

Abstract
RNA interference (RNAi) provides a powerful tool to silence specific gene expression and has been widely used to suppress host factors such as CCR5 and/or viral genes involved in HIV-1 replication. Newer nuclease-based gene-editing technologies, such as zinc finger nucleases (ZFN), transcription activator-like effector nucleases (TALEN) and the clustered regularly interspaced short palindromic repeats (CRISPR)/Cas9 system, also provide powerful tools to ablate specific genes. Because of differences in co-receptor usage and the high mutability of the HIV-1 genome, a combination of host factors and viral genes needs to be suppressed for effective prevention and treatment of HIV-1 infection. Whereas the continued presence of small interfering/short hairpin RNA (si/shRNA) mediators is needed for RNAi to be effective, the continued expression of nucleases in the gene-editing systems is undesirable. Thus, RNAi provides the only practical way for expression of multiple silencers in infected and uninfected cells, which is needed for effective prevention/treatment of infection. There have been several advances in the RNAi field in terms of si/shRNA design, targeted delivery to HIV-1 susceptible cells, and testing for efficacy in preclinical humanized mouse models. Here, we comprehensively review the latest advances in RNAi technology towards prevention and treatment of HIV-1.
Keywords: RNAi, si/shRNA, AgoshRNA, shRNA-miR, CCR5, HIV-1, lentiviral vector, humanized mice
Graphical Abstract
1. Introduction
Despite the availability of effective treatment, HIV-1 still remains a global epidemic, responsible for considerable morbidity and mortality. Although highly active antiretroviral therapy (HAART) can suppress viral replication to undetectable levels, it also has limitations, including high cost, patient compliance issues, the side effects of long-term therapy, as well as the emergence of drug resistance [1]. Moreover, although HAART extends the life of HIV-1-infected individuals, it does not offer a permanent cure, as interruption of therapy leads to rapid rebound of viremia from latent reservoirs [1]. Therefore, there is a need to develop more effective countermeasures against HIV-1 infection. Gene therapy has been thought to provide an attractive method for deriving HIV-1-resistant cells (reviewed in [2–4]). Interest in this field started following the identification of CCR5 as a major co-receptor for HIV-1 [5, 6] and the naturally occurring homozygous CCR5-Δ32 mutation (a 32-bp deletion in the single coding exon of the gene resulting in a frame-shift mutation that disrupts CCR5 expression on the cell surface) in humans, confers resistance to HIV-1 infection without other deleterious effects [7, 8]. The remarkable success in treating the so-called “Berlin Patient” has led to a resurgence of interest in gene therapy. In that case, an HIV-1-positive patient with lymphoma who had been transplanted with bone marrow from a CCR5-Δ32 homozygous donor became HIV-1 free, with no demonstrable virus 7 years after transplantation, showing the potential benefits of CCR5 disruption [9, 10]. However, due to the low frequency of CCR5-Δ32 homozygotes in the general population and the difficulties of identifying suitable HLA-matched donors, alternative methods to artificially disrupt CCR5 are being pursued. RNA interference (RNAi) and other gene-editing approaches provide a way to suppress CCR5 as well as viral genes.
RNA interference (RNAi) is a process in which small (~21 nt in length) double-stranded RNAs, either produced endogenously within mammalian cells (microRNA, also known as miRNA) or introduced exogenously into cells (small interfering RNA or siRNA), mediate sequence-specific suppression of gene expression (reviewed in [11–13]). The small RNA in the cytoplasm associates with a multi-protein complex called the RNA-induced silencing complex (RISC), consisting of Argonaute family (Ago1–4) and other proteins [14–16]. Upon RISC binding, one of the two strands of the small RNA is removed (by cleavage and degradation for Ago2-bound siRNA or unwinding by other Ago proteins) and the other so-called the “guide” strand directs the RISC to the corresponding messenger RNA (mRNA). If there is complete homology between the guide strand and the target mRNA, as is generally the case with siRNA, the small RNA-bound Ago-2 protein (which is the only Ago protein with slicer activity) cuts the mRNA at a site corresponding to positions 10–11 from the 5′ end of the guide strand [17, 18]. The mRNA is then rapidly degraded by exonucleases, leading to post-transcriptional gene silencing (PTGS). On the other hand, if there is only partial homology between the guide strand and the target mRNA as in miRNAs (generally between positions 2–8 at the 5′ end of the guide strand, which is called the “seed” sequence), the Ago proteins binding the mRNA and miRNA recruit a protein called GW182 (also known as TNRC6a, b, and c), which then leads to the recruitment of a series of proteins, including the Caf1–Ccr4 and Pan2–Pan-3 deadenylase complexes and the Dcp-1, Dcp-2 decapping complex, which eventually destabilize the mRNA by inducing deadenylation and decapping, resulting in PTGS (reviewed in [19]. Although siRNA is endogenously produced in plants and worms, mammalian cells generally only produce miRNAs and not siRNAs. The miRNAs (there are over 500 in humans) serve to rapidly regulate gene expression in response to environmental cues, which is important in almost every aspect of cellular physiology. Although siRNAs are not produced endogenously, artificial introduction of siRNAs has gained great attention because of their potential in therapeutics to silence selected cellular or viral genes in many diseases [20]. siRNAs can be either chemically synthesized and transfected into cells or they can be expressed within the cells using a DNA template to transcribe what is called a short hairpin RNA (shRNA, encoding the two complementary siRNA strands, which are separated by a non-homologous stem loop), generally under the control of Pol III promoters, which produce small RNAs like small nucleolar RNA (snoRNA). The shRNA is transcribed in the nucleus as stem loop RNA and exported to the cytoplasm to be further processed by Dicer into siRNA. The only difference between miRNA and shRNA biogenesis is that the former involves an additional step: the miRNA is transcribed as a long primary RNA (pri-miRNA), which is first processed by Drosha-DGCR8 in the nucleus into a pre-miRNA (with a stem loop structure resembling shRNA), and this molecule is then exported to the cytoplasm for Dicer processing into mature miRNA (Fig. 1). While exogenously introduced siRNA gets rapidly diluted, particularly in dividing cells, endogenously synthesized shRNA is more stable, particularly if expressed via genome-integrating lentiviral vectors [21]. Thus, for a chronic disease like HIV-1, shRNA is preferable for conferring long-term protection. Along these lines, attention has also focused on expressing shRNAs in autologous hematopoietic stem and progenitor cells (HSPCs), with the idea that when transplanted into humans, these cells can continuously generate HIV-1-resistant T cells and macrophages. Because introduced/produced si/shRNAs occur in abundance, they also have the potential to induce toxicities by competing with the endogenous miRNA machinery [22]. This interference can be avoided to a large extent by expressing shRNAs in the context of cellular miRNAs (discussed in detail later). In recent years, there have been several advances in the RNAi field in terms of si/shRNA design, delivery to HIV-1-susceptible cells, and the ability to test for efficacy in preclinical humanized mouse models. In this review, we will focus on the latest advances made in the RNAi field that, in our view, could facilitate HIV-1 therapy. However, this review is not intended to be a chronological or exhaustive description of all developments in the field, and several excellent reviews of the earlier literature are available [23–26].
Fig 1. si/shRNAs use the endogenous miRNA machinery for gene silencing.

While shRNAs mimic pre-miRNAs, siRNAs resemble mature miRNAs. Like pre-miRNAs, shRNAs are produced in the nucleus and are exported to the cytoplasm, where they are further processed by Dicer into mature siRNAs. Synthetic siRNAs, upon transfection, are loaded directly onto the RISC in the cytoplasm.
2. Need for multiple targets
Both viral genes and host genes essential for HIV-1 replication provide RNAi targets (Fig. 2). Since host factors are not subject to the variability associated with viral genes, they have been used as targets for RNAi. The major entry receptor for HIV-1 is the cell surface molecule, CD4. In addition, a co-receptor, primarily the C-C chemokine receptor 5 (CCR5) or the CXC chemokine receptor 4 (CXCR4), is also required for HIV-1 entry into cells. While primary infection occurs with CCR5-tropic virus, later during infection the virus may change into CXCR4-tropic (also known as T cell-tropic) virus, which is associated with rapid progression to full-blown AIDS. Although the receptor and co-receptors theoretically provide good RNAi targets, attention has focused on CCR5 because of its greater viability as a therapeutic target. For example, although initial proof-of-principle studies showed that suppressing CD4 by RNAi can effectively suppress viral replication [27], it does not provide a potential target for human therapy, because CD4 is essential for normal T cell receptor (TCR)-induced activation of CD4+ T cells, and its disruption can itself promote immunodeficiency. Similarly, CXCR4 is essential for retention of hematopoietic stem cells (HSCs) in bone marrow niches, and its disruption can have deleterious effects on T cell trafficking [28–31]. On the other hand, CCR5 provides a suitable target because: 1) primary infection with HIV-1 occurs with CCR5-tropic virus; 2) approximately 50% of infected individuals maintain CCR5-tropic virus throughout the course of the disease; and, more importantly, 3) spontaneous mutations of CCR5 that render the encoded protein nonfunctional occur infrequently, and such individuals have no apparent problems (except for a slightly increased susceptibility to West Nile virus infection) [9]. The spontaneous homozygous CCR5 (Δ32) mutation occurs in approximately 1% of Caucasians, and those individuals are highly resistant to HIV-1 infection. Moreover, even heterozygous mutations (which are seen in ~4–16% of Caucasians) confer partial resistance to HIV-1 by reducing the CCR5 levels in T cells [32].
Fig 2. RNAi targets for inhibition of HIV-1 replication.

HIV enters the cell by binding to cellular receptor (CD4) and co-receptor (CCR5 or CXCR4). Following entry and uncoating, the viral RNA genome is reverse transcribed and the proviral DNA integrated into the host genome. Proviral transcription generates viral mRNA that encodes viral proteins such as gag, pol, env etc. Thus, any region in the viral mRNA and/or mRNA for host genes involved in HIV-1 life cycle, such as CCR5 can serve as viable RNAi targets to suppress viral replication.
Even though CCR5 provides a good target, alone it is not ideal for suppressing HIV-1, because: 1) CCR5 disruption can only prevent infection and has no effect on already infected cells; 2) CCR5 disruption does not prevent cell–cell transmission; and 3) the virus can mutate to use alternative co-receptors to infect cells. Thus, a combination of cellular and viral targets appears to be necessary for effective control of HIV-1 replication. Many investigators have used this approach, as discussed below.
The viral genome itself provides a good target, because its disruption is not expected to have any unwanted effect on the host cells. Since all the viral proteins are produced using a single, full-length mRNA, targeting any region in the mRNA should be sufficient for RNAi-mediated cleavage. Correspondingly, many investigators have used different genes, including the viral gag, env, pol and nef, as RNAi targets. However, because the viral reverse transcriptase activity is highly error prone, the HIV-1 genome exhibits enormous variability [33]. The great majority of HIV-1 strains belong to group M, but within this group many subtypes (clades) exist that can be identified with different genetic signatures [34, 35]. Different clades dominate in different geographical locations but can also coexist. Furthermore, recombination may occur with different clades, and even within a given infected individual, many variants (quasispecies), differing in gene sequence, occur. Because RNAi is exquisitely sequence specific, even minor changes in the targeted area can affect efficacy. However, within a given gene, there are regions that are highly conserved, because they are essential for gene function, and any mutation within these regions imposes a fitness cost on the virus. Thus, it has been long emphasized that one should design RNAi to target highly conserved regions within the viral genome [36]. In addition, like combinatorial antiretroviral drugs, a combination of RNAi targets is expected to be necessary to reduce the chances of viral escape.
Mathematical modeling suggests that a combination of four RNAi targets may be sufficient to overcome escape. However, this requires all four shRNAs to be perfectly matched to each of the hundreds of circulating viral variants and the viral quasispecies present in patients. It has been estimated that simultaneous expression of seven shRNAs would cover nearly all HIV-1 strains by ensuring that at least four shRNAs are active against any given viral strain [37].
3. RNAi vs other gene editing technologies
Within the past few years, several novel gene-editing technologies have emerged: zinc finger nuclease (ZFN), transcription activator-like effector nuclease (TALEN), and the clustered regularly interspaced short palindromic repeats (CRISPR)/Cas9 system (reviewed in [38–41]. Unlike RNA interference, which requires the continued presence of effector moieties to maintain gene silencing, gene-editing technologies allow permanent disruption/deletion of the targeted gene after a single treatment. Moreover, these gene-editing techniques allow insertion of new DNA sequences at the edited sites. All the currently available gene-editing techniques use two components: one for specific DNA recognition and another for nuclease activity to induce double-stranded breaks at specific sites in the genomic DNA. Following induction of the double-stranded break, the DNA is repaired by either of two pathways: the error-prone non-homologous end joining (NHEJ) pathway, attended with small nucleotide additions or deletions (indels) that result in disruption of the reading frame and gene expression, or homologous recombination (when a DNA template with short homology to the two broken ends is also provided), which results in incorporation of the externally provided DNA at the cleavage site. Thus, these gene-editing systems can be used to knock out/delete genes and also to insert exotic DNA sequences at particular sites.
While ZFN and TALEN use DNA-binding motifs (for zinc finger proteins or transcription activator-like effector molecules, respectively) fused to the Folk1 endonuclease to mediate sequence-specific DNA cleavage, the most recent CRISPR/Cas9 system uses a short stretch of complementary RNA (that can be easily expressed using a U6 promoter) for DNA recognition together with Cas9 nuclease for DNA cleavage. Whereas ZFN/TALEN require 10 or 34 amino acids, respectively, to recognize a single nucleotide, CRISPR/Cas9 uses the Watson-Crick complementarity rule via a short single-guide RNA (sgRNA) molecule (a single RNA nucleotide for each DNA nucleotide) for DNA recognition. Thus, a major advantage is that, compared with ZFN/TALEN, which usually requires labor-intensive design and screening, CRSPR/Cas9 requires a much simpler design and a single cloning step. Because of the ease of usage, this system provides a tremendous opportunity for human gene therapy applications against a variety of diseases. Within 3 years of discovery, this technology has been used to knock out genes, create multiple gene knockout mice and monkeys, as well as knock in specific DNA sequences in a variety of systems (reviewed in [42, 43]). Many groups have successfully disrupted CCR5 using nucleases in various cell lines, primary T cells, HSPCs, as well as in humanized mice [2, 3, 44]. Notably, CCR5 modified (with ZFN) T cells has recently been tested in a clinical trial using a small cohort of 12 individuals [45]. Here, an adenovirus expressing CCR5 ZFN was used to transduce autologous CD4 T cells in vitro, and these cells were then reinfused into the patients. The gene-modified T cells appeared to have a survival advantage over unmodified cells during treatment interruption, suggesting HIV-1 resistance.
However, one major concern about gene editing for clinical use in humans is the high propensity for off-target effects. It has been well documented that off-target effects are a common problem with all editing techniques, particularly the CRISPR/Cas9 system [46–49]. Therefore, while the continued presence of RNAi mediators is needed to maintain suppression, continuous or even long-term exposure to gene-editing enzymes can be potentially harmful. Furthermore, off-target effects may occur anywhere in the genome, requiring labor-intensive next-generation sequencing for a thorough analysis of nonspecific effects. Therefore, although they may be superior approaches for knocking out dispensable host factors like CCR5 to confer HIV-1 resistance, they cannot readily be used to target viral genes in non-infected cells to preempt infection. Such an endeavor requires that the gene-editing systems be active permanently, which results in unacceptable levels of off-target effects and toxicity. Thus, these systems cannot currently be used to silence host factors and HIV-1 genes simultaneously, which will be required to confer effective resistance to the virus in a therapeutic setting. RNAi therefore appears to be the only system that can be used to silence host factors and HIV-1 genes at the same time, which is necessary for prevention as well as treatment.
4. Advances in si/shRNA design
The effector moiety of the double-stranded siRNA is the guide strand, which is complementary to the target mRNA. The passenger strand is not only nonfunctional but can also contribute to off-target effects. Off-target effects of siRNAs can result from sequence similarity of the guide or passenger strand to other mRNAs or even of the 5′ 2–8 nt to an mRNA 3′ untranslated region (UTR), in a similar manner as the miRNA seed sequence [50–53]. Other causes of off-target effects include the effect of certain motifs that could lead to toll-like receptor (TLR) activation, interferon production (GUCCUUCAA, UGUGU) [54, 55] or even cytotoxicity (UGGC) [56]. Therefore, siRNA design is governed by considerations of selective and efficient Ago loading of the guide strand and prevention of passenger strand loading. Since thermodynamic features at the ends of the siRNA determine strand separation, the 5′ end of the siRNA antisense (guide) strand should be less stable than the 3′ end for preferential loading of the guide strand [57, 58]. Structural studies of siRNA-bound Ago proteins showed that the 5′ end of the guide strand binds the so-called MID domain, and this binding affinity is greatest for U and A and 10 times less for G and C [59, 60]. With these considerations, the siRNA is generally designed to start with U or A, have a less-stable 5′ end compared with the 3′ end, have a G:C content of ~50%, and avoid certain motifs that induce toxicities. Using these criteria, several algorithms have been developed to roughly predict functionality by studying large sets of siRNAs [61–65].
Similar guidelines also apply to shRNA design. In addition, since shRNA is transcribed in the nucleus and exported to the cytoplasm by the same machinery that miRNA uses, shRNA, particularly in the abundant amounts expressed by Pol III promoters such as U6, can induce toxicity by saturating exportin 5, thereby blocking miRNA export [22]. Moreover, both siRNA and shRNA have the potential to compete with miRNA for binding to Dicer/Ago proteins. It should be noted that while the siRNA functionality critically depends on Ago2 mediated cleavage of the target mRNA (only Ago2 has slicer activity, not Agos 1, 3 and 4), siRNA is loaded on all Ago proteins. Therefore, siRNA (which is typically transfected or artificially produced within the cells in great abundance compared to miRNAs) can interfere with miRNA by saturating the different Ago proteins. However, certain pre-miRNAs (e.g. pre-miR-451) are not processed by Dicer, but by Ago2 into mature miRNAs, thereby assigning an essential function for Ago2 slicer activity in mammals. This has paved the way to designing Ago2-specific siRNAs that spare other Agos for miRNA function, thereby avoiding competition. These advances will be discussed in some detail below.
4.1 si/shRNA design
Early studies in Drosophila embryos using siRNAs of different length defined the optimal siRNA to be 19 nt long with 2-nt 3′ overhangs [66]. Although this has been considered the gold standard in the RNAi field, studies in mammalian cells suggest that the RNAi machinery is more flexible. Several novel RNAi-triggering structures that do not conform to the key features of classical siRNA in terms of overhang, length, or symmetry have been shown to have improved functionality, such as more potent gene-silencing activity, reduction of nonspecific responses and immune stimulation, or enhanced internalization over classical siRNAs (reviewed in [67]). These modifications include the 27 nt Dicer-substrate siRNA, which is processed by Dicer like the endogenous pre-miRNA [68], shorter than 19 bp siRNAs [69, 70], asymmetric siRNAs with shorter sense strand [71, 72] and triple-stranded RNA variant consisting of an intact 19-nt antisense strand and two contiguous 9–12-nt sense strands [73]. We have recently shown that introducing mismatches in the sense strand with respect to an intact anti-sense strand (similar to what is seen in natural miRNAs) can significantly enhance functionality and reduce off-target effects [74]. We also showed that this design could enhance the functionality of shRNAs, which are generally designed to generate perfectly complementary strands [75–77].
4.2 Ago2-specific siRNA
Mature miR-451 is expressed in an erythropoietic cell-restricted manner and is critical for erythrocyte differentiation. It is also dysregulated in many human malignancies, indicating that miR-451 might play a role in oncogenesis. Biogenesis of miR-451is unique in the sense that it uses a non-canonical pathway (reviewed in [78, 79]). Pri-mi451 is transcribed from an intergenic region adjacent to the protein-coding gene ERAL1 on chromosome 17. The pri-miR451 is processed by Drosha into a 42 nt pre-miR-451 and is exported to the cytoplasm. Unlike the canonical pre-miRNAs, pre-miR-451 is processed not by Dicer, but by Ago2 to generate mature miR-451. The differences between canonical and miR-451 biogenesis include: i) Processing by Drosha–DGCR8. While the canonical pri-miRNAs are processed into ~72-nt pre-miRNAs, pri-miR-451 is processed into a 42-nt pre-miRNA, which is not a suitable Dicer substrate. ii) Stem length. Unlike other pre-miRNAs in which the double-stranded stem is >19 nt, the pre-miR-451 stem is only 17 nt in length. iii) Complementarity. While the canonical miRNA stem contains regions with mismatches, the pre-miR-451 stem is perfectly complementary, allowing Ago2 to cleave the stem. iv) Processing by Ago2. While the single-stranded guide strand in canonical miRNA can be derived from either of the stem strands, the mature miR-451 is derived only from the 5′ arm of the stem plus loop sequence and part of the other stem arm. This is because the pre-miR-451 is processed by Ago2, whose slicer activity cuts the pre-miR-451 in the second arm corresponding to positions 10–11 from the 5′ end of pre-miR-451. By this cleavage, a 30-nt intermediate is generated that is further shortened by cellular RNAse into the 22-nt mature miR-451.
The miR-451 biogenesis pathway described above provides several advantages as a backbone for designing shRNAs: because only the guide strand but not the passenger strand is generated, it is likely to reduce off-target activity. Also because the pre-miRNA binds to Ago2 for processing, it may spare other Ago proteins to occupy other miRNAs, thereby reducing interference with cellular miRNA function. In addition to reducing off-target effects, this shRNA design may be particularly important in silencing genes in cells such as monocytes that lack a significant level of Dicer [80]. Several investigators have therefore tried to reprogram the miR-451 hairpin to silence target transcripts of choice. Earlier proof-of-principal studies expressed other miRNAs in a miR-451 background and showed effective function [81–83]. Later studies suggest that one can also design Dicer-independent shRNAs [84]. The Berkhout group found that reducing the shRNA stem length to <19nt and the loop to 4nt is enough to trigger Ago2 processing, bypassing the Dicer processing [85, 86] (Fig. 3). This shRNA design has been named AgoshRNA. In a further improvement of the design, the Berkhout group also showed the importance of a weak G-U or U-G base pair at the top of the hairpin stem and the importance of the 5′-end nucleotide and a bottom mismatch for efficient Ago2 processing [87]. Applying these principles previously tested anti-HIV-1 shRNA molecules could be converted into AgoshRNAs. Although the RNAi activity of AgoshRNA was diminished compared with regular shRNA, as expected, toxicity was much reduced [85]. On the other hand, we have shown that stabilizing the miR-451 backbone by making it completely complementary by Watson-Crick base pairing (converting the G:U wobble at positions 6 and 18 to G:C and reducing the stem length to 18 nt) can frequently increase RNAi activity compared with conventional shRNA [88].
Fig 3. Design of AgoshRNA.
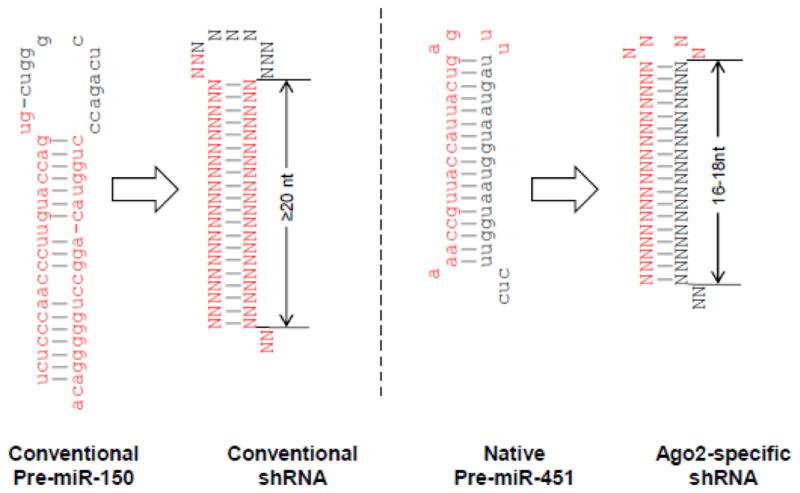
Conventional shRNA is designed to mimic conventional pre-miRNAs like miR-150 shown in the left panel. In contrast AgoshRNA is designed to mimic the pre-miR-451 as shown in the right panel. Conventional shRNA typically has a stem length of 19–22 nt and a larger loop (~10 nt) compared to AgoshRNA which has a 16–18 nt stem and a smaller (4 nt) loop. Whereas the conventional shRNA is processed by dicer and any of the 2 stem strands can give rise to guide strand of siRNA, AgoshRNA is processed by Ago2 and only the top strand along with the loop is loaded onto RISC (because of Ago2-mediated cleavage of AgoshRNA). Mature miRNA sequence is marked in red.
4.3 Multiplexed and miRNA-based shRNA
Because RNAi is exquisitely sequence specific, even minor changes in the targeted sequence can affect efficacy. This is especially pertinent in the case of a highly mutable virus such as HIV-1. To address this issue, as mentioned earlier, several aspects have to be emphasized: use of a combination of host and viral gene targets; targeting highly conserved regions that encode critical domains of the essential viral proteins, in which mutation comes at a fitness cost to the virus; and ideally expressing approximately seven host and conserved viral targets, which is expected to protect against nearly all HIV-1 strains by ensuring that at least four shRNAs are active against any given viral strain.
However, one potential problem of shRNAs expressed under control of the commonly used Pol III promoters is that the overexpressed shRNA can cause toxicities by competing with endogenous miRNAs for binding to exportin 5 for cytoplasmic transport [22]. Moreover, shRNAs in the cytoplasm that are processed by Dicer can also bind all Ago proteins, further compromising endogenous miRNA function [89]. Several studies suggest that expression of shRNAs in an endogenous miRNA backbone (shRNA-miRs) can avoid the toxicities caused by the saturation of RNAi machinery [90–92]. Many investigators have therefore tried to express multiple shRNAs, particularly shRNA-miRs. The Akkina group expressed two shRNAs targeting CCR5 and CXCR4 under the control of separate Pol III promoters, U6 and H1, and observed anti-HIV-1 effects in the macrophages derived from lentivirally transduced HSPCs [93]. Similarly, ter Brake et al. screened 86 highly conserved targets in the HIV-1 genome and identified 21 shRNAs with potent anti-HIV-1 activity. To avoid generation of escape mutants, they also expressed three shRNAs simultaneously in a lentiviral vector under the control of tandem H1 promoters [94]. However, it was noted that the expression of downstream shRNAs progressively declined. Another constraint in multiplexed shRNA expression using the same promoter is the instability of such vectors caused by rapid deletion of cassettes due to homologous recombination [76]. However, different Pol III promoters, such as U6, H1, and 7SK can be used to express multiple anti-HIV-1 shRNAs [76] (Fig. 4). Another strategy used to express multiple shRNAs is expression as an extended shRNA (eshRNA), in which two shRNAs on top of each other (total length of 42–43 nt) are expressed under the control of a Pol III promoter [95]. Activity declined however, with inclusion of additional shRNAs [25]. Similarly, long hairpin RNA (lhRNA) has a long-stemmed (60–70 nt) hairpin and is expressed under control of a Pol III promoter, with the idea that Dicer processing of the longer stem will result in the generation of overlapping shRNAs [96]. However, actual testing showed that the maximal activity was at the base of the stem and the efficacy progressively declined at distal stem regions [97, 98]. A combinatorial approach has also been used to express tat/rev shRNA along with a nucleolar TAR RNA decoy, and a CCR5-targeting ribozyme from independent Pol III promoters [99–101]. This design has been tested in a clinical trial. However, the frequency of gene marking in peripheral blood mononuclear cells (PBMCs) ranged from 0.02 to 0.32%, which was too low for clinical assessment of antiviral efficacy [102].
Fig 4. Multiplexed shRNA.

Multiple shRNAs can be expressed under control of different Pol III promoters (top). Multiplexed shRNAmiRs can be expressed in the context of polycistronic endogenous miRNA cluster (middle) or as multiple pri-miRNAs, each containing the minimal flanking sequence derived from different endogenous miRNAs (bottom). While the maximum number of shRNAs is limited to 4 in the polycistronic context, the number is virtually unlimited using different miRNA flanks.
Because some miRNA clusters are expressed in a polycistron, investigators have tried to convert them into anti-HIV-1 shRNAs (Fig. 4). Aagard et al. used a tri-cistronic miR-106b cluster encoded within the intron of the MCM7 gene to replace the three endogenous miRNAs with shRNAs targeting the tat and rev transcripts of HIV-1 expressed under the control of a Pol II U1 promoter [103]. This vector was further modified to include a nucleolar-localizing TAR RNA decoy and a nucleolar U5-targeting ribozyme and shown to strongly inhibit replication of the NL4-3 strain of HIV-1 in stably transduced lymphoblastoid CD4 T cell line, CEM [104]. Similarly, Liu et al. were able to express four anti-HIV-1 shRNAs in the polycistronic mir-17-92 background under control of the CMV promoter and showed that it conferred potent anti-HIV-1 activity in vitro [105]. Although polycistronic pri-miRNA shuttles represent a promising combinatorial RNAi approach, the efficiency of processing of shRNAs from these pri-miRNA precursors is variable [96, 106]. Another limitation is that, so far, polycistronic shRNA-miR expression has been limited to a maximum of four shRNA-miRs, although, as noted earlier, it has been estimated that seven shRNA-miRs may be ideal for covering all variants. Moreover, although shRNA-miR expression under a polycistronic miRNA background was described in 2011, so far this has not been tested for in vivo efficacy in humanized mouse models.
To address the limitations of current multiplexing approaches, we used a simple strategy of using minimal flanking sequences from multiple endogenous miRNAs to co-express a large number of shRNA-miRs [107]. Although Drosha cleavage was previously reported to occur ~11 nt from the lower stem-ssRNA junction [108], we have recently found that the microprocessor measures the distances from both the lower and upper stem-ssRNA junctions to determine the cleavage site in human cells, and optimal distance from both structures is critical to the precision of Drosha processing [109]. However, the optimal flanking sequence required to ensure Drosha processing of primary shRNA-miRs was not previously known. By a systematic analysis of the different flanking sequence lengths for different endogenous miRNA backbones, we determined that incorporation of ~30 nt of flanking sequence may be enough to ensure processing of different shRNA-miRs. We then cloned multiple shRNAs in tandem, each containing a minimal flanking sequence from a different miRNA (to avoid deletion by homologous recombination) and expressed them from a lentiviral vector under the control of the Pol II EF-1α promoter. Deep sequencing of transduced cells showed accurate processing of individual shRNA-miRs. Moreover, unlike lshRNAs, the expression levels of individual shRNAs did not progressively decrease with the distance from the promoter. We used this system to express one shRNA-miR targeting CCR5 and six shRNA-miRs targeting different regions in the HIV-1 genome. The seven shRNA-miR-transduced populations of T cells were protected against both R5-tropic and X4-tropic HIV-1 infection. Additionally, when T cells from HIV-1-seropositive individuals were transduced and transplanted into NOD/SCID/IL-2R γc−/− mice (Hu-PBL model), efficient suppression of endogenous HIV-1 replication with restoration of CD4 T cell counts was observed [107]. Therefore, this strategy for multiplexing shRNA appears to provide a promising gene therapeutic approach for HIV-1 infection.
5. Humanized mouse model to test anti-HIV therapy
Since HIV infects only human cells, humanized mouse models engrafted with functional human cells have become important preclinical tools for evaluating novel anti-HIV strategies. The development of IL2rγ-deficient mice in the Rag−/− and the SCID/NOD backgrounds enabled easy generation of humanized mice (reviewed in [110]). These mice lack endogenous murine T, B, and NK cells and also exhibit defects in cytokine signaling. The mice can be engrafted with human T cells to create a simple Hu-PBL model in which T cells expand rapidly because of xenogenic activation. The Hu-PBL model is amenable to HIV infection and provides a quick way to assess the antiviral potential of drugs or T cell gene-modification strategies. However, the lack of de novo hematopoiesis in this model is a limitation. The time frame of experimental utility is also limited in this model because the animals develop a lethal graft-versus-host disease (GVHD) after a few weeks. In contrast to Hu-PBL model, complete human adaptive immune system can be reconstituted in these immunodeficient mice by transplanting them with human CD34 hematopoietic stem progenitor cells (HSPCs). Unlike in the PBL model, here the T and B cells are not activated (naïve), and therefore HIV infection in this model is more akin to human infection. Thus, these mice provide an ideal small animal model to test HSPC-based gene therapy approaches. In the earlier version of the Hu-HSPC model, CD34+ HSPCs were injected into irradiated newborn or adult IL2rγ−/− mouse strains via intravenous, intrahepatic, or intracardiac injections. Although a full complement of hematopoietic cell lineages were generated, including T cells, which are susceptible to HIV infection, there was a lack of human HLA-restriction, because the T cells developed in the mouse thymic environment. A more advanced version of the model is the so-called BLT mouse, in which the irradiated NOD/SCID/IL2rγ−/− mice are transplanted with human fetal liver and thymus tissue under the renal capsule followed by injection of CD34+ HSPCs isolated from fetal liver [111]. This allows a full complement of the human immune system, including T and B cells, NK cells, macrophages, and dendritic cells, to be efficiently reconstituted. However, both T and B cell responses against many human pathogens are still suboptimal in these mice, probably due to the absence of critical species-specific cytokines or factors that are important in cellular development and survival. Technical advances like replacing mouse cytokine genes with corresponding human genes or engineering IL2rγ−/− strains of NOD/SCID or Rag−/− mice to transgenically express human class I or class II molecules [112, 113] have resulted in improved reconstitution and immune responses. In fact, these mice transgenic for HLA-DR0401 molecules (DRAG mice) reconstituted with HLA-II-matched human HSPCs could mount robust cellular and humoral immune responses in response to infectious or vaccine challenge. Furthermore, unlike BLT mice, human cells can be generated in the DRAG mice by simple i.v. infusion of CD34+ HSPCs without human fetal liver and thymus transplants [113, 114]. Overall, with the ongoing efforts at further refining the human environment, Hu-mice are likely to emerge as an ideal tool for preclinical testing of human vaccines and therapeutics.
6. RNAi therapy for HIV in humanized mice
Both shRNA and siRNAs have been used to achieve RNAi-mediated inhibition of HIV-1. Significant advances have been made in using them to inhibit HIV-1 in terms of design, delivery and testing efficacy in humanized mice models. However, for both synthetic siRNA and expressed shRNA, many challenges will have to be overcome for widespread clinical use as a treatment modality for HIV-1 infection.
Rational design of siRNAs has become possible because of the rapid advances in our understanding of RNAi biology, but using them as antiviral drugs for HIV-1 remains a challenge because of difficulties with in vivo distribution, cellular internalization, and cytosolic release. Naked siRNAs are not only susceptible to body fluid nucleases, they are also rapidly eliminated by the kidneys. Chemical modifications of siRNAs (such as 2′-O methyl substitution, addition of a phosphorothioate linkage at the 3′ end and the use of locked nucleic acids) have been used to increase siRNA stability, both in vitro and in vivo [115, 116]. Other important considerations for systemic use of siRNA are the avoidance of undesirable off-target or immune-stimulatory effects, which can be achieved to a large extent by judicious choice of target sequences. By far the biggest hurdle for successful use of RNAi as therapy is delivery to the desired cells and tissues. The high molecular weight and anionic charge of siRNAs precludes entry by simple diffusion; furthermore, to be functional the molecules have to escape from the endosomal/lysomal compartment into the cytosol. Delivery is a particularly difficult challenge for HIV-1 infection because the uptake of siRNA by virus-susceptible T cells and macrophages is extremely inefficient. Thus, many different strategies are being tested to improve the efficiency of siRNA delivery into these cells.
In general, non-specific carriers, such as cell-penetrating peptides, lipids, or polymers, have not proved useful for delivery of siRNA to primary immune cells. However, two recent studies have reported success in the delivery of siRNA to T cells with a new generation of cationic dendrimers [117]. Carbosilane dendriplexes conjugated to a siRNA targeting the HIV Nef molecule were shown to be efficiently incorporated into primary CD4+ T cells in vitro and could effectively reduce viral replication. Zhou et al. were able to suppress HIV viral loads by several orders of magnitude in humanized Rag2−/− γc−/− mice using cationic poly(amidoamine) (PAMAMM) dendrimers for therapeutic delivery of a cocktail of Dicer substrate siRNAs targeting viral and cellular transcripts [118].
Delivery approaches specifically tailored to T cells and monocytes/macrophages are also being developed. Targeting siRNA to cells and tissues of interest has the advantage of improving their therapeutic index while at the same time minimizing the potential unintended off-target effects at other sites. Antibodies and aptamers are being investigated as targeting reagents to induce siRNA internalization into HIV-susceptible cells. The first proof of concept study to show that the strategy could work used an antibody Fab fragment directed at the HIV envelope as the targeting ligand. The antibody F105 was fused to protamine to bind and transport the siRNA cargo. The antibody protamine reagent F105-P was able to deliver siRNAs and silence gene expression specifically in HIV-1-infected cells [119]. This report was soon followed by a study from our group that showed for the first time the feasibility of siRNA for treatment of HIV-1 in vivo in a humanized mouse model. Here, to deliver siRNA to T cells, a single-chain antibody to the T cell surface protein CD7 was fused to nine arginine residues (the positive charge on Rs allows negatively charged siRNA to bind). Twice-a-week i.v. injection of antiviral siRNAs bound to CD7-9R was enough to suppress CD4 decline and substantially reduce the plasma viral load compared with controls in HIV-1-infected humanized mice [120]. Similarly, nanoparticles coated with an antibody directed at LFA-1 (an integrin molecule expressed on a broader spectrum of HIV-1-susceptible cell types, including T cells, macrophages, and dendritic cells) can deliver siRNA and suppress HIV infection in humanized mice [121].
In another approach, aptamers have been used for cell-specific targeting of anti HIV-1 siRNAs [122, 123]. Aptamers are short nucleic acids, akin to antibodies, that bind to target molecules with high specificity and affinity. The advantages over antibody-based targeting are their lack of immunogenicity and amenability to further improvements, such as further truncations to obtain minimal aptamer sequences or chemical modifications for improved activity or stability. Aptamers are selected by screening large libraries of DNA or RNA sequences for high-affinity binding to immobilized proteins in a process called “systematic evolution of ligands by exponential enrichment” (SELEX) or a more sophisticated whole-cell-based SELEX approach for selection of cell-internalizing aptamers. Aptamers can be conjugated to siRNA by a simple in vitro T7 transcription reaction that covalently joins the aptamer to one strand of the siRNA. More recently, a more flexible aptamer–“sticky bridge” siRNA conjugation has been developed in which the aptamers are generated with a pair of complementary GC-rich sticky-bridge sequences through which the siRNA(s) of choice can be non-covalently assembled [124]. Aptamers specific for HIV-1 gp120, the CD4 receptor, or recently the CCR5 receptor have been shown to be capable of delivering chimeric siRNA molecules into T cells or HIV-infected cells to effectively suppress viral replication. In one study, siRNA targeting viral tat/rev was fused to a gp120 aptamer. When this reagent was administered to humanized mice infected with HIV-1 (by weekly injection for 5 weeks), it significantly suppressed viral load and prevented the CD4 T cell decline seen with control mice [125]. A cell-specific RNA aptamer directed at the major HIV co-receptor CCR5 was used to knock down the cellular factor transportin 3 (TNPO3) to inhibit HIV replication in primary human PBMCs [126].
Although these studies show the feasibility of siRNA treatment during acute HIV infection, siRNA as a drug treatment approach in humans appears unlikely to be successful, considering the chronic nature of infection and the need for repeated treatment. The only potentially practical application of synthetic siRNA for HIV would be as a topical vaginal microbicide to prevent mucosal transmission [127, 128]. In fact, the feasibility of this approach has been demonstrated in humanized mice, which were treated intravaginally with a CD4 aptamer conjugated to siRNAs targeting CCR5 as well as viral gag and vif. When applied 2 days before vaginal HIV-1 challenge, the infection was completely blocked. However, applying earlier than 2 days before challenge appeared to reduce effectiveness
The shRNA approach is the more practical way forward for HIV therapy. In particular, shRNA delivery via lentiviral vectors holds great promise, because shRNA continues to be produced following vector integration into the host genome, and thus a single treatment will have lasting effect. The concept here is that the modified cells, even at low-to-moderate levels of gene modification, have a selective survival advantage over unmodified cells, allowing them to proliferate and expand over time and, as a consequence, minimize viral loads and reduce viral reservoirs [129]. In fact, based on mathematical modeling it has been estimated that gene marking of ~10–20% in HSPCs would be sufficient to have an impact on viral load and CD4+ lymphocyte counts [130]. Two approaches can be used to achieve this: transduction of T cells to render them HIV-resistant and transduction of HSPCs that enables continuous generation of HIV-resistant lymphoid and myeloid cell progeny (Fig. 5). In both cases, transduction can be accomplished ex vivo with cells that are isolated from patients (following apheresis for T cells and isolation of HSPCs from bone marrow or GMCSF-mobilized blood cells). The cells are transduced with a lentiviral vector encoding shRNAs and transplanted back to the patient. Proof-of-principle of this approach has been shown in humanized mice infected with HIV-1. Anderson et al. developed a lentiviral vector to express a rev/tat shRNA, a TAR decoy, and a CCR5 ribozyme. When CD34+ HSPCs transduced with the combination vector were injected into SCID-hu thy/liv grafts, isolated T cells from the graft were shown to resist HIV infection in vitro [131]. In another study, Shimizu et al. generated HIV-1-resistant humanized mice using human CD34+ HSPCs transduced with a lentiviral vector encoding a potent shRNA targeting CCR5. The T cells from these mice were resistant to CCR5-tropic virus when tested in vitro [132]. The same vector has also been shown to result in stable shRNA expression (after transfusion of transduced CD34+ cells) in non-human primates for extended times, albeit at low levels [133]. The same group also generated humanized mice using HSPC transduced with lentivirus encoding shRNAs targeting CCR5 and HIV-1 LTR and showed that the mice were resistant to challenge with both R5 and X4 tropic virus [134]. This CCR5 shRNA has also been combined with the C46 antiviral peptide (an HIV-1 entry inhibitor derived from the C-terminal heptad repeat of HIV-1 gp41, modified for expression on the cell surface) in a lentiviral vector (LVsh5/C46) to provide additional protection. Upon HIV-1 challenge, humanized BLT mice generated with LVsh5/C46-transduced CD34+ cells were protected from CD4 T cell decline and had lower viral loads compared with control mice [135]. Similarly, the Berkhout group generated humanized mice with CD34+ cells transduced with lentivirus expressing three shRNAs targeting the viral integrase, protease, and tat/rev genes (shPol1, shPol47 and shR/T5), and found that the transduced GFP+-sorted T cells were resistant to HIV infection ex vivo [136]. Walker et al. used a lentiviral vector expressing TRIM5α, CCR5 shRNA, and a TAR decoy to provide combinatorial anti-HIV activity [137]. When CD34+ cells were transduced with this vector and transplanted to NSG mice, after challenge with either X4- or R5-tropic virus, the progressive CD4 T cell decline observed in control mice was completely prevented and, in fact, the gene-modified cells showed a selective survival advantage. However, plasma viremia was not reduced, probably because of continuous generation of HIV-susceptible unmodified T cells.
Fig 5. Schematic of potential RNAi therapy strategy for humans.

Two approaches can be used for human therapy. Either autologous CD4 T cells (purified from PBMC obtained by apheresis) or CD34+ HSPC (purified from bone marrow or blood after mobilization by GM-CSF treatment) can be in vitro modified to express shRNA (generally by transduction with lentivirus encoding CCR5 and antiviral shRNAs) and reinfused into the patients. Modified CD4 T cells provide a one-time source of HIV-1 resistant T cells. HSPC modification is expected to result in the continuous generation of HIV-1 resistant progeny T cells and macrophages.
One of the main hurdles in stem cell-based gene therapy for HIV-1 is the low frequency of gene-modified cells that is typically generated. To overcome this, attempts have been made to select for modified cells. The endogenous enzyme o-6-methylguanine-DNA methytransferase (MGMT) is required for repairing DNA damage caused by alkylating agents such as BCNU (carmustine). O6-benzylguanine (O6-BG) deactivates endogenous MGMT so that cells cannot repair BCNU-induced DNA damage resulting in cell death. However, gene-modified cells that express a modified MGMT (MGMTP140K) are not sensitive to O6-BG treatment and therefore can repair DNA damage from BCNU treatment and survive. Thus, treatment with O6-BG/BCNU can be used to select gene-modified cells. Chung et al used a lentiviral vector encoding anti-HIV shRNAs that also expressed MGMTP140K. Humanized mice were generated using CD34+ cells transduced with this lentiviral vector. Seven weeks following transplantation, the mice were treated with O6-BG and BCNU weekly for 3 weeks. This treatment resulted in an increase in the frequency of the modified genes by 10–15 fold, suggesting that this is a robust system [104, 138].
Lentiviral treatment of PBMCs has also been shown to be feasible for therapy in the PBL model. Suzuki et al. transduced human PBMCs with lentivirus encoding shRNAs targeting the promoter region of the HIV LTR (which mediates transcriptional gene silencing by inducing epigenetic changes) and infused these cells into NOD/SCID mice deficient for Janus kinase 3 (NOJ mice). These mice when infected with HIV-1 were resistant to CD4 T cell depletion and had a reduced viral load compared with controls [139]. We have transduced PBMCs from HIV-seropositive individuals with a lentiviral vector expressing seven shRNAs and shown its effectiveness in suppressing endogenous virus activation in hu-PBL mice [107].
7. Safety of lentiviral vectors for human therapy
Murine leukemia virus (MLV)-based γ-retroviral and HIV-based lentiviral vectors have been used for human gene therapy (reviewed in [140]). However, the use of earlier generations of retroviral vectors in resulted in severe adverse effects, including leukemia, myelodysplasia, and lymphoma, in gene therapy trials for diseases such as X-linked severe combined immunodeficiency, chronic granulomatous disease, and Wiskott-Aldrich syndrome [141–143]. This was caused by the transcriptional activation of nearby proto-oncogenes by the powerful enhancer elements contained within the γ-retroviral long terminal repeats (LTRs) of the vector. On the other hand, the newer self-inactivating (SIN) vectors, in which the absence of enhancer elements in their LTRs makes them less able to transactivate endogenous genes after integration, are safer [144]. Even so, from a perspective of safety, lentiviral vectors may be preferable primarily because the insertion site selection of lentiviral vectors differs from that of γ-retroviral vectors, probably due to differences between the integrases of the two viruses [145, 146]. In contrast to retroviral vectors which preferentially integrate near host cell promoters and enhancers, the lentiviral vectors integrate somewhat randomly throughout the genome and thus less likely to activate oncogenes. In fact, lentiviral vectors have been used recently in clinical trials and have so far proven safe with no reported malignancies [143, 147, 148].
8. Human clinical trials
Bone marrow transplantation necessitates the use of preconditioning regimens (radio- or chemotherapy) to ablate the endogenous hematopoietic cells and free the niche, which is required for successful engraftment of gene-modified stem cells. Since HIV-1 is controllable with HAART, ablative regimens are unacceptable in HIV-1-positive individuals without other diseases, such as cancer, in which bone marrow transplantation is indicated. Thus, most of the clinical trials using gene-modified HSPCs have been done in HIV-1 patients who develop lymphomas. Even here, ethical consideration dictates the use of unmodified HSPCs as well. Hence, combinations of unmodified and gene-modified cells are generally used, which significantly dilutes the modified cell numbers, making assessment of the anti-HIV-1 efficacy of the RNAi difficult. In the first study of HSPC-based gene therapy, four HIV-1 patients with AIDS-related non-Hodgkin’s lymphoma were given gene-modified HSPCs in addition to unmodified cells [99]. To derive gene-modified cells, HSPCs were transduced with a lentiviral vector encoding three RNA-based anti-HIV-1 moieties (tat/rev short-hairpin RNA, TAR decoy, and CCR5 ribozyme). The treatment was well tolerated, the transfected cells were successfully engrafted in all four infused patients, and persistent expression of the introduced siRNA and ribozyme vector expression in multiple cell lineages was observed at low levels (0.1–0.3% of PBMCs) for up to 24 months. However, because of the low numbers of modified cells, the anti-HIV-1 efficacy could not be determined. Further trials using the same vector and combination chemotherapy have been initiated (NCT02337985, NCT01961063).
Other studies under clinical trial include the LVsh5/C46 vector referred to earlier, which is being evaluated in a phase 1/2 clinical trial in which autologous hematopoietic cells are transduced ex vivo, followed by infusion back into HIV-1+ subjects (NCT01734850, NCT02378922). Another trial (NCT02343666) using the LVsh5/C46 vector also aims to select for gene-modified cells using the O6-BG/BCNU treatment system described earlier.
9. Conclusion and perspectives
In recent years, significant advances have been made in si/shRNA design to increase effectiveness and prevent undesirable effects. Since gene therapy is not risk free and involves expensive procedures, it is not currently being pursued to prevent infection in uninfected individuals. Current research efforts have therefore aimed at protecting uninfected cells and combating infected cells in already infected individuals. Under these circumstances, targeting CCR5 alone in infected subjects is probably not the best approach, since it will not protect against emergence of X4-tropic virus. Also, considering the immense propensity of the HIV-1 genome for mutations, multiple shRNAs targeting both dispensable host factors and viral genes will be necessary for any effective treatment approach. However, so far the only viable host factor that can be targeted (without adverse effects) is CCR5, and thus targeting viral genes at the same time becomes extremely important. Although newer gene-editing techniques enable easy and permanent disruption of CCR5, they do not provide an easy way to disable HIV-1 viral genes, which is necessary to protect uninfected cells. Such an endeavor requires that the gene-editing nucleases to be permanently expressed in the cells, which could cause an unacceptably high level of off-target effects. Thus, the only currently available, practical, and non-toxic way to confer the potential to suppress HIV-1 in infected and uninfected cells is via expression of multiple miR-based shRNAs. In this regard, notable advancements have been achieved in the use of polycistronic miRNA design to express up to four shRNAs and, more importantly, in the use of different endogenous miRNA-based flanking sequences to express a virtually unlimited number of shRNAs.
The most practical way to confer long-term HIV-1 resistance is the infusion of HSPCs transduced with shRNA-expressing lentiviral vectors, with the idea that the stem cells provide a source for continuous generation of HIV-1-resistant T and myeloid cell progeny. One major current limitation of this approach is the low level of gene marking generally achieved in clinical trials (generally a few percent of progeny cells). Two recent developments show promise in overcoming this shortcoming. By using highly purified and homogenous batches of lentiviral vectors, optimized culture conditions, vector exposure, and a dose-adjusted myeloablative regimen before HSPC infusion, Naldini’s group was able to obtain high levels of stable and long-term (>1 yr) gene marking in 50–80% of progeny cells [149, 150]. Although the above approach was used in treating leukodystrophy and Wiskott-Aldrich syndrome patients, in general it provides reassurance of the viability of HSPC-based gene therapy in humans and should be followed in gene therapy trials for HIV-1. The second approach is to enrich for transduced stem cells using the MGMT/O6-BG/BCNU treatment mentioned earlier, which resulted in >10-fold enrichment in monkey and mouse models and is under clinical trial for HIV-1 gene therapy. Considering the advances mentioned above and others that are expected, RNAi-based gene therapy is a promising approach for treating HIV-1.
Table 1.
RNAi approached tested for HIV inhibition in vivo
| Host/viral RNAi Targets | Vehicle | Model | Route of Delivery | Result | Reference |
|---|---|---|---|---|---|
|
| |||||
| HIV tat/rev siRNA, TNPO3 siRNA and CD4 siRNA | Cationic poly(amidoamine(PAMAMM) dendrimers | HIV infected Rag2−/− γc−/− humanized mice | Intravenous | Reduction in plasma viremia, inhibition of CD4 decline | 118 |
| HIV-gag/c-myc, MDM2 and VEGF siRNAs | HIV-envelope antibody F105 fused to Protamine | WT mice with HIV-env expressing B6 melanoma tumors | Intratumoral/ Intravenous | Reduction in tumor size | 119 |
| HIV tat, Vif siRNA and CCR5siRNAs | CD7scFv/9R | HIV infected BLT mice | Intravenous | Reduction in plasma viremia, inhibition of CD4 decline | 120 |
| CCR5 siRNA | LFA-1 antibody coated nanoparticles | HIV infected BLT mice | Intravenous | Reduction in plasma viremia, inhibition of CD4 decline | 121 |
| HIV tat/rev siRNA | Gp120 aptamer/siRNA chimera | HIV infected Rag2−/−γc−/− humanized mice | Intravenous | Reduction in plasma viremia, inhibition of CD4 decline | 125 |
| HIV gag/vif siRNA | CD4 aptamer/siRNA chimera | HIV infected BLT | Intravaginal | Reduction in viremia | 127 |
| CCR5 shRNA LTR shRNA | Lentiviral transduction of HSPC | HIV infected BLT mice | HSPC transduction | Reduction in plasma viremia, inhibition of CD4 decline | 134 |
| CCR5 shRNA and C46 peptide | Lentiviral transduction of HSPC | HIV infected BLT mice | HSPC transduction | Reduction in plasma viremia, inhibition of CD4 decline | 135 |
| CCR5 shRNA, TRIM5α and TAR decoy | Lentiviral transduction of HSPC | Rag1−/−γc−/− mice | HSPC transduction | Inhibition of CD4 decline | 137 |
| LTR shRNA | Lentiviral transduction of PBMC | Hu-PBL mice | Transplantaion of transduced PMBC | Reduction in plasma viremia, inhibition of CD4 decline | 139 |
| 7 ShRNAs targeting CCR5 and HIV Gag Env, Tat, Pol, Vif, | Lentiviral transduction of PBMC | Hu-PBL mice | Transplantaion of transduced PMBC | Reduction in plasma viremia, inhibition of CD4 decline | 107 |
| shRNA targeting an overlapping region in Tat and Rev, TAR decoy RNA and CCR5 Ribozyme | Lentiviral transduction of HSPCs | Pilot Human Clinical trial NTC00569985 | HSPC transfer | Gene marking in PBMCs for upto 6 months | 99 |
Acknowledgments
This work was supported by NIH grant R21HL116268 to P.S. and R21AI108259 to MN.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Carr A. Toxicity of antiretroviral therapy and implications for drug development. Nat Rev Drug Discov. 2003;2:624–634. doi: 10.1038/nrd1151. [DOI] [PubMed] [Google Scholar]
- 2.Cornu TI, Mussolino C, Bloom K, Cathomen T. Editing CCR5: a novel approach to HIV gene therapy. Adv Exp Med Biol. 2015;848:117–130. doi: 10.1007/978-1-4939-2432-5_6. [DOI] [PubMed] [Google Scholar]
- 3.Drake MJ, Bates P. Application of gene-editing technologies to HIV-1. Curr Opin HIV AIDS. 2015;10:123–127. doi: 10.1097/COH.0000000000000139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gu WG. Genome editing-based HIV therapies. Trends Biotechnol. 2015;33:172–179. doi: 10.1016/j.tibtech.2014.12.006. [DOI] [PubMed] [Google Scholar]
- 5.Alkhatib G, Combadiere C, Broder CC, Feng Y, Kennedy PE, Murphy PM, Berger EA. CC CKR5: a RANTES, MIP-1alpha, MIP-1beta receptor as a fusion cofactor for macrophage-tropic HIV-1. Science. 1996;272:1955–1958. doi: 10.1126/science.272.5270.1955. [DOI] [PubMed] [Google Scholar]
- 6.Deng H, Liu R, Ellmeier W, Choe S, Unutmaz D, Burkhart M, Di Marzio P, Marmon S, Sutton RE, Hill CM, Davis CB, Peiper SC, Schall TJ, Littman DR, Landau NR. Identification of a major co-receptor for primary isolates of HIV-1. Nature. 1996;381:661–666. doi: 10.1038/381661a0. [DOI] [PubMed] [Google Scholar]
- 7.Samson M, Libert F, Doranz BJ, Rucker J, Liesnard C, Farber CM, Saragosti S, Lapoumeroulie C, Cognaux J, Forceille C, Muyldermans G, Verhofstede C, Burtonboy G, Georges M, Imai T, Rana S, Yi Y, Smyth RJ, Collman RG, Doms RW, Vassart G, Parmentier M. Resistance to HIV-1 infection in caucasian individuals bearing mutant alleles of the CCR-5 chemokine receptor gene. Nature. 1996;382:722–725. doi: 10.1038/382722a0. [DOI] [PubMed] [Google Scholar]
- 8.Liu R, Paxton WA, Choe S, Ceradini D, Martin SR, Horuk R, MacDonald ME, Stuhlmann H, Koup RA, Landau NR. Homozygous defect in HIV-1 coreceptor accounts for resistance of some multiply-exposed individuals to HIV-1 infection. Cell. 1996;86:367–377. doi: 10.1016/s0092-8674(00)80110-5. [DOI] [PubMed] [Google Scholar]
- 9.Allers K, Schneider T. CCR5Delta32 mutation and HIV infection: basis for curative HIV therapy. Curr Opin Virol. 2015;14:24–29. doi: 10.1016/j.coviro.2015.06.007. [DOI] [PubMed] [Google Scholar]
- 10.Hutter G, Nowak D, Mossner M, Ganepola S, Mussig A, Allers K, Schneider T, Hofmann J, Kucherer C, Blau O, Blau IW, Hofmann WK, Thiel E. Long-term control of HIV by CCR5 Delta32/Delta32 stem-cell transplantation. N Engl J Med. 2009;360:692–698. doi: 10.1056/NEJMoa0802905. [DOI] [PubMed] [Google Scholar]
- 11.Vaishnaw AK, Gollob J, Gamba-Vitalo C, Hutabarat R, Sah D, Meyers R, de Fougerolles T, Maraganore J. A status report on RNAi therapeutics. Silence. 2010;1:14. doi: 10.1186/1758-907X-1-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shankar P, Manjunath N, Lieberman J. The prospect of silencing disease using RNA interference. JAMA. 2005;293:1367–1373. doi: 10.1001/jama.293.11.1367. [DOI] [PubMed] [Google Scholar]
- 13.Meister G, Tuschl T. Mechanisms of gene silencing by double-stranded RNA. Nature. 2004;431:343–349. doi: 10.1038/nature02873. [DOI] [PubMed] [Google Scholar]
- 14.Meister G. Argonaute proteins: functional insights and emerging roles. Nat Rev Genet. 2013;14:447–459. doi: 10.1038/nrg3462. [DOI] [PubMed] [Google Scholar]
- 15.Hutvagner G, Simard MJ. Argonaute proteins: key players in RNA silencing. Nat Rev Mol Cell Biol. 2008;9:22–32. doi: 10.1038/nrm2321. [DOI] [PubMed] [Google Scholar]
- 16.Hock J, Meister G. The Argonaute protein family. Genome Biol. 2008;9:210. doi: 10.1186/gb-2008-9-2-210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Matranga C, Tomari Y, Shin C, Bartel DP, Zamore PD. Passenger-strand cleavage facilitates assembly of siRNA into Ago2-containing RNAi enzyme complexes. Cell. 2005;123:607–620. doi: 10.1016/j.cell.2005.08.044. [DOI] [PubMed] [Google Scholar]
- 18.Rand TA, Petersen S, Du F, Wang X. Argonaute2 cleaves the anti-guide strand of siRNA during RISC activation. Cell. 2005;123:621–629. doi: 10.1016/j.cell.2005.10.020. [DOI] [PubMed] [Google Scholar]
- 19.Jonas S, Izaurralde E. Towards a molecular understanding of microRNA-mediated gene silencing. Nat Rev Genet. 2015;16:421–433. doi: 10.1038/nrg3965. [DOI] [PubMed] [Google Scholar]
- 20.Wittrup A, Lieberman J. Knocking down disease: a progress report on siRNA therapeutics. Nat Rev Genet. 2015;16:543–552. doi: 10.1038/nrg3978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Subramanya S, Kim SS, Manjunath N, Shankar P. RNA interference-based therapeutics for human immunodeficiency virus HIV-1 treatment: synthetic siRNA or vector-based shRNA? Expert Opin Biol Ther. 2010;10:201–213. doi: 10.1517/14712590903448158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Grimm D, Streetz KL, Jopling CL, Storm TA, Pandey K, Davis CR, Marion P, Salazar F, Kay MA. Fatality in mice due to oversaturation of cellular microRNA/short hairpin RNA pathways. Nature. 2006;441:537–541. doi: 10.1038/nature04791. [DOI] [PubMed] [Google Scholar]
- 23.Zhou J, Neff CP, Liu X, Zhang J, Li H, Smith DD, Swiderski P, Aboellail T, Huang Y, Du Q, Liang Z, Peng L, Akkina R, Rossi JJ. Systemic administration of combinatorial dsiRNAs via nanoparticles efficiently suppresses HIV-1 infection in humanized mice. Molecular therapy : the journal of the American Society of Gene Therapy. 2011;19:2228–2238. doi: 10.1038/mt.2011.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Eekels JJ, Berkhout B. Toward a durable treatment of HIV-1 infection using RNA interference. Prog Mol Biol Transl Sci. 2011;102:141–163. doi: 10.1016/B978-0-12-415795-8.00001-5. [DOI] [PubMed] [Google Scholar]
- 25.Liu YP, Berkhout B. Lentiviral delivery of RNAi effectors against HIV-1. Curr Top Med Chem. 2009;9:1130–1143. doi: 10.2174/156802609789630866. [DOI] [PubMed] [Google Scholar]
- 26.Manjunath N, Wu H, Subramanya S, Shankar P. Lentiviral delivery of short hairpin RNAs. Adv Drug Deliv Rev. 2009;61:732–745. doi: 10.1016/j.addr.2009.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Novina CD, Murray MF, Dykxhoorn DM, Beresford PJ, Riess J, Lee SK, Collman RG, Lieberman J, Shankar P, Sharp PA. siRNA-directed inhibition of HIV-1 infection. Nat Med. 2002;8:681–686. doi: 10.1038/nm725. [DOI] [PubMed] [Google Scholar]
- 28.Chung SH, Seki K, Choi BI, Kimura KB, Ito A, Fujikado N, Saijo S, Iwakura Y. CXC chemokine receptor 4 expressed in T cells plays an important role in the development of collagen-induced arthritis. Arthritis Res Ther. 2010;12:R188. doi: 10.1186/ar3158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nagasawa T. The chemokine CXCL12 and regulation of HSC and B lymphocyte development in the bone marrow niche. Adv Exp Med Biol. 2007;602:69–75. doi: 10.1007/978-0-387-72009-8_9. [DOI] [PubMed] [Google Scholar]
- 30.Dar A, Goichberg P, Shinder V, Kalinkovich A, Kollet O, Netzer N, Margalit R, Zsak M, Nagler A, Hardan I, Resnick I, Rot A, Lapidot T. Chemokine receptor CXCR4-dependent internalization and resecretion of functional chemokine SDF-1 by bone marrow endothelial and stromal cells. Nat Immunol. 2005;6:1038–1046. doi: 10.1038/ni1251. [DOI] [PubMed] [Google Scholar]
- 31.Burger JA, Peled A. CXCR4 antagonists: targeting the microenvironment in leukemia and other cancers. Leukemia. 2009;23:43–52. doi: 10.1038/leu.2008.299. [DOI] [PubMed] [Google Scholar]
- 32.Lai Y. CCR5-targeted hematopoietic stem cell gene approaches for HIV disease: current progress and future prospects. Curr Stem Cell Res Ther. 2012;7:310–317. doi: 10.2174/157488812800793108. [DOI] [PubMed] [Google Scholar]
- 33.Lloyd SB, Kent SJ, Winnall WR. The high cost of fidelity. AIDS Res Hum Retroviruses. 2014;30:8–16. doi: 10.1089/aid.2013.0153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tebit DM, Nankya I, Arts EJ, Gao Y. HIV diversity, recombination and disease progression: how does fitness “fit” into the puzzle? AIDS Rev. 2007;9:75–87. [PubMed] [Google Scholar]
- 35.Thomson MM, Najera R. Molecular epidemiology of HIV-1 variants in the global AIDS pandemic: an update. AIDS Rev. 2005;7:210–224. [PubMed] [Google Scholar]
- 36.Knoepfel SA, Centlivre M, Liu YP, Boutimah F, Berkhout B. Selection of RNAi-based inhibitors for anti-HIV gene therapy. World J Virol. 2012;1:79–90. doi: 10.5501/wjv.v1.i3.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.McIntyre GJ, Groneman JL, Yu YH, Tran A, Applegate TL. Multiple shRNA combinations for near-complete coverage of all HIV-1 strains. AIDS Res Ther. 2011;8:1. doi: 10.1186/1742-6405-8-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gersbach CA, Perez-Pinera P. Activating human genes with zinc finger proteins, transcription activator-like effectors and CRISPR/Cas9 for gene therapy and regenerative medicine. Expert Opin Ther Targets. 2014;18:835–839. doi: 10.1517/14728222.2014.913572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Niu J, Zhang B, Chen H. Applications of TALENs and CRISPR/Cas9 in human cells and their potentials for gene therapy. Mol Biotechnol. 2014;56:681–688. doi: 10.1007/s12033-014-9771-z. [DOI] [PubMed] [Google Scholar]
- 40.Zhang F, Wen Y, Guo X. CRISPR/Cas9 for genome editing: progress, implications and challenges. Hum Mol Genet. 2014;23:R40–46. doi: 10.1093/hmg/ddu125. [DOI] [PubMed] [Google Scholar]
- 41.Manjunath N, Yi G, Dang Y, Shankar P. Newer gene editing technologies toward HIV gene therapy. Viruses. 2013;5:2748–2766. doi: 10.3390/v5112748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Riordan SM, Heruth DP, Zhang LQ, Ye SQ. Application of CRISPR/Cas9 for biomedical discoveries. Cell Biosci. 2015;5:33. doi: 10.1186/s13578-015-0027-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sternberg SH, Doudna JA. Expanding the Biologist’s Toolkit with CRISPR-Cas9. Mol Cell. 2015;58:568–574. doi: 10.1016/j.molcel.2015.02.032. [DOI] [PubMed] [Google Scholar]
- 44.Hutter G, Bodor J, Ledger S, Boyd M, Millington M, Tsie M, Symonds G. CCR5 Targeted Cell Therapy for HIV and Prevention of Viral Escape. Viruses. 2015;7:4186–4203. doi: 10.3390/v7082816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tebas P, Stein D, Tang WW, Frank I, Wang SQ, Lee G, Spratt SK, Surosky RT, Giedlin MA, Nichol G, Holmes MC, Gregory PD, Ando DG, Kalos M, Collman RG, Binder-Scholl G, Plesa G, Hwang WT, Levine BL, June CH. Gene editing of CCR5 in autologous CD4 T cells of persons infected with HIV. N Engl J Med. 2014;370:901–910. doi: 10.1056/NEJMoa1300662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Courtney DG, Moore JE, Atkinson SD, Maurizi E, Allen EH, Pedrioli DM, McLean WH, Nesbit MA, Moore CB. CRISPR/Cas9 DNA cleavage at SNP-derived PAM enables both in vitro and in vivo KRT12 mutation-specific targeting. Gene Ther. 2015 doi: 10.1038/gt.2015.82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Koo T, Lee J, Kim JS. Measuring and Reducing Off-Target Activities of Programmable Nucleases Including CRISPR-Cas9. Mol Cells. 2015;38:475–481. doi: 10.14348/molcells.2015.0103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pauwels K, Podevin N, Breyer D, Carroll D, Herman P. Engineering nucleases for gene targeting: safety and regulatory considerations. N Biotechnol. 2014;31:18–27. doi: 10.1016/j.nbt.2013.07.001. [DOI] [PubMed] [Google Scholar]
- 49.Mali P, Esvelt KM, Church GM. Cas9 as a versatile tool for engineering biology. Nat Methods. 2013;10:957–963. doi: 10.1038/nmeth.2649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jackson AL, Bartz SR, Schelter J, Kobayashi SV, Burchard J, Mao M, Li B, Cavet G, Linsley PS. Expression profiling reveals off-target gene regulation by RNAi. Nat Biotechnol. 2003;21:635–637. doi: 10.1038/nbt831. [DOI] [PubMed] [Google Scholar]
- 51.Jackson AL, Burchard J, Schelter J, Chau BN, Cleary M, Lim L, Linsley PS. Widespread siRNA “off-target” transcript silencing mediated by seed region sequence complementarity. RNA. 2006;12:1179–1187. doi: 10.1261/rna.25706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Scacheri PC, Rozenblatt-Rosen O, Caplen NJ, Wolfsberg TG, Umayam L, Lee JC, Hughes CM, Shanmugam KS, Bhattacharjee A, Meyerson M, Collins FS. Short interfering RNAs can induce unexpected and divergent changes in the levels of untargeted proteins in mammalian cells. Proc Natl Acad Sci U S A. 2004;101:1892–1897. doi: 10.1073/pnas.0308698100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Birmingham A, Anderson EM, Reynolds A, Ilsley-Tyree D, Leake D, Fedorov Y, Baskerville S, Maksimova E, Robinson K, Karpilow J, Marshall WS, Khvorova A. 3′ UTR seed matches, but not overall identity, are associated with RNAi off-targets. Nat Methods. 2006;3:199–204. doi: 10.1038/nmeth854. [DOI] [PubMed] [Google Scholar]
- 54.Hornung V, Guenthner-Biller M, Bourquin C, Ablasser A, Schlee M, Uematsu S, Noronha A, Manoharan M, Akira S, de Fougerolles A, Endres S, Hartmann G. Sequence-specific potent induction of IFN-alpha by short interfering RNA in plasmacytoid dendritic cells through TLR7. Nat Med. 2005;11:263–270. doi: 10.1038/nm1191. [DOI] [PubMed] [Google Scholar]
- 55.Judge AD, Sood V, Shaw JR, Fang D, McClintock K, MacLachlan I. Sequence-dependent stimulation of the mammalian innate immune response by synthetic siRNA. Nat Biotechnol. 2005;23:457–462. doi: 10.1038/nbt1081. [DOI] [PubMed] [Google Scholar]
- 56.Fedorov Y, Anderson EM, Birmingham A, Reynolds A, Karpilow J, Robinson K, Leake D, Marshall WS, Khvorova A. Off-target effects by siRNA can induce toxic phenotype. RNA. 2006;12:1188–1196. doi: 10.1261/rna.28106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Khvorova A, Reynolds A, Jayasena SD. Functional siRNAs and miRNAs exhibit strand bias. Cell. 2003;115:209–216. doi: 10.1016/s0092-8674(03)00801-8. [DOI] [PubMed] [Google Scholar]
- 58.Schwarz DS, Hutvagner G, Du T, Xu Z, Aronin N, Zamore PD. Asymmetry in the assembly of the RNAi enzyme complex. Cell. 2003;115:199–208. doi: 10.1016/s0092-8674(03)00759-1. [DOI] [PubMed] [Google Scholar]
- 59.Frank F, Sonenberg N, Nagar B. Structural basis for 5′-nucleotide base-specific recognition of guide RNA by human AGO2. Nature. 2010;465:818–822. doi: 10.1038/nature09039. [DOI] [PubMed] [Google Scholar]
- 60.Parker JS, Roe SM, Barford D. Structural insights into mRNA recognition from a PIWI domain-siRNA guide complex. Nature. 2005;434:663–666. doi: 10.1038/nature03462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Vert JP, Foveau N, Lajaunie C, Vandenbrouck Y. An accurate and interpretable model for siRNA efficacy prediction. BMC Bioinformatics. 2006;7:520. doi: 10.1186/1471-2105-7-520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Shabalina SA, Spiridonov AN, Ogurtsov AY. Computational models with thermodynamic and composition features improve siRNA design. BMC Bioinformatics. 2006;7:65. doi: 10.1186/1471-2105-7-65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Reynolds A, Leake D, Boese Q, Scaringe S, Marshall WS, Khvorova A. Rational siRNA design for RNA interference. Nat Biotechnol. 2004;22:326–330. doi: 10.1038/nbt936. [DOI] [PubMed] [Google Scholar]
- 64.Ui-Tei K, Naito Y, Takahashi F, Haraguchi T, Ohki-Hamazaki H, Juni A, Ueda R, Saigo K. Guidelines for the selection of highly effective siRNA sequences for mammalian and chick RNA interference. Nucleic Acids Res. 2004;32:936–948. doi: 10.1093/nar/gkh247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Henschel A, Buchholz F, Habermann B. DEQOR: a web-based tool for the design and quality control of siRNAs. Nucleic Acids Res. 2004;32:W113–120. doi: 10.1093/nar/gkh408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Elbashir SM, Martinez J, Patkaniowska A, Lendeckel W, Tuschl T. Functional anatomy of siRNAs for mediating efficient RNAi in Drosophila melanogaster embryo lysate. EMBO J. 2001;20:6877–6888. doi: 10.1093/emboj/20.23.6877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Chang CI, Kim HA, Dua P, Kim S, Li CJ, Lee DK. Structural diversity repertoire of gene silencing small interfering RNAs. Nucleic Acid Ther. 2011;21:125–131. doi: 10.1089/nat.2011.0286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kim DH, Behlke MA, Rose SD, Chang MS, Choi S, Rossi JJ. Synthetic dsRNA Dicer substrates enhance RNAi potency and efficacy. Nat Biotechnol. 2005;23:222–226. doi: 10.1038/nbt1051. [DOI] [PubMed] [Google Scholar]
- 69.Li Z, Fortin Y, Shen SH. Forward and robust selection of the most potent and noncellular toxic siRNAs from RNAi libraries. Nucleic Acids Res. 2009;37:e8. doi: 10.1093/nar/gkn953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Chu CY, Rana TM. Potent RNAi by short RNA triggers. RNA. 2008;14:1714–1719. doi: 10.1261/rna.1161908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Sun X, Rogoff HA, Li CJ. Asymmetric RNA duplexes mediate RNA interference in mammalian cells. Nat Biotechnol. 2008;26:1379–1382. doi: 10.1038/nbt.1512. [DOI] [PubMed] [Google Scholar]
- 72.Chang CI, Yoo JW, Hong SW, Lee SE, Kang HS, Sun X, Rogoff HA, Ban C, Kim S, Li CJ, Lee DK. Asymmetric shorter-duplex siRNA structures trigger efficient gene silencing with reduced nonspecific effects. Molecular therapy : the journal of the American Society of Gene Therapy. 2009;17:725–732. doi: 10.1038/mt.2008.298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Bramsen JB, Laursen MB, Damgaard CK, Lena SW, Babu BR, Wengel J, Kjems J. Improved silencing properties using small internally segmented interfering RNAs. Nucleic Acids Res. 2007;35:5886–5897. doi: 10.1093/nar/gkm548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Wu H, Ma H, Ye C, Ramirez D, Chen S, Montoya J, Shankar P, Wang XA, Manjunath N. Improved siRNA/shRNA functionality by mismatched duplex. PloS one. 2011;6:e28580. doi: 10.1371/journal.pone.0028580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Moore CB, Guthrie EH, Huang MT, Taxman DJ. Short hairpin RNA (shRNA): design, delivery, and assessment of gene knockdown. Methods Mol Biol. 2010;629:141–158. doi: 10.1007/978-1-60761-657-3_10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.ter Brake O, Hooft Kt, Liu YP, Centlivre M, von Eije KJ, Berkhout B. Lentiviral vector design for multiple shRNA expression and durable HIV-1 inhibition. Molecular therapy : the journal of the American Society of Gene Therapy. 2008;16:557–564. doi: 10.1038/sj.mt.6300382. [DOI] [PubMed] [Google Scholar]
- 77.Taxman DJ, Livingstone LR, Zhang J, Conti BJ, Iocca HA, Williams KL, Lich JD, Ting JP, Reed W. Criteria for effective design, construction, and gene knockdown by shRNA vectors. BMC Biotechnol. 2006;6:7. doi: 10.1186/1472-6750-6-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Liu YP, Karg M, Harwig A, Herrera-Carrillo E, Jongejan A, van Kampen A, Berkhout B. Mechanistic insights on the Dicer-independent AGO2-mediated processing of AgoshRNAs. RNA Biol. 2015;12:92–100. doi: 10.1080/15476286.2015.1017204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Yang JS, Lai EC. Dicer-independent, Ago2-mediated microRNA biogenesis in vertebrates. Cell Cycle. 2010;9:4455–4460. doi: 10.4161/cc.9.22.13958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Coley W, Van Duyne R, Carpio L, Guendel I, Kehn-Hall K, Chevalier S, Narayanan A, Luu T, Lee N, Klase Z, Kashanchi F. Absence of DICER in monocytes and its regulation by HIV-1. J Biol Chem. 2010;285:31930–31943. doi: 10.1074/jbc.M110.101709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Yang JS, Maurin T, Robine N, Rasmussen KD, Jeffrey KL, Chandwani R, Papapetrou EP, Sadelain M, O’Carroll D, Lai EC. Conserved vertebrate mir-451 provides a platform for Dicer-independent, Ago2-mediated microRNA biogenesis. Proc Natl Acad Sci U S A. 2010;107:15163–15168. doi: 10.1073/pnas.1006432107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Cifuentes D, Xue H, Taylor DW, Patnode H, Mishima Y, Cheloufi S, Ma E, Mane S, Hannon GJ, Lawson ND, Wolfe SA, Giraldez AJ. A novel miRNA processing pathway independent of Dicer requires Argonaute2 catalytic activity. Science. 2010;328:1694–1698. doi: 10.1126/science.1190809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Cheloufi S, Dos Santos CO, Chong MM, Hannon GJ. A dicer-independent miRNA biogenesis pathway that requires Ago catalysis. Nature. 2010;465:584–589. doi: 10.1038/nature09092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Berkhout B, Liu YP. Towards improved shRNA and miRNA reagents as inhibitors of HIV-1 replication. Future Microbiol. 2014;9:561–571. doi: 10.2217/fmb.14.5. [DOI] [PubMed] [Google Scholar]
- 85.Liu YP, Karg M, Herrera-Carrillo E, Berkhout B. Towards Antiviral shRNAs Based on the AgoshRNA Design. PloS one. 2015;10:e0128618. doi: 10.1371/journal.pone.0128618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Liu YP, Schopman NC, Berkhout B. Dicer-independent processing of short hairpin RNAs. Nucleic Acids Res. 2013;41:3723–3733. doi: 10.1093/nar/gkt036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Herrera-Carrillo E, Harwig A, Liu YP, Berkhout B. Probing the shRNA characteristics that hinder Dicer recognition and consequently allow Ago-mediated processing and AgoshRNA activity. RNA. 2014;20:1410–1418. doi: 10.1261/rna.043950.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Ma H, Zhang J, Wu H. Designing Ago2-specific siRNA/shRNA to Avoid Competition with Endogenous miRNAs. Mol Ther Nucleic Acids. 2014;3:e176. doi: 10.1038/mtna.2014.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Grimm D, Wang L, Lee JS, Schurmann N, Gu S, Borner K, Storm TA, Kay MA. Argonaute proteins are key determinants of RNAi efficacy, toxicity, and persistence in the adult mouse liver. The Journal of clinical investigation. 2010;120:3106–3119. doi: 10.1172/JCI43565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Wu P, Wilmarth MA, Zhang F, Du G. miRNA and shRNA expression vectors based on mRNA and miRNA processing. Methods Mol Biol. 2013;936:195–207. doi: 10.1007/978-1-62703-083-0_16. [DOI] [PubMed] [Google Scholar]
- 91.McBride JL, Boudreau RL, Harper SQ, Staber PD, Monteys AM, Martins I, Gilmore BL, Burstein H, Peluso RW, Polisky B, Carter BJ, Davidson BL. Artificial miRNAs mitigate shRNA-mediated toxicity in the brain: implications for the therapeutic development of RNAi. Proc Natl Acad Sci U S A. 2008;105:5868–5873. doi: 10.1073/pnas.0801775105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Boudreau RL, Martins I, Davidson BL. Artificial microRNAs as siRNA shuttles: improved safety as compared to shRNAs in vitro and in vivo. Molecular therapy : the journal of the American Society of Gene Therapy. 2009;17:169–175. doi: 10.1038/mt.2008.231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Anderson J, Akkina R. CXCR4 and CCR5 shRNA transgenic CD34+ cell derived macrophages are functionally normal and resist HIV-1 infection. Retrovirology. 2005;2:53. doi: 10.1186/1742-4690-2-53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.ter Brake O, Konstantinova P, Ceylan M, Berkhout B. Silencing of HIV-1 with RNA interference: a multiple shRNA approach. Molecular therapy : the journal of the American Society of Gene Therapy. 2006;14:883–892. doi: 10.1016/j.ymthe.2006.07.007. [DOI] [PubMed] [Google Scholar]
- 95.Liu YP, Haasnoot J, Berkhout B. Design of extended short hairpin RNAs for HIV-1 inhibition. Nucleic Acids Res. 2007;35:5683–5693. doi: 10.1093/nar/gkm596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Saayman S, Arbuthnot P, Weinberg MS. Deriving four functional anti-HIV siRNAs from a single Pol III-generated transcript comprising two adjacent long hairpin RNA precursors. Nucleic Acids Res. 2010;38:6652–6663. doi: 10.1093/nar/gkq460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Saayman S, Barichievy S, Capovilla A, Morris KV, Arbuthnot P, Weinberg MS. The efficacy of generating three independent anti-HIV-1 siRNAs from a single U6 RNA Pol III-expressed long hairpin RNA. PloS one. 2008;3:e2602. doi: 10.1371/journal.pone.0002602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Barichievy S, Saayman S, von Eije KJ, Morris KV, Arbuthnot P, Weinberg MS. The inhibitory efficacy of RNA POL III-expressed long hairpin RNAs targeted to untranslated regions of the HIV-1 5′ long terminal repeat. Oligonucleotides. 2007;17:419–431. doi: 10.1089/oli.2007.0095. [DOI] [PubMed] [Google Scholar]
- 99.DiGiusto DL, Krishnan A, Li L, Li H, Li S, Rao A, Mi S, Yam P, Stinson S, Kalos M, Alvarnas J, Lacey SF, Yee JK, Li M, Couture L, Hsu D, Forman SJ, Rossi JJ, Zaia JA. RNA-based gene therapy for HIV with lentiviral vector-modified CD34(+) cells in patients undergoing transplantation for AIDS-related lymphoma. Sci Transl Med. 2010;2:36ra43. doi: 10.1126/scitranslmed.3000931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.DiGiusto DL, Stan R, Krishnan A, Li H, Rossi JJ, Zaia JA. Development of hematopoietic stem cell based gene therapy for HIV-1 infection: considerations for proof of concept studies and translation to standard medical practice. Viruses. 2013;5:2898–2919. doi: 10.3390/v5112898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Li MJ, Kim J, Li S, Zaia J, Yee JK, Anderson J, Akkina R, Rossi JJ. Long-term inhibition of HIV-1 infection in primary hematopoietic cells by lentiviral vector delivery of a triple combination of anti-HIV shRNA, anti-CCR5 ribozyme, and a nucleolar-localizing TAR decoy. Molecular therapy : the journal of the American Society of Gene Therapy. 2005;12:900–909. doi: 10.1016/j.ymthe.2005.07.524. [DOI] [PubMed] [Google Scholar]
- 102.Chung J, Scherer LJ, Gu A, Gardner AM, Torres-Coronado M, Epps EW, Digiusto DL, Rossi JJ. Optimized lentiviral vectors for HIV gene therapy: multiplexed expression of small RNAs and inclusion of MGMT(P140K) drug resistance gene. Molecular therapy : the journal of the American Society of Gene Therapy. 2014;22:952–963. doi: 10.1038/mt.2014.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Aagaard LA, Zhang J, von Eije KJ, Li H, Saetrom P, Amarzguioui M, Rossi JJ. Engineering and optimization of the miR-106b cluster for ectopic expression of multiplexed anti-HIV RNAs. Gene Ther. 2008;15:1536–1549. doi: 10.1038/gt.2008.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Chung J, Zhang J, Li H, Ouellet DL, DiGiusto DL, Rossi JJ. Endogenous MCM7 microRNA cluster as a novel platform to multiplex small interfering and nucleolar RNAs for combinational HIV-1 gene therapy. Hum Gene Ther. 2012;23:1200–1208. doi: 10.1089/hum.2012.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Liu YP, Haasnoot J, ter Brake O, Berkhout B, Konstantinova P. Inhibition of HIV-1 by multiple siRNAs expressed from a single microRNA polycistron. Nucleic Acids Res. 2008;36:2811–2824. doi: 10.1093/nar/gkn109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Ely A, Naidoo T, Arbuthnot P. Efficient silencing of gene expression with modular trimeric Pol II expression cassettes comprising microRNA shuttles. Nucleic Acids Res. 2009;37:e91. doi: 10.1093/nar/gkp446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Choi JG, Bharaj P, Abraham S, Ma H, Yi G, Ye C, Dang Y, Manjunath N, Wu H, Shankar P. Multiplexing seven miRNA-Based shRNAs to suppress HIV replication. Molecular therapy : the journal of the American Society of Gene Therapy. 2015;23:310–320. doi: 10.1038/mt.2014.205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Han J, Lee Y, Yeom KH, Nam JW, Heo I, Rhee JK, Sohn SY, Cho Y, Zhang BT, Kim VN. Molecular basis for the recognition of primary microRNAs by the Drosha-DGCR8 complex. Cell. 2006;125:887–901. doi: 10.1016/j.cell.2006.03.043. [DOI] [PubMed] [Google Scholar]
- 109.Ma H, Wu Y, Choi JG, Wu H. Lower and upper stem-single-stranded RNA junctions together determine the Drosha cleavage site. Proc Natl Acad Sci U S A. 2013;110:20687–20692. doi: 10.1073/pnas.1311639110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Shultz LD, Brehm MA, Garcia-Martinez JV, Greiner DL. Humanized mice for immune system investigation: progress, promise and challenges. Nat Rev Immunol. 2012;12:786–798. doi: 10.1038/nri3311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Wege AK, Melkus MW, Denton PW, Estes JD, Garcia JV. Functional and phenotypic characterization of the humanized BLT mouse model. Curr Top Microbiol Immunol. 2008;324:149–165. doi: 10.1007/978-3-540-75647-7_10. [DOI] [PubMed] [Google Scholar]
- 112.Shultz LD, Saito Y, Najima Y, Tanaka S, Ochi T, Tomizawa M, Doi T, Sone A, Suzuki N, Fujiwara H, Yasukawa M, Ishikawa F. Generation of functional human T-cell subsets with HLA-restricted immune responses in HLA class I expressing NOD/SCID/IL2r gamma(null) humanized mice. Proc Natl Acad Sci U S A. 2010;107:13022–13027. doi: 10.1073/pnas.1000475107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Danner R, Chaudhari SN, Rosenberger J, Surls J, Richie TL, Brumeanu TD, Casares S. Expression of HLA class II molecules in humanized NOD.Rag1KO.IL2RgcKO mice is critical for development and function of human T and B cells. PloS one. 2011;6:e19826. doi: 10.1371/journal.pone.0019826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Allam A, Majji S, Peachman K, Jagodzinski L, Kim J, Ratto-Kim S, Wijayalath W, Merbah M, Kim JH, Michael NL, Alving CR, Casares S, Rao M. TFH cells accumulate in mucosal tissues of humanized-DRAG mice and are highly permissive to HIV-1. Sci Rep. 2015;5:10443. doi: 10.1038/srep10443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Ku SH, Jo SD, Lee YK, Kim K, Kim SH. Chemical and structural modifications of RNAi therapeutics. Adv Drug Deliv Rev. 2015 doi: 10.1016/j.addr.2015.10.015. [DOI] [PubMed] [Google Scholar]
- 116.Lundin KE, Hojland T, Hansen BR, Persson R, Bramsen JB, Kjems J, Koch T, Wengel J, Smith CI. Biological activity and biotechnological aspects of locked nucleic acids. Adv Genet. 2013;82:47–107. doi: 10.1016/B978-0-12-407676-1.00002-0. [DOI] [PubMed] [Google Scholar]
- 117.Perise-Barrios AJ, Jimenez JL, Dominguez-Soto A, de la Mata FJ, Corbi AL, Gomez R, Munoz-Fernandez MA. Carbosilane dendrimers as gene delivery agents for the treatment of HIV infection. J Control Release. 2014;184:51–57. doi: 10.1016/j.jconrel.2014.03.048. [DOI] [PubMed] [Google Scholar]
- 118.Zhou J, Rossi JJ. Current progress in the development of RNAi-based therapeutics for HIV-1. Gene Ther. 2011;18:1134–1138. doi: 10.1038/gt.2011.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Song E, Zhu P, Lee SK, Chowdhury D, Kussman S, Dykxhoorn DM, Feng Y, Palliser D, Weiner DB, Shankar P, Marasco WA, Lieberman J. Antibody mediated in vivo delivery of small interfering RNAs via cell-surface receptors. Nat Biotechnol. 2005;23:709–717. doi: 10.1038/nbt1101. [DOI] [PubMed] [Google Scholar]
- 120.Kumar P, Ban HS, Kim SS, Wu H, Pearson T, Greiner DL, Laouar A, Yao J, Haridas V, Habiro K, Yang YG, Jeong JH, Lee KY, Kim YH, Kim SW, Peipp M, Fey GH, Manjunath N, Shultz LD, Lee SK, Shankar P. T cell-specific siRNA delivery suppresses HIV-1 infection in humanized mice. Cell. 2008;134:577–586. doi: 10.1016/j.cell.2008.06.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Kim SS, Peer D, Kumar P, Subramanya S, Wu H, Asthana D, Habiro K, Yang YG, Manjunath N, Shimaoka M, Shankar P. RNAi-mediated CCR5 silencing by LFA-1-targeted nanoparticles prevents HIV infection in BLT mice. Molecular therapy : the journal of the American Society of Gene Therapy. 2010;18:370–376. doi: 10.1038/mt.2009.271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Takahashi M, Burnett JC, Rossi JJ. Aptamer-siRNA chimeras for HIV. Adv Exp Med Biol. 2015;848:211–234. doi: 10.1007/978-1-4939-2432-5_11. [DOI] [PubMed] [Google Scholar]
- 123.Zhou J, Rossi J. Cell-type-specific aptamer and aptamer-small interfering RNA conjugates for targeted human immunodeficiency virus type 1 therapy. J Investig Med. 2014;62:914–919. doi: 10.1097/JIM.0000000000000103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Zhou J, Neff CP, Swiderski P, Li H, Smith DD, Aboellail T, Remling-Mulder L, Akkina R, Rossi JJ. Functional in vivo delivery of multiplexed anti-HIV-1 siRNAs via a chemically synthesized aptamer with a sticky bridge. Molecular therapy : the journal of the American Society of Gene Therapy. 2013;21:192–200. doi: 10.1038/mt.2012.226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Neff CP, Zhou J, Remling L, Kuruvilla J, Zhang J, Li H, Smith DD, Swiderski P, Rossi JJ, Akkina R. An aptamer-siRNA chimera suppresses HIV-1 viral loads and protects from helper CD4(+) T cell decline in humanized mice. Sci Transl Med. 2011;3:66ra66. doi: 10.1126/scitranslmed.3001581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Zhou J, Satheesan S, Li H, Weinberg MS, Morris KV, Burnett JC, Rossi JJ. Cell-specific RNA aptamer against human CCR5 specifically targets HIV-1 susceptible cells and inhibits HIV-1 infectivity. Chem Biol. 2015;22:379–390. doi: 10.1016/j.chembiol.2015.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Wheeler LA, Trifonova R, Vrbanac V, Basar E, McKernan S, Xu Z, Seung E, Deruaz M, Dudek T, Einarsson JI, Yang L, Allen TM, Luster AD, Tager AM, Dykxhoorn DM, Lieberman J. Inhibition of HIV transmission in human cervicovaginal explants and humanized mice using CD4 aptamer-siRNA chimeras. The Journal of clinical investigation. 2011;121:2401–2412. doi: 10.1172/JCI45876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Wheeler LA, Vrbanac V, Trifonova R, Brehm MA, Gilboa-Geffen A, Tanno S, Greiner DL, Luster AD, Tager AM, Lieberman J. Durable knockdown and protection from HIV transmission in humanized mice treated with gel-formulated CD4 aptamer-siRNA chimeras. Molecular therapy : the journal of the American Society of Gene Therapy. 2013;21:1378–1389. doi: 10.1038/mt.2013.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Wolstein O, Boyd M, Millington M, Impey H, Boyer J, Howe A, Delebecque F, Cornetta K, Rothe M, Baum C, Nicolson T, Koldej R, Zhang J, Keech N, Camba Colon J, Breton L, Bartlett J, An DS, Chen IS, Burke B, Symonds GP. Preclinical safety and efficacy of an anti-HIV-1 lentiviral vector containing a short hairpin RNA to CCR5 and the C46 fusion inhibitor. Mol Ther Methods Clin Dev. 2014;1:11. doi: 10.1038/mtm.2013.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Murray JM, Fanning GC, Macpherson JL, Evans LA, Pond SM, Symonds GP. Mathematical modelling of the impact of haematopoietic stem cell-delivered gene therapy for HIV. J Gene Med. 2009;11:1077–1086. doi: 10.1002/jgm.1401. [DOI] [PubMed] [Google Scholar]
- 131.Anderson J, Li MJ, Palmer B, Remling L, Li S, Yam P, Yee JK, Rossi J, Zaia J, Akkina R. Safety and efficacy of a lentiviral vector containing three anti-HIV genes--CCR5 ribozyme, tat-rev siRNA, and TAR decoy--in SCID-hu mouse-derived T cells. Molecular therapy : the journal of the American Society of Gene Therapy. 2007;15:1182–1188. doi: 10.1038/sj.mt.6300157. [DOI] [PubMed] [Google Scholar]
- 132.Shimizu S, Hong P, Arumugam B, Pokomo L, Boyer J, Koizumi N, Kittipongdaja P, Chen A, Bristol G, Galic Z, Zack JA, Yang O, Chen IS, Lee B, An DS. A highly efficient short hairpin RNA potently down-regulates CCR5 expression in systemic lymphoid organs in the hu-BLT mouse model. Blood. 2010;115:1534–1544. doi: 10.1182/blood-2009-04-215855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.An DS, Donahue RE, Kamata M, Poon B, Metzger M, Mao SH, Bonifacino A, Krouse AE, Darlix JL, Baltimore D, Qin FX, Chen IS. Stable reduction of CCR5 by RNAi through hematopoietic stem cell transplant in non-human primates. Proc Natl Acad Sci U S A. 2007;104:13110–13115. doi: 10.1073/pnas.0705474104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Ringpis GE, Shimizu S, Arokium H, Camba-Colon J, Carroll MV, Cortado R, Xie Y, Kim PY, Sahakyan A, Lowe EL, Narukawa M, Kandarian FN, Burke BP, Symonds GP, An DS, Chen IS, Kamata M. Engineering HIV-1-resistant T-cells from short-hairpin RNA-expressing hematopoietic stem/progenitor cells in humanized BLT mice. PloS one. 2012;7:e53492. doi: 10.1371/journal.pone.0053492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Burke BP, Levin BR, Zhang J, Sahakyan A, Boyer J, Carroll MV, Colon JC, Keech N, Rezek V, Bristol G, Eggers E, Cortado R, Boyd MP, Impey H, Shimizu S, Lowe EL, Ringpis GE, Kim SG, Vatakis DN, Breton LR, Bartlett JS, Chen IS, Kitchen SG, An DS, Symonds GP. Engineering Cellular Resistance to HIV-1 Infection In Vivo Using a Dual Therapeutic Lentiviral Vector. Mol Ther Nucleic Acids. 2015;4:e236. doi: 10.1038/mtna.2015.10. [DOI] [PubMed] [Google Scholar]
- 136.Centlivre M, Legrand N, Klamer S, Liu YP, Jasmijn von Eije K, Bohne M, Rijnstra ES, Weijer K, Blom B, Voermans C, Spits H, Berkhout B. Preclinical in vivo evaluation of the safety of a multi-shRNA-based gene therapy against HIV-1. Mol Ther Nucleic Acids. 2013;2:e120. doi: 10.1038/mtna.2013.48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Walker JE, Chen RX, McGee J, Nacey C, Pollard RB, Abedi M, Bauer G, Nolta JA, Anderson JS. Generation of an HIV-1-resistant immune system with CD34(+) hematopoietic stem cells transduced with a triple-combination anti-HIV lentiviral vector. J Virol. 2012;86:5719–5729. doi: 10.1128/JVI.06300-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Kiem HP, Wu RA, Sun G, von Laer D, Rossi JJ, Trobridge GD. Foamy combinatorial anti-HIV vectors with MGMTP140K potently inhibit HIV-1 and SHIV replication and mediate selection in vivo. Gene Ther. 2010;17:37–49. doi: 10.1038/gt.2009.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Suzuki K, Hattori S, Marks K, Ahlenstiel C, Maeda Y, Ishida T, Millington M, Boyd M, Symonds G, Cooper DA, Okada S, Kelleher AD. Promoter Targeting shRNA Suppresses HIV-1 Infection In vivo Through Transcriptional Gene Silencing. Mol Ther Nucleic Acids. 2013;2:e137. doi: 10.1038/mtna.2013.64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Persons DA. Lentiviral vector gene therapy: effective and safe? Molecular therapy : the journal of the American Society of Gene Therapy. 2010;18:861–862. doi: 10.1038/mt.2010.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Aiuti A, Bacchetta R, Seger R, Villa A, Cavazzana-Calvo M. Gene therapy for primary immunodeficiencies: Part 2. Curr Opin Immunol. 2012;24:585–591. doi: 10.1016/j.coi.2012.07.012. [DOI] [PubMed] [Google Scholar]
- 142.Stein S, Ott MG, Schultze-Strasser S, Jauch A, Burwinkel B, Kinner A, Schmidt M, Kramer A, Schwable J, Glimm H, Koehl U, Preiss C, Ball C, Martin H, Gohring G, Schwarzwaelder K, Hofmann WK, Karakaya K, Tchatchou S, Yang R, Reinecke P, Kuhlcke K, Schlegelberger B, Thrasher AJ, Hoelzer D, Seger R, von Kalle C, Grez M. Genomic instability and myelodysplasia with monosomy 7 consequent to EVI1 activation after gene therapy for chronic granulomatous disease. Nat Med. 2010;16:198–204. doi: 10.1038/nm.2088. [DOI] [PubMed] [Google Scholar]
- 143.Hacein-Bey-Abina S, Pai SY, Gaspar HB, Armant M, Berry CC, Blanche S, Bleesing J, Blondeau J, de Boer H, Buckland KF, Caccavelli L, Cros G, De Oliveira S, Fernandez KS, Guo D, Harris CE, Hopkins G, Lehmann LE, Lim A, London WB, van der Loo JC, Malani N, Male F, Malik P, Marinovic MA, McNicol AM, Moshous D, Neven B, Oleastro M, Picard C, Ritz J, Rivat C, Schambach A, Shaw KL, Sherman EA, Silberstein LE, Six E, Touzot F, Tsytsykova A, Xu-Bayford J, Baum C, Bushman FD, Fischer A, Kohn DB, Filipovich AH, Notarangelo LD, Cavazzana M, Williams DA, Thrasher AJ. A modified gamma-retrovirus vector for X-linked severe combined immunodeficiency. N Engl J Med. 2014;371:1407–1417. doi: 10.1056/NEJMoa1404588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Fischer A, Hacein-Bey-Abina S, Cavazzana-Calvo M. Gene therapy of primary T cell immunodeficiencies. Gene. 2013;525:170–173. doi: 10.1016/j.gene.2013.03.092. [DOI] [PubMed] [Google Scholar]
- 145.Montini E, Cesana D, Schmidt M, Sanvito F, Bartholomae CC, Ranzani M, Benedicenti F, Sergi LS, Ambrosi A, Ponzoni M, Doglioni C, Di Serio C, von Kalle C, Naldini L. The genotoxic potential of retroviral vectors is strongly modulated by vector design and integration site selection in a mouse model of HSC gene therapy. The Journal of clinical investigation. 2009;119:964–975. doi: 10.1172/JCI37630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Felice B, Cattoglio C, Cittaro D, Testa A, Miccio A, Ferrari G, Luzi L, Recchia A, Mavilio F. Transcription factor binding sites are genetic determinants of retroviral integration in the human genome. PloS one. 2009;4:e4571. doi: 10.1371/journal.pone.0004571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Cicalese MP, Aiuti A. Clinical applications of gene therapy for primary immunodeficiencies. Hum Gene Ther. 2015;26:210–219. doi: 10.1089/hum.2015.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Coci EG, Maetzig T, Zychlinski D, Rothe M, Suerth JD, Klein C, Schambach A. Novel self-inactivating vectors for reconstitution of Wiskott-Aldrich syndrome. Curr Gene Ther. 2015;15:245–254. doi: 10.2174/1566523215666150126120327. [DOI] [PubMed] [Google Scholar]
- 149.Biffi A, Montini E, Lorioli L, Cesani M, Fumagalli F, Plati T, Baldoli C, Martino S, Calabria A, Canale S, Benedicenti F, Vallanti G, Biasco L, Leo S, Kabbara N, Zanetti G, Rizzo WB, Mehta NA, Cicalese MP, Casiraghi M, Boelens JJ, Del Carro U, Dow DJ, Schmidt M, Assanelli A, Neduva V, Di Serio C, Stupka E, Gardner J, von Kalle C, Bordignon C, Ciceri F, Rovelli A, Roncarolo MG, Aiuti A, Sessa M, Naldini L. Lentiviral hematopoietic stem cell gene therapy benefits metachromatic leukodystrophy. Science. 2013;341:1233158. doi: 10.1126/science.1233158. [DOI] [PubMed] [Google Scholar]
- 150.Aiuti A, Biasco L, Scaramuzza S, Ferrua F, Cicalese MP, Baricordi C, Dionisio F, Calabria A, Giannelli S, Castiello MC, Bosticardo M, Evangelio C, Assanelli A, Casiraghi M, Di Nunzio S, Callegaro L, Benati C, Rizzardi P, Pellin D, Di Serio C, Schmidt M, Von Kalle C, Gardner J, Mehta N, Neduva V, Dow DJ, Galy A, Miniero R, Finocchi A, Metin A, Banerjee PP, Orange JS, Galimberti S, Valsecchi MG, Biffi A, Montini E, Villa A, Ciceri F, Roncarolo MG, Naldini L. Lentiviral hematopoietic stem cell gene therapy in patients with Wiskott-Aldrich syndrome. Science. 2013;341:1233151. doi: 10.1126/science.1233151. [DOI] [PMC free article] [PubMed] [Google Scholar]