang SL, Bottorff DA, Ebinu JO, Dickie P, Ostergaard HL, Stone JC. RasGRP is essential for mouse thymocyte differentiation and TCR signaling. Nat Immunol. 2000;1:317–321. doi: 10.1038/79766. [DOI] [PubMed] [Google Scholar]
- 21.Coughlin JJ, Stang SL, Dower NA, Stone JC. RasGRP1 and RasGRP3 regulate B cell proliferation by facilitating B cell receptor-Ras signaling. J Immunol. 2005;175:7179–7184. doi: 10.4049/jimmunol.175.11.7179. [DOI] [PubMed] [Google Scholar]
- 22.Coughlin JJ, Stang SL, Dower NA, Stone JC. The role of RasGRPs in regulation of lymphocyte proliferation. Immunol Lett. 2006;105:77–82. doi: 10.1016/j.imlet.2006.01.005. [DOI] [PubMed] [Google Scholar]
- 23.Bartlett A, Buhlmann JE, Stone J, Lim B, Barrington RA. Multiple checkpoint breach of B cell tolerance in Rasgrp1-deficient mice. J Immunol. 2013;191:3605–3613. doi: 10.4049/jimmunol.1202892. [DOI] [PubMed] [Google Scholar]
- 24.Priatel JJ, Teh SJ, Dower NA, Stone JC, Teh HS. RasGRP1 transduces low-grade TCR signals which are critical for T cell development, homeostasis, and differentiation. Immunity. 2002;17:617–627. doi: 10.1016/s1074-7613(02)00451-x. [DOI] [PubMed] [Google Scholar]
- 25.Brasch F, Birzele J, Ochs M, Guttentag SH, Schoch OD, Boehler A, Beers MF, Muller KM, Hawgood S, Johnen G. Surfactant proteins in pulmonary alveolar proteinosis in adults. Eur Respir J. 2004;24:426–435. doi: 10.1183/09031936.04.00076403. [DOI] [PubMed] [Google Scholar]
- 26.Yoshida M, Ikegami M, Reed JA, Chroneos ZC, Whitsett JA. GM-CSF regulates protein and lipid catabolism by alveolar macrophages. Am J Physiol Lung Cell Mol Physiol. 2001;280:L379–386. doi: 10.1152/ajplung.2001.280.3.L379. [DOI] [PubMed] [Google Scholar]
- 27.Bonfield TL, Raychaudhuri B, Malur A, Abraham S, Trapnell BC, Kavuru MS, Thomassen MJ. PU.1 regulation of human alveolar macrophage differentiation requires granulocyte-macrophage colony-stimulating factor. Am J Physiol Lung Cell Mol Physiol. 2003;285:L1132–1136. doi: 10.1152/ajplung.00216.2003. [DOI] [PubMed] [Google Scholar]
- 28.Bonfield TL, Farver CF, Barna BP, Malur A, Abraham S, Raychaudhuri B, Kavuru MS, Thomassen MJ. Peroxisome proliferator-activated receptor-gamma is deficient in alveolar macrophages from patients with alveolar proteinosis. Am J Respir Cell Mol Biol. 2003;29:677–682. doi: 10.1165/rcmb.2003-0148OC. [DOI] [PubMed] [Google Scholar]
- 29.Thomassen MJ, Barna BP, Malur AG, Bonfield TL, Farver CF, Malur A, Dalrymple H, Kavuru MS, Febbraio M. ABCG1 is deficient in alveolar macrophages of GM-CSF knockout mice and patients with pulmonary alveolar proteinosis. J Lipid Res. 2007;48:2762–2768. doi: 10.1194/jlr.P700022-JLR200. [DOI] [PubMed] [Google Scholar]
- 30.Tafuri A, Shahinian A, Bladt F, Yoshinaga SK, Jordana M, Wakeham A, Boucher LM, Bouchard D, Chan VS, Duncan G, Odermatt B, Ho A, Itie A, Horan T, Whoriskey JS, Pawson T, Penninger JM, Ohashi PS, Mak TW. ICOS is essential for effective T-helper-cell responses. Nature. 2001;409:105–109. doi: 10.1038/35051113. [DOI] [PubMed] [Google Scholar]
- 31.Warnatz K, Bossaller L, Salzer U, Skrabl-Baumgartner A, Schwinger W, van der Burg M, van Dongen JJ, Orlowska-Volk M, Knoth R, Durandy A, Draeger R, Schlesier M, Peter HH, Grimbacher B. Human ICOS deficiency abrogates the germinal center reaction and provides a monogenic model for common variable immunodeficiency. Blood. 2006;107:3045–3052. doi: 10.1182/blood-2005-07-2955. [DOI] [PubMed] [Google Scholar]
- 32.Choi YS, Kageyama R, Eto D, Escobar TC, Johnston RJ, Monticelli L, Lao C, Crotty S. ICOS receptor instructs T follicular helper cell versus effector cell differentiation via induction of the transcriptional repressor Bcl6. Immunity. 2011;34:932–946. doi: 10.1016/j.immuni.2011.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Suzuki T, Sakagami T, Rubin BK, Nogee LM, Wood RE, Zimmerman SL, Smolarek T, Dishop MK, Wert SE, Whitsett JA, Grabowski G, Carey BC, Stevens C, van der Loo JC, Trapnell BC. Familial pulmonary alveolar proteinosis caused by mutations in CSF2RA. J Exp Med. 2008;205:2703–2710. doi: 10.1084/jem.20080990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Martinez-Moczygemba M, Doan ML, Elidemir O, Fan LL, Cheung SW, Lei JT, Moore JP, Tavana G, Lewis LR, Zhu Y, Muzny DM, Gibbs RA, Huston DP. Pulmonary alveolar proteinosis caused by deletion of the GM-CSFRalpha gene in the X chromosome pseudoautosomal region 1. J Exp Med. 2008;205:2711–2716. doi: 10.1084/jem.20080759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dirksen U, Nishinakamura R, Groneck P, Hattenhorst U, Nogee L, Murray R, Burdach S. Human pulmonary alveolar proteinosis associated with a defect in GM-CSF/IL-3/IL-5 receptor common beta chain expression. J Clin Invest. 1997;100:2211–2217. doi: 10.1172/JCI119758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nei T, Urano S, Motoi N, Takizawa J, Kaneko C, Kanazawa H, Tazawa R, Nakagaki K, Akagawa KS, Akasaka K, Ichiwata T, Azuma A, Nakata K. IgM-type GM-CSF autoantibody is etiologically a bystander but associated with IgG-type autoantibody production in autoimmune pulmonary alveolar proteinosis. Am J Physiol Lung Cell Mol Physiol. 2012;302:L959–964. doi: 10.1152/ajplung.00378.2011. [DOI] [PubMed] [Google Scholar]
- 37.Seymour JF, Doyle IR, Nakata K, Presneill JJ, Schoch OD, Hamano E, Uchida K, Fisher R, Dunn AR. Relationship of anti-GM-CSF antibody concentration, surfactant protein A and B levels, and serum LDH to pulmonary parameters and response to GM-CSF therapy in patients with idiopathic alveolar proteinosis. Thorax. 2003;58:252–257. doi: 10.1136/thorax.58.3.252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kobayashi A, Takanezawa Y, Hirata T, Shimizu Y, Misasa K, Kioka N, Arai H, Ueda K, Matsuo M. Efflux of sphingomyelin, cholesterol, and phosphatidylcholine by ABCG1. J Lipid Res. 2006;47:1791–1802. doi: 10.1194/jlr.M500546-JLR200. [DOI] [PubMed] [Google Scholar]
- 39.Malur A, Huizar I, Wells G, Barna BP, Malur AG, Thomassen MJ. Lentivirus-ABCG1 instillation reduces lipid accumulation and improves lung compliance in GM-CSF knock-out mice. Biochem Biophys Res Commun. 2011;415:288–293. doi: 10.1016/j.bbrc.2011.10.043. [DOI] [PubMed] [Google Scholar]
- 40.Baldan A, Tarr P, Lee R, Edwards PA. ATP-binding cassette transporter G1 and lipid homeostasis. Curr Opin Lipidol. 2006;17:227–232. doi: 10.1097/01.mol.0000226113.89812.bb. [DOI] [PubMed] [Google Scholar]
- 41.Malur A, Baker AD, McCoy AJ, Wells G, Barna BP, Kavuru MS, Malur AG, Thomassen MJ. Restoration of PPARgamma reverses lipid accumulation in alveolar macrophages of GM-CSF knockout mice. Am J Physiol Lung Cell Mol Physiol. 2011;300:L73–80. doi: 10.1152/ajplung.00128.2010. [DOI] [PubMed] [Google Scholar]
- 42.Out R, Hoekstra M, Hildebrand RB, Kruit JK, Meurs I, Li Z, Kuipers F, Van Berkel TJ, Van Eck M. Macrophage ABCG1 deletion disrupts lipid homeostasis in alveolar macrophages and moderately influences atherosclerotic lesion development in LDL receptor-deficient mice. Arterioscler Thromb Vasc Biol. 2006;26:2295–2300. doi: 10.1161/01.ATV.0000237629.29842.4c. [DOI] [PubMed] [Google Scholar]
- 43.Baldan A, Tarr P, Vales CS, Frank J, Shimotake TK, Hawgood S, Edwards PA. Deletion of the transmembrane transporter ABCG1 results in progressive pulmonary lipidosis. J Biol Chem. 2006;281:29401–29410. doi: 10.1074/jbc.M606597200. [DOI] [PubMed] [Google Scholar]
- 44.Carey B, Trapnell BC. The molecular basis of pulmonary alveolar proteinosis. Clin Immunol. 2010;135:223–235. doi: 10.1016/j.clim.2010.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chen GH, Olszewski MA, McDonald RA, Wells JC, Paine R, 3rd, Huffnagle GB, Toews GB. Role of granulocyte macrophage colony-stimulating factor in host defense against pulmonary Cryptococcus neoformans infection during murine allergic bronchopulmonary mycosis. The American journal of pathology. 2007;170:1028–1040. doi: 10.2353/ajpath.2007.060595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Paine R, 3rd, Preston AM, Wilcoxen S, Jin H, Siu BB, Morris SB, Reed JA, Ross G, Whitsett JA, Beck JM. Granulocyte-macrophage colony-stimulating factor in the innate immune response to Pneumocystis carinii pneumonia in mice. J Immunol. 2000;164:2602–2609. doi: 10.4049/jimmunol.164.5.2602. [DOI] [PubMed] [Google Scholar]
- 47.Deepe GS, Jr., Gibbons R, Woodward E. Neutralization of endogenous granulocyte-macrophage colony-stimulating factor subverts the protective immune response to Histoplasma capsulatum. J Immunol. 1999;163:4985–4993. [PubMed] [Google Scholar]
- 48.Beers MF, Atochina EN, Preston AM, Beck JM. Inhibition of lung surfactant protein B expression during Pneumocystis carinii pneumonia in mice. The Journal of laboratory and clinical medicine. 1999;133:423–433. doi: 10.1016/s0022-2143(99)90019-7. [DOI] [PubMed] [Google Scholar]
- 49.Chen GH, Teitz-Tennenbaum S, Neal LM, Murdock BJ, Malachowski AN, Dils AJ, Olszewski MA, Osterholzer JJ. Local GM-CSF-Dependent Differentiation and Activation of Pulmonary Dendritic Cells and Macrophages Protect against Progressive Cryptococcal Lung Infection in Mice. J Immunol. 2016;196:1810–1821. doi: 10.4049/jimmunol.1501512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Odegard JM, Marks BR, DiPlacido LD, Poholek AC, Kono DH, Dong C, Flavell RA, Craft J. ICOS-dependent extrafollicular helper T cells elicit IgG production via IL-21 in systemic autoimmunity. J Exp Med. 2008;205:2873–2886. doi: 10.1084/jem.20080840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Frey O, Meisel J, Hutloff A, Bonhagen K, Bruns L, Kroczek RA, Morawietz L, Kamradt T. Inducible costimulator (ICOS) blockade inhibits accumulation of polyfunctional T helper 1/T helper 17 cells and mitigates autoimmune arthritis. Annals of the rheumatic diseases. 2010;69:1495–1501. doi: 10.1136/ard.2009.119164. [DOI] [PubMed] [Google Scholar]
- 52.Ansari MJ, Fiorina P, Dada S, Guleria I, Ueno T, Yuan X, Trikudanathan S, Smith RN, Freeman G, Sayegh MH. Role of ICOS pathway in autoimmune and alloimmune responses in NOD mice. Clin Immunol. 2008;126:140–147. doi: 10.1016/j.clim.2007.07.019. [DOI] [PubMed] [Google Scholar]
- 53.Johnston RJ, Poholek AC, DiToro D, Yusuf I, Eto D, Barnett B, Dent AL, Craft J, Crotty S. Bcl6 and Blimp-1 are reciprocal and antagonistic regulators of T follicular helper cell differentiation. Science. 2009;325:1006–1010. doi: 10.1126/science.1175870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Nurieva RI, Chung Y, Martinez GJ, Yang XO, Tanaka S, Matskevitch TD, Wang YH, Dong C. Bcl6 mediates the development of T follicular helper cells. Science. 2009;325:1001–1005. doi: 10.1126/science.1176676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Smith C, Buhlmann JE, Wang X, Bartlett A, Lim B, Barrington RA. CD275-independent IL-17 producing T follicular helper-like cells in lymphopenic autoimmune-prone mice. J Immunol. 2016 doi: 10.4049/jimmunol.1402193. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]