

Abstract
Gordon Holmes syndrome (GHS) is a distinct phenotype of autosomal recessive cerebellar ataxia, characterized by ataxia, dementia, reproductive defects and hypogonadism; it has been recently found to be associated with RNF216 mutation. We performed whole-exome sequencing and filtered the resulting novel variants by the coordinates of the shared autozygome. We identified a novel splicing variant in RNF216 that is likely to abolish the canonical splice site at the junction of exon/intron 13 (NM_207111.3:c.2061G>A). We herein report two patients with GHS caused by a novel RNF216 mutation as the first follow up report on RNF216-related GHS, and show interfamilial variability of phenotype supporting the previously reported RNF216-related cases.
Key words: cerebellar ataxia, hypogonadotropic hypogonadism, RNF216, Gordon Holmes
Introduction
Autosomal recessive cerebellar ataxias are a clinically and genetically heterogeneous group of neurological disorders. A distinct phenotype within this group is cerebellar ataxia associated with hypogonadotropic hypogonadism; this association has been found in two diseases, Gordon Holmes (GHS) and Boucher Neuhauser syndromes.1 GHS (MIM 212840) is a rare phenotype, first described in 1907 by Holmes,2 and comprises adolescence or early adulthood age of onset, isolated idiopathic hypogonadotropic hypogonadism and progressive neurodegenerative manifestations including cognitive impairment, dysarthria, cerebellar ataxia. Recently, mutations in genes involved in ubiquination (RNF216, OTUD4, STUB1) were identified in families with GHS.3,4 Margolin et al. identified mutations in the RNF216 gene either alone or in combination (digenic mutations) with mutations in OTUD4 as a cause of GHS.3 We report two patients with GHS caused by a novel RNF216 mutation as the first follow up report on RNF216-related GHS, and show interfamilial variability of phenotype supporting the previously reported RNF216-related cases. Our study includes 2 out of 9 brothers of a consanguineous Middle Eastern parents (Figure 1), presented with poor development of secondary sexual characteristics and ataxia.
Figure 1.
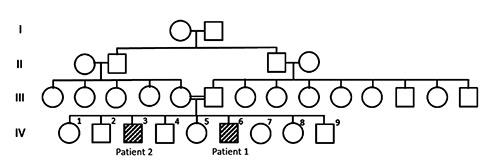
Family pedigree.
Case Report #1
A 26-year-old male, presented at the age of 20 with poor development of secondary sexual characteristics, independent for his activities of daily living (ADLs). His examination revealed poor facial hair growth, gynecomastia hypogenitalism, and mild cerebellar ataxia. No nystagmus or cognitive impairment were noted. His examinations showed low serum testosterone level of 0.12 nmol/L (reference range for his age is 9.9-27.8 nmol), free androgen index 1.4% (14.8-94.8) and low luteinizing hormone level. Evaluation for bone growth using hand X-ray showed that the metacarpal heads and growth plates of the phalanges are not fused yet. The carpal bones are well ossified. The bone age is about 14 years to 15 years, chronological age at time of the study was 20 years (Figure 2). Brain magnetic resonance imaging (MRI) revealed mild cerebellar atrophy, significant subcortical white matter confluent patchy areas of hyperintensity on T2-weighted imaging and fluid-attenuated inversion recovery (FLAIR) imaging, and partial empty sella with no mass lesions identified (Figure 3).
Figure 2.
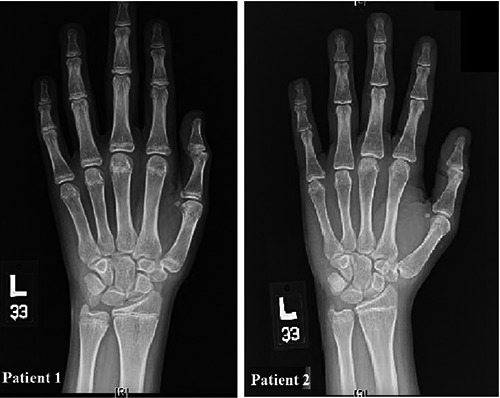
Hand X-ray showed delayed bone growth. Patient 1: The chronological age of this patient is 20 years. The metacarpal heads and growth plates of the phalanges are not fused yet. The carpal bones are well ossified. The bone age of this patient is about 14 years to 15 years. Patient 2: The chronological age for the patient is 25 years. The growth plate of the distal radius and ulna are not fused yet where it normally starts at the age of 17 to 19 and maximally should be fused by the age of 20.
Figure 3.
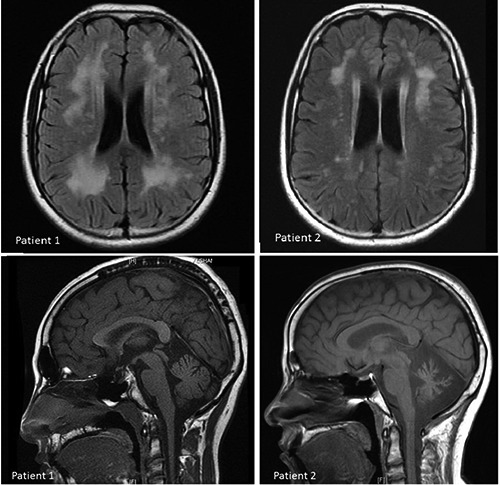
Brain magnetic resonance imaging. Patient 1: mild cerebellar atrophy, mild cortical atrophy, significant bilateral confluent white matter abnormalities and partial empty sella with no mass lesions identified. Patient 2: significant cerebellum atrophy, mild cortical atrophy, mild white matter changes and normal pituitary gland.
Case Report #2
A 31 year old male, developed ataxic gait at the age of 24 with features of poor development of secondary sexual characteristics. His disease progressively worsened with clear cognitive deterioration and need for support in daily activities to the degree that he became totally dependent. His examination revealed no facial hair, hypogenitalism, gynecomastia, cognitive impairment, dysarthria, broken saccadic eye movement, appendicular and truncal cerebellar ataxia, and exaggerated deep tendon reflexes. No nystagmus was noted. His workup showed low luteinizing hormone level, low testosterone level of 0.44 nmol/L (9.9-27.8 nmol), free androgen index 1.5% (14.8-94.8). X-ray study of his hands at the age of 25 years showed that the growth plate of the distal radius and ulna are not fused yet where it normally starts at the age of 17 to 19 and maximally should be fused by the age of 20 (Figure 2). Brain MRI revealed significant cerebellar atrophy, subcortical white matter patchy areas of hyperintensity on T2-weighted imaging and fluid-attenuated inversion recovery (FLAIR) imaging, and normal pituitary gland (Figure 3).
Results
Reproductive endocrine evaluation in both patients revealed only low levels of LH and testosterone, no other pituitary hormones were affected. Cerebrospinal fluid (CSF) analysis, evoked potential and metabolic workup were normal.
Both patients were started on testosterone injection 250 mcg IM every 3 weeks. Both showed good response to treatment regarding secondary sexual characteristics, in the form of facial, axillary and pubic hair growth, and deepening of voice. There was no change in their neurological symptoms with treatment.
Genetic analysis
The shared autozygome of the two individuals was checked against known disease genes but no good candidate was identified (RNF216 had not been identified). Therefore, we proceeded with whole-exome sequencing and filtered the resulting novel variants by the coordinates of the shared autozygome. This resulted in the identified of a novel splicing variant in RNF216 that is likely to abolish the canonical splice site at the junction of exon/intron 13 (NM_207111.3:c.2061G>A). This variant was absent in ~600 ethnically matched exomes, in the 1000 genomes and ExAC browser. Homozygosity for this mutation was confirmed by Sanger sequencing in both patients.5
Discussion
RNF216 (ring finger protein 216) is an E3 ubiquitin ligase involved in regulation of autophagy witch is a cellular process concerned with cellular homeostasis via the degradation of the cell own cytosolic components or protein aggregates.6 Margolin et al. identified digenic homozygous mutations in RNF216 and OTUD4 in three affected siblings of a consanguineous family originally from the Middle East, as well as a compound heterozygous truncating and missense mutations in RNF216 and single mutations in another family.3 Homozygous STUB1 mutation have recently been reported in a consanguineous family with GHS thus expanding the genetic heterogeneity of this condition.7 The phenotype of RNF216-mediated neurodegeneration have been expanded recently to include Huntington-like disorder after identifying of RNF216 mutation in Belgian families presented initially with prominent chorea, behavioral problems, severe dementia and Low gonadotropin serum levels.8 The novel mutation we identified in this report further expands the allelic heterogeneity of this neurodegenerative disease. Our patients presented in their early adulthood with idiopathic hypogonadotropic hypogonadism and no other pituitary abnormalities. We observed in patient 1, normal cognitive functions and only mild cerebellar ataxia consistent with the radiological evidence of mild cerebellar atrophy, although the white matter involvement was significant. In contrast, patient 2 suffered from a more progressive phenotype manifesting as dysarthria, cerebellar ataxia and cognitive impairment with radiological evidence of prominent cerebellar atrophy but paradoxically milder picture of leukodystrophy in comparison with patient 1 (Figure 3). We compared the clinical features of our patients to other patients with GHS caused by RNF216 mutation reported by Margolin et al. and we noticed that they share same age of onset, cerebellar ataxia, lack of nystagmus, cerebellar atrophy, white matter changes and normal pituitary appearance, of note that our first patient who had partial empty sella which had not been reported previously.
Conclusions
In summary, we report two patients with GHS caused by a novel RNF216 mutation and show Interfamilial variability of phenotype supporting the previously reported RNF216-related cases.
References
- 1.Synofzik M, Gonzalez MA, Lourenco CM, et al. PNPLA6 mutations cause Boucher-Neuhauser and Gordon Holmes syndromes as part of a broad neurodegenerative spectrum. Brain 2014;137:69-77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Holmes G. A form of familial degeneration of the cerebellum. Brain 1907;30:466-89. [Google Scholar]
- 3.Margolin DH, Kousi M, Chan YM, et al. Ataxia, dementia, and hypogonadotropism caused by disordered ubiquitination. N Engl J Med 2013;368:1992-2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shi CH, Schisler JC, Rubel CE, et al. Ataxia and hypogonadism caused by the loss of ubiquitin ligase activity of the U box protein CHIP. Hum Mol Genet 2014;23:1013-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Alazami AM, Patel N, Shamseldin HE, et al. Accelerating novel candidate gene discovery in neurogenetic disorders via whole-exome sequencing of prescreened multiplex consanguineous families. Cell Rep 2015;10(148-61. [DOI] [PubMed] [Google Scholar]
- 6.Xu C, Feng K, Zhao X, et al. Regulation of autophagy by E3 ubiquitin ligase RNF216 through BECN1 ubiquitination. Autophagy 2014;10:2239-50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Heimdal K, Sanchez-Guixe M, Aukrust I, et al. STUB1 mutations in autosomal recessive ataxias - evidence for mutation-specific clinical heterogeneity. Orphanet J Rare Dis 2014;9:146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Santens P, Van Damme T, Steyaert W, et al. RNF216 mutations as a novel cause of autosomal recessive Huntington-like disorder. Neurology 2015;84:1760-6. [DOI] [PubMed] [Google Scholar]