

Abstract
Glomerular mesangial cell (GMC) proliferation and death are involved in the pathogenesis of glomerular disorders. The mechanisms that control GMC survival are poorly understood, but may include signal transduction pathways that are modulated by changes in intracellular Ca2+ ([Ca2+]i) concentration. In this study, we investigated whether activation of the canonical transient receptor potential (TRPC) 6 channels and successive [Ca2+]i elevation alter neonatal GMC survival. Hyperforin (HF)-induced TRPC6 channel activation increased [Ca2+]i concentration, inhibited proliferation, and triggered apoptotic cell death in primary neonatal pig GMCs. HF-induced neonatal GMC apoptosis was not associated with oxidative stress. However, HF-induced TRPC6 channel activation stimulated nuclear translocation of the nuclear factor of activated T-cells, cytoplasmic 1 (NFATc1). HF also increased cell death surface receptor Fas ligand (FasL) level and caspase-8 activity in the cells; effects mitigated by [Ca2+]i chelator BAPTA, calcineurin/NFAT inhibitor VIVIT, and TRPC6 channel knockdown. Accordingly, HF-induced neonatal GMC apoptosis was attenuated by BAPTA, VIVIT, Fas blocking antibody, and a caspase-3/7 inhibitor. These findings suggest that TRPC6 channel-dependent [Ca2+]i elevation and the ensuing induction of the calcineurin/NFAT, FasL/Fas, and caspase signaling cascades promote neonatal pig GMC apoptosis.
Glomerular mesangial cell (GMC) proliferation and death are involved in the maintenance of glomerular integrity and pathophysiological mechanisms that underlie kidney dysfunctions1. GMC apoptosis contributes to the resolution of glomerular hypercellularity, a common characteristic of proliferative glomerulonephritis2. Extracellular matrix accumulation, GMC apoptosis, and glomerular sclerosis are associated with proteinuria and hypertension in diabetic nephropathy1,3,4. Mesangial integrity is also altered in childhood nephrotic syndrome5. The mechanisms that regulate GMC survival are unresolved, but may include signal transduction pathways that are modulated by changes in intracellular Ca2+ ([Ca2+]i) concentration6.
Ion channels, including voltage-dependent Ca2+, Ca2+-activated K+, Ca2+-activated Cl−, and transient receptor potential channels are functionally expressed in GMCs7,8. These channels control [Ca2+]i concentration and hence, Ca2+-sensitive cellular events, including contraction, proliferation, and apoptosis7,8. The canonical transient receptor potential (TRPC) 6 has been implicated in glomerular pathophysiology9,10. TRPC6 channel activation alters podocyte survival and actin cytoskeleton dynamics9,10. Focal segmental glomerulosclerosis, an important cause of nephrotic syndrome is associated with TRPC6 channel gain of function mutations and succeeding elevation in TRPC6-dependent Ca2+ influx in podocytes11,12. An increase in Ca2+ influx elicited by angiotensin II-induced TRPC6 channel activation in podocytes has also been reported in diabetic nephropathy13. By contrast, TRPC6 channel expression and angiotensin II-induced [Ca2+]i elevation are downregulated in high glucose-challenged GMCs14. Studies have also shown that TRPC6-mediated [Ca2+]i elevation regulates angiotensin II- and phenylephrine-induced proliferation and chronic hypoxia-induced actin assembly and reorganization in GMCs15,16,17. However, the downstream targets that link TRPC6-dependent Ca2+ signaling to cellular events in GMCs are poorly understood.
The nuclear factor of activated T cells (NFAT) family of transcription factors includes four members whose activations are regulated by calcineurin, a Ca2+-dependent protein phosphatase18,19,20. NFATs control transcription of a variety of genes, including those involved in cell differentiation, growth, and death18,19,20. In cardiac cells and podocytes, NFATs are targets of TRPC6-dependent [Ca2+]i elevation21,22,23,24. However, whether NFATs are downstream effectors of TRPC6 channel activation in GMCs is unclear. Given that NFAT-regulated genes control cell survival18,19,20, we examined whether a direct activation of TRPC6 channels alters neonatal GMC survival via NFAT signaling pathway. Our data suggest that hyperforin (HF)-induced TRPC6 activation inhibits proliferation and promotes apoptosis of primary neonatal pig GMCs. We also show that TRPC6-mediated neonatal GMC apoptosis is associated with an induction of the cell death surface receptor Fas ligand (FasL) and caspase-8 by NFATc1. Collectively, we provide a novel insight into the mechanisms by which TRPC6 channel-dependent [Ca2+]i elevation and sequential activation of the calcineurin/NFAT and FasL/Fas signaling pathways stimulate neonatal pig GMC apoptosis.
Results
HF-induced TRPC6 channel activation elevates [Ca2+]i in neonatal GMCs
TRPC6 channels regulate [Ca2+]i concentration in rat and human GMCs14,15,16,17. To confirm that activation of TRPC6 channels stimulates Ca2+ influx in neonatal pig GMCs, we studied the effect of HF, a TRPC6 channel activator25,26,27,28,29,30 on [Ca2+]i concentration in the cells. First, we examined whether HF stimulates Ca2+ release from intracellular Ca2+ stores in the cells. In the absence of extracellular Ca2+, HF did not alter basal [Ca2+]i in the cells (Fig. 1a). However, successive re-addition of extracellular Ca2+ in the continued presence of HF resulted in an increase in [Ca2+]i by 186.7 ± 3.4 nM (n = 3; Fig. 1a). By contrast, in the absence of extracellular Ca2+, protonophore carbonyl cyanide m-chlorophenyl hydrazine (CCCP) elevated [Ca2+]i by 64.4 ± 9.2 nM (n = 5; Fig. 1a) in the cells. Restoration of extracellular Ca2+ in the presence of CCCP did not stimulate a further increase in [Ca2+]i (29.2 ± 6.0 nM; n = 5; P < 0.05 vs. absence of extracellular Ca2+; Fig. 1a). Next, we knocked down TRPC6 channels and examined the effect of HF on [Ca2+]i concentration in the cells. As shown in Fig. 1b,c, TRPC6 siRNA reduced TRPC6 protein expression by ~66%. HF increased [Ca2+]i by ~197 nM in control cells (Fig. 1d,e). However, TRPC6 channel knockdown significantly reduced HF-induced [Ca2+]i elevation (Fig. 1d,e). These data indicate that HF does not stimulate Ca2+ release from intracellular organelles, but elevates [Ca2+]i by activating membrane-resident TRPC6 channels in neonatal pig GMCs.
Figure 1. HF-induced TRPC6 channel activation elevates [Ca2+]i in neonatal GMCs.

(a) Exemplar traces showing the effects of HF and CCCP on [Ca2+]i concentration in the absence and presence of extracellular Ca2+ in neonatal pig GMCs. The bathing solution was supplemented with EGTA (0.1 mM) and nimodipine (1 μM) to chelate Ca2+ and block L-type Ca2+ channels, respectively. (b,c) Western blot image and mean data (n = 3 each) confirming siRNA-mediated knockdown of TRPC6 channels. (d,e) Traces and mean data indicating that TRPC6 channel knockdown attenuates HF (10 μM)-induced [Ca2+]i elevation in neonatal GMCs (n = 4 each). *P < 0.05 vs. control siRNA.
TRPC6 channel activation inhibits proliferation and promotes apoptosis of neonatal GMC
Changes in [Ca2+]i regulate several intracellular signaling pathways that promote GMC proliferation and death6,31. Data here indicate that HF stimulates [Ca2+]i elevation via TRPC6 channels (Fig. 1). To study the effect of HF on GMC survival, we measured the time-course of proliferation and apoptotic death in the cells. The CellPlayer caspase-3/7 green fluorescence reagent is non-fluorescent in non-apoptotic cells. However, activation of caspase activity during apoptosis increases the fluorescent signal. As shown in Fig. 2a,b, HF inhibited neonatal pig GMC confluence in a time- and concentration-dependent manner. At the same time, HF increased caspase-3/7 activity in the cells (Fig. 2c,d). At 1 and 3 μM, HF inhibited GMC proliferation but did not significantly induce apoptosis (Fig. 2b,d). To confirm that TRPC6 channels mediate HF-induced neonatal pig GMC apoptosis, we measured caspase-3/7 activity in cells transfected with a non-targeting control and TRPC6 channel siRNAs. As shown in Fig. 2e,f, knockdown of TRPC6 channels attenuated HF-induced caspase-3/7 activation in the cells. Together, these data suggest that HF-induced TRPC6 channel activation inhibits proliferation and stimulates apoptosis of neonatal pig GMCs.
Figure 2. TRPC6 channel activation inhibits proliferation and promotes apoptosis of neonatal GMCs.

(a) Phase contrast images (segmentation mask illustrated in goldenrod; HF, 10 μM) and (b) Cell growth curves showing time- and concentration-dependent anti-proliferative effect of HF (n = 4 each) in neonatal pig GMCs. (c) Images (green fluorescent staining of nuclear DNA in apoptotic cells; HF, 10 μM) and (d) Kinetic curves demonstrating that HF (n = 4 each) induces time- and concentration-dependent increase in caspase-3/7 activity in neonatal pig GMCs. (e) Images and (f) Kinetic curves illustrating that TRPC6 channel knockdown attenuates HF (10 μM; n = 4 each)-induced caspase-3/7 activity in neonatal pig GMCs. #P < 0.05 vs. control (DMSO; 12–36 hr); *P < 0.05 vs. control (DMSO; 10–36 hr); $P < 0.05 vs. control (DMSO; 6–36 hr); **P < 0.05 vs. control (DMSO; 10–36 hr); ***P < 0.05 vs. control (DMSO; 2–36 hr); %P < 0.05 vs. control siRNA (20–36 hr); Scale bars = 100 μm.
HF-induced neonatal GMC apoptosis is not associated with oxidative stress
To investigate the role of oxidative stress in HF-induced neonatal GMC apoptosis, we first measured reactive oxygen species (ROS) generation in control and neonatal pig GMCs treated with HF. Confocal microscopy showed that pyocyanin, a superoxide inducer stimulates superoxide generation in neonatal pig GMCs (Fig. 3a). By contrast, neither DMSO (control) nor HF caused significant superoxide generation in the cells (Fig. 3a). Thiobarbituric acid reactive substance (TBARS) assay indicated that unlike oxidant tert-butyl hydroperoxide (TBHP), HF did not increase malondialdehyde (MDA; an end product of lipid peroxidation) in the cells (Fig. 3b). Whereas HF did not alter H2O2 level, TBHP elevated H2O2 production in the cells (Fig. 3c). Next, we examined whether inhibition of oxidative stress mitigates HF-induced neonatal pig GMC apoptosis. As shown in Fig. 3d,e, TBHP promoted rapid apoptosis of the cells. TEMPOL, a cell permeable ROS scavenger essentially abolished TBHP-induced GMC apoptosis (Fig. 3d,e). By contrast, TEMPOL did not alter HF-induced apoptosis of the cells (Fig. 3f,g). These findings indicate that HF-induced neonatal pig GMC apoptosis is not associated with oxidative stress.
Figure 3. HF-induced neonatal GMC apoptosis is not associated with oxidative stress.

(a) Representative confocal images showing that superoxide production was detected in GMCs treated with ROS inducer pyocyanin (10 μM; 8 hr)-, but not in HF (10 μM; 8 hr)-treated cells (images were obtained from 3 coverslips per experimental condition). (b) Mean data illustrating that unlike tert-butyl hydroperoxide (TBHP; 10 μM; 8 hr), HF (10 μM; 8 hr) does not increase MDA production in neonatal pig GMCs (n = 4 each). (c) Mean data showing cellular H2O2 concentration in DMSO (control)-, HF (10 μM; 8 hr)- and TBHP (10 μM; 8 hr)-treated cells (n = 5 each). (d) Images (green fluorescent staining of nuclear DNA in apoptotic cells) and (e) Kinetic curves indicating that TBHP (10 μM; n = 5 each)-induced caspase-3/7 activity in neonatal pig GMCs is inhibited by TEMPOL. (f) Images and (g) Kinetic curves demonstrating that HF (10 μM; n = 4 each)-induced caspase-3/7 activity in neonatal pig GMCs is unaltered by TEMPOL. *P < 0.05 vs. DMSO and HF; #P < 0.05 vs. Control (PBS; 6–18 hr); **P < 0.05 vs. TBHP (6–18 hr); Scale bars = 100 μm.
TRPC6 channel-dependent [Ca2+]i elevation stimulates nuclear translocation of NFATc1 in neonatal GMCs
To examine whether HF-induced [Ca2+]i elevation via TRPC6 channels stimulates nuclear translocation of NFATc1 in neonatal GMCs, we studied the localization of a GFP-NFATc1 cDNA clone that was transiently transfected into neonatal pig GMCs. Unlike DMSO (control)-treated cells, GFP-NFATc1 showed predominant nuclear localization in HF-treated cells (Fig. 4a,b). GFP-NFATc1 nuclear localization in the cells was significantly reduced by BAPTA, an [Ca2+]i chelator and VIVIT, a calcineurin/NFAT inhibitor (Fig. 4a,b). Also, GFP-NFATc1 nuclear localization was attenuated by TRPC6 channel knockdown (Fig. 4a,c). These data suggest that HF-induced [Ca2+]i elevation via TRPC6 channels elicits nuclear translocation of NFATc1 in neonatal pig GMCs.
Figure 4. TRPC6 channel-dependent [Ca2+]i elevation stimulates nuclear translocation of NFATc1 in neonatal GMCs.

(a) Confocal microscopy images, and (b,c) Mean data showing that HF (10 μM; 1 hr) stimulates nuclear translocation of GFP-tagged NFATc1 in neonatal pig GMCs and that BAPTA (1 μM), VIVIT (1 μM), and TRPC6 channel knockdown inhibit HF-induced NFATc1 nuclear translocation (n = 9, DMSO, HF, BAPTA + HF, and VIVIT + HF; n = 10, control siRNA + DMSO; n = 12 control siRNA + HF and TRPC6 siRNA + HF image fields from 3 coverslips per experimental condition). Cells were pretreated with blockers/inhibitors for ~30 min before the addition of HF. *P < 0.05 vs. DMSO; #P < 0.05 vs. HF; **P < 0.05 vs. control siRNA + DMSO; ##P < 0.05 vs. control siRNA + HF; Sacle bar = 20 μm. White arrows indicate NFATc1 nuclear localization.
TRPC6 channel-mediated [Ca2+]i elevation increases FasL level and caspase-8 activity in neonatal GMCs
FasL/Fas interaction at the cell surface is a major contributor to death-inducing signaling pathways in many cell types, including GMCs32,33. FasL/Fas interaction triggers caspase-8 and -3 activation32. Our data indicate that HF-induced TRPC6 activation stimulates NFATc1 nuclear localization in neonatal pig GMCs (Fig. 4). Given that NFATs are involved in transcriptional regulation of FasL expression34,35, we reasoned that TRPC6-depedent NFATc1 activation may elevate membrane FasL level and stimulate caspase-8 activity in neonatal GMCs. Hence, we used a porcine FasL ELISA kit to quantify the level of FasL in neonatal pig GMC lysates. We also measured caspase-8 activity in the cell lysates. As shown in Fig. 5a, FasL level was increased in neonatal pig GMCs treated with HF. HF-induced increase in FasL was diminished in cells pretreated with BAPTA and VIVIT (Fig. 5a). siRNA-mediated knockdown of TRPC6 channels also reduced HF-induced increase in FasL level (Fig. 5b). Similarly, HF treatment elevated caspase-8 activity in neonatal pig GMCs (Fig. 5c). HF-induced elevation in caspase-8 activity was significantly attenuated by BAPTA, VIVIT, and TRPC6 channel knockdown (Fig. 5c,d). To examine the role of FasL/Fas activation in HF-induced caspase-8 activity, we blocked FasL/Fas interaction with a Fas blocking antibody (Fas BA). As shown in Fig. 5c, there was no significant change in caspase-8 activity in GMCs, pretreated with Fas BA before HF application. These findings suggest that TRPC6-dependent [Ca2+]i elevation in neonatal pig GMCs increases FasL and caspase-8 activity via calcineurin/NFAT signaling. Our data also demonstrate that FasL/Fas interaction regulates HF-induced caspase-8 activation in the cells.
Figure 5. TRPC6 channel-mediated [Ca2+]i elevation increases FasL level and caspase-8 activity in neonatal GMCs.

(a) Mean data obtained from porcine FasL ELISA assay demonstrating that HF (10 μM; 8 hr) elevates FasL level in neonatal pig GMCs and that HF-induced FasL is abrogated by BAPTA (1 μM) and VIVIT (1 μM); n = 6 each. (b) Mean data showing that HF (10 μM; 8 hr)-induced FasL elevation in neonatal pig GMCs is reduced by siRNA-mediated TRPC6 channel knockdown; n = 6 each. (c) Mean data obtained from caspase-8 colorimetric assay indicating that HF (10 μM; 8 hr) increases caspase-8 activity in neonatal GMCs, and that BAPTA (1 μM), VIVIT (1 μM), and Fas blocking antibody (Fas BA; 10 μg/mL; n = 4 each) essentially abolish HF-induced increase in caspase-8 activity. (d) Mean data showing that HF (10 μM; 8 hr)-induced caspase-8 activity in neonatal pig GMCs is attenuated by siRNA-mediated TRPC6 channel knockdown (n = 4, control siRNA and control siRNA + HF; n = 5, TRPC6 siRNA + HF). Cells were pretreated with blockers/inhibitors for ~30 min before the addition of HF. Data are expressed as fold change relative to the control. *P < 0.05 vs. DMSO; #P < 0.05 vs. HF; **P < 0.05 vs. control siRNA; ##P < 0.05 vs. control siRNA + HF.
TRPC6 channel-dependent [Ca2+]i elevation and successive activation of the NFAT and FasL/Fas signaling pathways trigger neonatal GMC apoptosis
To confirm that TRPC6 channel-dependent [Ca2+]i elevation and successive activation of the extrinsic apoptotic signaling pathway induce neonatal GMC apoptosis, we measured HF-induced caspase-3/7 activation in neonatal pig GMCs pretreated with DMSO (control), BAPTA, and inhibitors of NFAT, Fas, and caspase-3/7. As shown in Fig. 6, HF-induced caspase-3/7 activation was mitigated by BAPTA, VIVIT, FK506 (a calcineurin inhibitor), Fas BA, and Ac-DEVD-CHO (a caspase-3/7 inhibitor). Together, these data demonstrate that an elevation in [Ca2+]i elicited by HF-induced TRPC6 channel activation, and sequential stimulation of the calcineurin/NFAT, FasL/Fas, and caspase signaling pathways promote neonatal GMC apoptosis (Fig. 7).
Figure 6. TRPC6 channel-dependent [Ca2+]i elevation and successive activation of the NFAT and FasL/Fas signaling pathways trigger neonatal GMC apoptosis.

(a) Representative microplate graphs and (b) Kinetic curves showing HF-induced caspase-3/7 activity in control and BAPTA (1 μM; n = 10)-, FK506 (100 nM; n = 10)-, VIVIT (1 μM; n = 10)-, Ac-DEVD-CHO (50 μM; n = 10)-, and Fas BA (10 μg/mL; n = 5)-treated GMCs. Cells were pretreated with blockers/inhibitors for ~30 min before the addition of HF. *P < 0.05 vs. control (8–36 hr); #P < 0.05 vs. control (10–36 hr).
Figure 7. Schematic diagram illustrating hypothetical mechanisms by which an elevation in [Ca2+]i as a result of TRPC6 channel activation, and subsequent induction of the calcineurin/NFAT, FasL/Fas, and caspase signaling cascades promote neonatal GMC apoptosis.
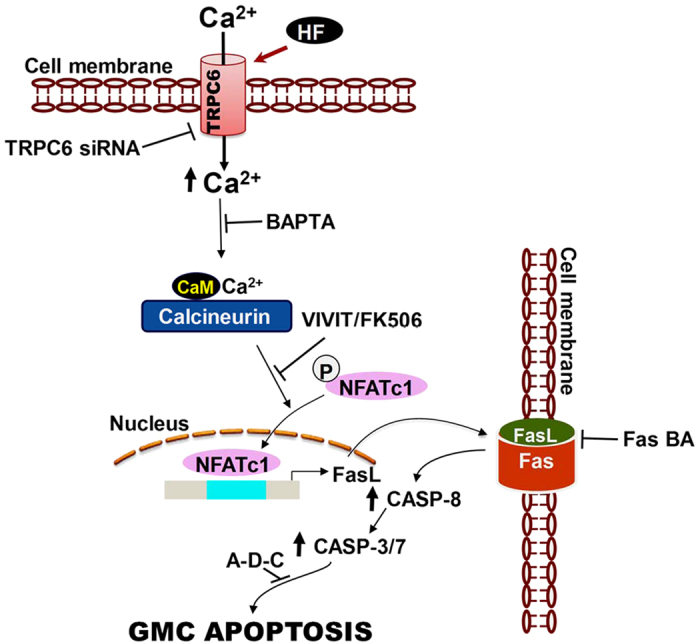
CASP (caspase); Ac-DEVD-CHO (A-D-C); BA (blocking antibody); CaM (calmodulin).
Discussion
The fine-tuning of the expression and function of ion channels and second messengers that maintain [Ca2+]i concentration underlies a variety of signaling events that control cell survival6. HF, a phloroglucinol derivative and selective TRPC6 channel activator elevated [Ca2+]i in neuronal, vascular smooth muscle, and dermal cells and cardiac fibroblast25,26,27,28. HF impedes tumor cell growth by inducing caspase-dependent apoptosis36,37. However, modulation of GMC survival by HF-induced TRPC6 activation has not been previously investigated. In this study, we show for the first time that activation of TRPC6 channels by HF increases [Ca2+]i concentration and induces NFAT-, FasL/Fas-, and caspase-dependent neonatal pig GMC apoptosis.
The control of mesangial [Ca2+]i homeostasis involves TRPC channel-mediated signal transduction pathways8,38. TRPC channels, comprising of seven members (TRPC1-7), can assemble as homo- or hetero-tetramers to form non-selective cation channels that are activated by multiple signaling modalities, including membrane stretch, G-protein-coupled receptor stimulation, and [Ca2+]i store depletion39,40. Previous studies have shown that TRPC6 channels participate in GMC proliferation associated with angiotensin II- and phenylephrine-induced store- and receptor-operated Ca2+ entry in rat and human GMC lines15,16. Here, simultaneous measurement of cell confluence metrics and caspase-3/7 activity in real-time indicated that a direct activation of TRPC6 channels inhibits proliferation and triggers apoptosis of primary neonatal pig GMCs. The conflicting data on the effect of TRPC6 activation on GMC growth may be related to kidney maturation or the different mechanisms of channel activation. Whether [Ca2+]i elevation induces GMC proliferation or death may also depend on downstream cellular signaling pathways that are activated6.
To investigate the mechanisms that mediate HF-induced neonatal GMC apoptosis, we tested the hypothesis that HF promotes oxidative stress in the cells. Our data show that at the concentration that elicited apoptotic cell death, HF did not stimulate lipid peroxidation and superoxide and H2O2 production in the cells. Furthermore, at the concentration that abrogated TBHP-induced apoptosis, ROS scavenger TEMPOL did not alter HF-induced apoptosis of the cells. Hence, oxidative damage is an unlikely mechanism of HF-induced neonatal pig GMC apoptosis.
The NFAT family of transcription factors are important regulators of genes that promote differentiation, proliferation and death in a broad range of cells18,19,20. In resting cells, phosphorylated NFATs are mostly localized in the cytoplasm, and are activated by calcineurin, a Ca2+/calmodulin (CaM)-dependent serine/threonine phosphatase18,19,20. Following an elevation in [Ca2+]i, the Ca2+-CaM complex activates calcineurin. NFAT proteins are then directly dephosphorylated by activated calcineurin, resulting in their nuclear translocation and induction of NFAT-mediated gene transcription18,19,20. In cardiac cells, NFAT stimulation and the resultant pathological hypertrophy are associated with [Ca2+]i increase caused by the activation of TRPC1, TRPC3, TRPC4, and TRPC6 channels22,41,42. In the kidney, TRPC6 channel-mediated activation of NFATc1-dependent gene transcription is involved in glomerulosclerosis23,24. Tumor necrosis factor- and angiotensin II-induced activation of NFATc1 can also increase TRPC6 channel expression in podocytes21,43. Here, we show for the first time that activation of TRPC6 channels in neonatal GMCs stimulates NFATc1 nuclear translocation in a Ca2+-dependent fashion. HF-induced apoptotic cell death was also reduced in neonatal GMCs pretreated with calcineurin/NFAT inhibitors FK506 and VIVIT. These findings indicate that NFAT is a downstream target of TRPC6 channel-dependent [Ca2+]i elevation in GMCs. The NFAT family consists of four Ca2+/calcineurin-responsive members NFATc1, NFATc2, NFATc3, and NFATc418,19,20. Thus, activation of multiple NFAT members by TRPC6-mediated [Ca2+]i elevation in GMCs is possible, but not demonstrated in this study.
Activation of the cell death surface receptor, Fas by its cognate ligand FasL is a major contributor to the extrinsic pathway of programmed cell death32,34,44. FasL/Fas interaction recruits the adapter protein Fas-associated protein with death domain, which in turn employs its death effector domain to induce procaspase-8 and -1045,46,47. Formation of the death-inducing signaling complex as a result of clustering and subsequent autocatalysis of procaspase-8 and -10 triggers apoptosis via downstream executioner caspases, including caspase-3 and -745,47,48. The expression of FasL is controlled by transcription factors such as nuclear factor-kappa B, early growth response protein, and NFAT34,35. NFAT-induced FasL expression has been implicated in Jurkat T, breast cancer, cardiac, neuronal, and Leydig cell apoptosis49,50,51,52, but whether NFAT regulates cell death induced by the FasL/Fas pathway in GMCs was unclear. Data here show that TRPC6 activation elevates membrane FasL level and caspase-8 activity in neonatal pig GMCs, and these effects were essentially abolished by [Ca2+]i chelation, calcineurin/NFAT inhibition, and TRPC6 channel knockdown. These data suggest the involvement of TRPC6-dependent calcineurin/NFAT activation in FasL and caspase-8 induction in neonatal pig GMCs. HF-induced caspase-3/7 activity was also attenuated by [Ca2+]i chelation, calcineurin/NFAT inhibition, and Fas blockade. Together, these findings indicate that TRPC6 channel-mediated [Ca2+]i elevation and subsequent activation of calcineurin/NFAT, FasL/Fas, and caspase cascades result in neonatal pig GMC apoptosis.
Studies have suggested that HF exhibit protonophore activity, and may stimulate Ca2+ release from the mitochondria, independently of TRPC6 channels53,54,55. Protonophores, including CCCP, inhibit mitochondrial Ca2+ uptake and induce mitochondrial Ca2+ release, thereby increasing [Ca2+]i56,57. Here, we show that in neonatal pig GMCs, CCCP elevated [Ca2+]i which was independent of extracellular Ca2+, suggesting Ca2+ release from the intracellular stores. By contrast, HF did not stimulate Ca2+ release from the intracellular stores but induced nimodipine-insensitive and TRPC6-dependent Ca2+ influx in the cells. HF-induced NFATc1 nuclear translocation, FasL and caspase 8 activation, and apoptosis of neonatal pig GMCs were also dependent on TRPC6 channels. These data suggest that TRPC6 channels mediate the effects of HF reported in the present study. Perhaps, the molecular mechanisms underlying the pharmacological actions of HF is dependent on cell type. Also, data in this study were derived from primary neonatal GMCs. Thus, TRPC6 channel-mediated intracellular signaling may differ in adult GMCs.
Whereas the contribution of TRPC6 channels to podocyte pathology is well known, their role in mesangial pathophysiology is poorly understood. Given the involvement of GMC apoptosis in glomerulopathy, we propose that mesangial derangement promoted by TRPC6-dependent [Ca2+]i elevation and subsequent cell death may be of importance in renal dysfunctions, especially in immature kidneys.
Methods
Primary GMC culture
Animal welfare and use in this study were in compliance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals. All experimental protocols were approved by and carried out in accordance with the guidelines and regulations of the Institutional Animal Care and Use Committee of the University of Tennessee Health Science Center (UTHSC). Male newborn pigs (1.5–2 kg) were purchased from Nichols Hog Farm (Olive Branch, MS) and maintained at the UTHSC Comparative Medicine Department. Renal glomeruli were isolated from the pigs (<1-week-old) by serial sieving of renal cortical homogenates using sterile stainless steel meshes. GMCs were then cultured from decapsulated glomeruli as we have previously described58.
Western immunoblotting
Cultured GMCs were homogenized in ice-cold RIPA buffer supplemented with a protease inhibitor cocktail (Thermo Scientific, Rockford, IL). Following isolation, proteins were separated and detected as we have previously described58,59.
siRNA and cDNA transfection
GMC suspensions were electroporated in single cuvettes containing pre-warmed culture medium and a pool of 2 target-specific TRPC6 siRNAs or a non-targeting control siRNA (Sigma-Aldrich, St. Louis, MO). An electrical field to induce siRNA transfection was applied using the ECM 830 square wave electroporation system (Harvard Apparatus, Holliston, MA). Ten minutes after electroporation, the cells were plated for ~72 hours. Western immunoblotting was used to confirm efficient knockdown. GFP-tagged human nuclear factor of activated T-cells, cytoplasmic 1 (NFATc1; OriGene Technologies, Rockville, MD) cDNA clone (400 ng/mL) was transfected into GMCs using the TurboFect transfection reagent (Life Technologies, Grand Island, NY). Transfected cells were maintained in 5% CO2 humidified incubator for 48 h at 37 °C. Images were acquired using a Zeiss laser-scanning confocal microscope.
[Ca2+]i imaging
Changes in [Ca2+]i concentrations were measured in GMCs loaded with fura-2-acetoxymethyl ester using a ratiometric fluorescence system (Ionoptix Corp., Milton, MA, USA) as we have previously described58,59. To examine whether HF and CCCP induce Ca2+ release from intracellular stores, cells were incubated in Ca2+ free modified Krebs’ solution58,59 supplemented with 0.1 mM EGTA (a calcium chelator) and L-type Ca2+ channel blocker nimodipine (1 μM).
Live content microscopy of GMCs
Real-time growth and kinetic quantification of apoptosis in GMCs seeded in microplates (5 × 103 cells/well) were performed using CellPlayer caspase-3/7 reagent (Essen BioScience, Ann Arbor, MI) and the IncuCyte ZOOM live content microscopy system (Essen BioScience). Cell growth and kinetic activation of caspase-3/7 were monitored in real-time and automatically acquired at two-hourly intervals by the IncuCyte interface and software.
Reactive oxygen species (ROS) determination
Lipid peroxidation was evaluated in cell lysates using the TBARS kit (Thermo Scientific, Rockford, IL, Cat. No. KA1381). Superoxide generation was examined using a superoxide fluorogenic kit that specifically detects superoxide levels in live cells (Enzo Life Sciences, Farmingdale, NY; Cat. No. ENZ-51012). The fluorescence generated by this probe was visualized and documented using a Zeiss laser-scanning confocal microscope. H2O2 concentration in the cells was measured using a colorimetric detection kit (Arbor Assay, Ann Arbor, MI, Cat. No. K034-H1).
FasL and caspase-8 assays
GMCs were cultured in 6-well microplates as described above. Membrane FasL concentration and caspase-8 activity were quantified in cell lysates using the porcine FasL ELISA (NeoScientific, Woburn, MA, Cat. No. PF0058) and FLICE/caspase-8 colorimetric assay (BioVision, Inc. Milpitas, CA, Cat. No. K113-25) kits. Data obtained from the cell lysates were normalized to protein concentrations.
Antibodies and chemicals
Mouse monoclonal anti-actin (Cat. No. ab3280) and rabbit polyclonal anti-TRPC6 (Cat. No. ab81111) primary antibodies and HRP-conjugated anti-rabbit and anti-mouse secondary antibodies were purchased from Abcam (Cambridge, MA). DMSO, hyperforin, and tert-Butyl hydroperoxide were purchased from Sigma-Aldrich. BAPTA, FK506, VIVIT, Ac-DEVD-CHO, Fas blocking antibody, ionomycin, pluronic F-127, and Fura-2 AM were purchased from Assay Biotechnology (Sunnyvale, CA), Cell Signaling Technology (Danvers, MA), Cayman Chemical Company (Ann Arbor, MI), Biotium Inc. (Hayward, CA), ProSpec-Tany TechnoGene Ltd (Rehovot, Israel), Cayman Chemical Company, Anaspec (Fremont, CA), and Life Technologies, respectively.
Data analysis
All data are expressed as mean ± standard error of mean (SEM). Statistical significance was determined using Student’s t-tests for paired or unpaired data and analysis of variance with Bonferroni post hoc test for multiple comparisons. A P value < 0.05 was considered significant.
Additional Information
How to cite this article: Soni, H. and Adebiyi, A. TRPC6 channel activation promotes neonatal glomerular mesangial cell apoptosis via calcineurin/NFAT and FasL/Fas signaling pathways. Sci. Rep. 6, 29041; doi: 10.1038/srep29041 (2016).
Acknowledgments
This work was supported by the NIH grant R01DK101668 to A.A.
Footnotes
Author Contributions H.S. and A.A. designed and conducted the experiments and analyzed the data. A.A. conceived the study, directed the project, and wrote the manuscript.
References
- Schlondorff D. & Banas B. The mesangial cell revisited: no cell is an island. J. Am. Soc. Nephrol. 20, 1179–1187 (2009). [DOI] [PubMed] [Google Scholar]
- Baker A. J. et al. Mesangial cell apoptosis: the major mechanism for resolution of glomerular hypercellularity in experimental mesangial proliferative nephritis. J Clin. Invest 94, 2105–2116 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mauer S. M. et al. Structural-functional relationships in diabetic nephropathy. J. Clin. Invest 74, 1143–1155 (1984). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steffes M. W., Osterby R., Chavers B. & Mauer S. M. Mesangial expansion as a central mechanism for loss of kidney function in diabetic patients. Diabetes 38, 1077–1081 (1989). [DOI] [PubMed] [Google Scholar]
- Habib R. Nephrotic syndrome in the 1st year of life. Pediatr. Nephrol. 7, 347–353 (1993). [DOI] [PubMed] [Google Scholar]
- Saleh H., Schlatter E., Lang D., Pauels H. G. & Heidenreich S. Regulation of mesangial cell apoptosis and proliferation by intracellular Ca2+ signals. Kidney Int. 58, 1876–1884 (2000). [DOI] [PubMed] [Google Scholar]
- Stockand J. D. & Sansom S. C. Glomerular mesangial cells: electrophysiology and regulation of contraction. Physiol Rev. 78, 723–744 (1998). [DOI] [PubMed] [Google Scholar]
- Ma R., Pluznick J. L. & Sansom S. C. Ion channels in mesangial cells: function, malfunction, or fiction. Physiology. (Bethesda.) 20, 102–111 (2005). [DOI] [PubMed] [Google Scholar]
- Dryer S. E. & Reiser J. TRPC6 channels and their binding partners in podocytes: role in glomerular filtration and pathophysiology. Am. J Physiol Renal Physiol 299, F689–F701 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlondorff J. S. & Pollak M. R. TRPC6 in glomerular health and disease: what we know and what we believe. Semin. Cell Dev. Biol. 17, 667–674 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiser J. et al. TRPC6 is a glomerular slit diaphragm-associated channel required for normal renal function. Nat. Genet. 37, 739–744 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winn M. P. et al. A mutation in the TRPC6 cation channel causes familial focal segmental glomerulosclerosis. Science 308, 1801–1804 (2005). [DOI] [PubMed] [Google Scholar]
- Ilatovskaya D. V. et al. Podocyte injury in diabetic nephropathy: implications of angiotensin II-dependent activation of TRPC channels. Sci. Rep 5, 17637 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham S. et al. Downregulation of TRPC6 protein expression by high glucose, a possible mechanism for the impaired Ca2+ signaling in glomerular mesangial cells in diabetes. Am. J. Physiol Renal Physiol 293, F1381–F1390 (2007). [DOI] [PubMed] [Google Scholar]
- Qiu G. & Ji Z. AngII-induced glomerular mesangial cell proliferation inhibited by losartan via changes in intracellular calcium ion concentration. Clin. Exp. Med 14, 169–176 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong F. et al. Alpha1-Adrenergic Receptor Activation Stimulates Calcium Entry and Proliferation via TRPC6 Channels in Cultured Human Mesangial Cells. Cell Physiol Biochem. 36, 1928–1938 (2015). [DOI] [PubMed] [Google Scholar]
- Liao C. et al. The upregulation of TRPC6 contributes to Ca2+ signaling and actin assembly in human mesangial cells after chronic hypoxia. Biochem. Biophys. Res. Commun. 421, 750–756 (2012). [DOI] [PubMed] [Google Scholar]
- Rao A., Luo C. & Hogan P. G. Transcription factors of the NFAT family: regulation and function. Annu. Rev. Immunol. 15, 707–747 (1997). [DOI] [PubMed] [Google Scholar]
- Horsley V. & Pavlath G. K. NFAT: ubiquitous regulator of cell differentiation and adaptation. J Cell Biol. 156, 771–774 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Im S. H. & Rao A. Activation and deactivation of gene expression by Ca2+/calcineurin-NFAT-mediated signaling. Mol. Cells 18, 1–9 (2004). [PubMed] [Google Scholar]
- Nijenhuis T. et al. Angiotensin II contributes to podocyte injury by increasing TRPC6 expression via an NFAT-mediated positive feedback signaling pathway. Am. J Pathol. 179, 1719–1732 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuwahara K. et al. TRPC6 fulfills a calcineurin signaling circuit during pathologic cardiac remodeling. J Clin. Invest 116, 3114–3126 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlondorff J., Del C. D., Carrasquillo R., Lacey V. & Pollak M. R. TRPC6 mutations associated with focal segmental glomerulosclerosis cause constitutive activation of NFAT-dependent transcription. Am. J Physiol Cell Physiol 296, C558–C569 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y. et al. Activation of NFAT signaling in podocytes causes glomerulosclerosis. J Am. Soc. Nephrol. 21, 1657–1666 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding Y. et al. Reactive oxygen species-mediated TRPC6 protein activation in vascular myocytes, a mechanism for vasoconstrictor-regulated vascular tone. J Biol. Chem. 286, 31799–31809 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leuner K. et al. Hyperforin–a key constituent of St. John’s wort specifically activates TRPC6 channels. FASEB J. 21, 4101–4111 (2007). [DOI] [PubMed] [Google Scholar]
- Muller M. et al. Specific TRPC6 channel activation, a novel approach to stimulate keratinocyte differentiation. J Biol. Chem. 283, 33942–33954 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikeda K. et al. Roles of transient receptor potential canonical (TRPC) channels and reverse-mode Na+/Ca2+ exchanger on cell proliferation in human cardiac fibroblasts: effects of transforming growth factor beta1. Cell Calcium 54, 213–225 (2013). [DOI] [PubMed] [Google Scholar]
- Leuner K. et al. Hyperforin modulates dendritic spine morphology in hippocampal pyramidal neurons by activating Ca2+-permeable TRPC6 channels. Hippocampus 23, 40–52 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leuner K. et al. Simple 2,4-diacylphloroglucinols as classic transient receptor potential-6 activators–identification of a novel pharmacophore. Mol. Pharmacol. 77, 368–377 (2010). [DOI] [PubMed] [Google Scholar]
- Whiteside C., Munk S., Zhou X., Miralem T. & Templeton D. M. Chelation of intracellular calcium prevents mesangial cell proliferative responsiveness. J Am. Soc. Nephrol. 9, 14–25 (1998). [DOI] [PubMed] [Google Scholar]
- Nagata S. Fas ligand-induced apoptosis. Annu. Rev. Genet. 33, 29–55 (1999). [DOI] [PubMed] [Google Scholar]
- Boyle J. J. Human macrophages kill human mesangial cells by Fas-L-induced apoptosis when triggered by antibody via CD16. Clin. Exp. Immunol. 137, 529–537 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinkoski M. J. & Green D. R. Fas ligand, death gene. Cell Death. Differ. 6, 1174–1181 (1999). [DOI] [PubMed] [Google Scholar]
- Kavurma M. M. & Khachigian L. M. Signaling and transcriptional control of Fas ligand gene expression. Cell Death. Differ. 10, 36–44 (2003). [DOI] [PubMed] [Google Scholar]
- Merhi F. et al. Hyperforin inhibits Akt1 kinase activity and promotes caspase-mediated apoptosis involving Bad and Noxa activation in human myeloid tumor cells. PLoS. One. 6, e25963 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hostanska K., Reichling J., Bommer S., Weber M. & Saller R. Hyperforin a constituent of St John’s wort (Hypericum perforatum L.) extract induces apoptosis by triggering activation of caspases and with hypericin synergistically exerts cytotoxicity towards human malignant cell lines. Eur. J Pharm. Biopharm. 56, 121–132 (2003). [DOI] [PubMed] [Google Scholar]
- Graham S. et al. Abundance of TRPC6 protein in glomerular mesangial cells is decreased by ROS and PKC in diabetes. Am. J. Physiol Cell Physiol 301, C304–C315 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clapham D. E. TRP channels as cellular sensors. Nature 426, 517–524 (2003). [DOI] [PubMed] [Google Scholar]
- Vazquez G., Wedel B. J., Aziz O., Trebak M. & Putney J. W. Jr. The mammalian TRPC cation channels. Biochim. Biophys. Acta 1742, 21–36 (2004). [DOI] [PubMed] [Google Scholar]
- Ohba T. et al. Upregulation of TRPC1 in the development of cardiac hypertrophy. J Mol. Cell Cardiol. 42, 498–507 (2007). [DOI] [PubMed] [Google Scholar]
- Makarewich C. A. et al. Transient receptor potential channels contribute to pathological structural and functional remodeling after myocardial infarction. Circ. Res. 115, 567–580 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abkhezr M. et al. Pleiotropic signaling evoked by tumor necrosis factor in podocytes. Am. J Physiol Renal Physiol 309, F98–108 (2015). [DOI] [PubMed] [Google Scholar]
- Nagata S. & Golstein P. The Fas death factor. Science 267, 1449–1456 (1995). [DOI] [PubMed] [Google Scholar]
- Kischkel F. C. et al. Death receptor recruitment of endogenous caspase-10 and apoptosis initiation in the absence of caspase-8. J Biol. Chem. 276, 46639–46646 (2001). [DOI] [PubMed] [Google Scholar]
- Boldin M. P. et al. A novel protein that interacts with the death domain of Fas/APO1 contains a sequence motif related to the death domain. J Biol. Chem. 270, 7795–7798 (1995). [DOI] [PubMed] [Google Scholar]
- Chinnaiyan A. M., O’Rourke K., Tewari M. & Dixit V. M. FADD, a novel death domain-containing protein, interacts with the death domain of Fas and initiates apoptosis. Cell 81, 505–512 (1995). [DOI] [PubMed] [Google Scholar]
- Kischkel F. C. et al. Cytotoxicity-dependent APO-1 (Fas/CD95)-associated proteins form a death-inducing signaling complex (DISC) with the receptor. EMBO J 14, 5579–5588 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalivendi S. V. et al. Doxorubicin activates nuclear factor of activated T-lymphocytes and Fas ligand transcription: role of mitochondrial reactive oxygen species and calcium. Biochem. J 389, 527–539 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chai W. R., Wang Q. & Gao H. B. NFAT2 is implicated in corticosterone-induced rat Leydig cell apoptosis. Asian J Androl 9, 623–633 (2007). [DOI] [PubMed] [Google Scholar]
- Srivastava R. K., Sasaki C. Y., Hardwick J. M. & Longo D. L. Bcl-2-mediated drug resistance: inhibition of apoptosis by blocking nuclear factor of activated T lymphocytes (NFAT)-induced Fas ligand transcription. J Exp. Med 190, 253–265 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jayanthi S. et al. Calcineurin/NFAT-induced up-regulation of the Fas ligand/Fas death pathway is involved in methamphetamine-induced neuronal apoptosis. Proc. Natl. Acad. Sci. USA 102, 868–873 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tu P., Gibon J. & Bouron A. The TRPC6 channel activator hyperforin induces the release of zinc and calcium from mitochondria. J Neurochem. 112, 204–213 (2010). [DOI] [PubMed] [Google Scholar]
- Friedland K. & Harteneck C. Hyperforin: To Be or Not to Be an Activator of TRPC(6). Rev. Physiol Biochem. Pharmacol. 169, 1–24 (2015). [DOI] [PubMed] [Google Scholar]
- Sell T. S., Belkacemi T., Flockerzi V. & Beck A. Protonophore properties of hyperforin are essential for its pharmacological activity. Sci. Rep 4, 7500 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babcock D. F., Herrington J., Goodwin P. C., Park Y. B. & Hille B. Mitochondrial participation in the intracellular Ca2+ network. J Cell Biol. 136, 833–844 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh C. et al. Modulation of calcium signalling by mitochondria. Biochim. Biophys. Acta 1787, 1374–1382 (2009). [DOI] [PubMed] [Google Scholar]
- Adebiyi A., Soni H., John T. A. & Yang F. Lipid rafts are required for signal transduction by angiotensin II receptor type 1 in neonatal glomerular mesangial cells. Exp. Cell Res. 324, 92–104 (2014). [DOI] [PubMed] [Google Scholar]
- Adebiyi A. RGS2 regulates urotensin II-induced intracellular Ca2+ elevation and contraction in glomerular mesangial cells. J. Cell Physiol 229, 502–511 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]