

Abstract
It has been demonstrated that Src could modulate NMDA receptor, and PAR1 could also affect NMDAR signaling. However, whether PAR1 could regulate NMDAR through Src under ICH has not yet been investigated. In this study, we demonstrated the role of Src-PSD95-GluN2A signaling cascades in rat ICH model and in vitro thrombin challenged model. Using the PAR1 agonist SFLLR, antagonist RLLFS and Src inhibitor PP2, electrophysiological analysis showed that PAR1 regulated NMDA-induced whole-cell currents (INMDA) though Src in primary cultured neurons. Both in vivo and in vitro results showed the elevated phosphorylation of tyrosine in Src and GluN2A and enhanced interaction of the Src-PSD95-GluN2A under model conditions. Treatment with the PAR1 antagonist RLLFS, AS-PSD95 (Antisense oligonucleotide against PSD95) and Src inhibitor PP2 inhibited the interaction among Src-PSD95-GluN2A, and p-Src, p-GluN2A. Co-application of SFLLR and AS-PSD95, PP2, or MK801 (NMDAR inhibitor) abolished the effect of SF. In conclusion, our results demonstrated that activated thrombin receptor PAR1 induced Src activation, enhanced the interaction among Src-PSD95-GluN2A signaling modules, and up-regulated GluN2A phosphorylation after ICH injury. Elucidation of such signaling cascades would possibly provide novel targets for ICH treatment.
Thrombin, a serine protease generated by the cleavage of prothrombin, is an essential component of the coagulation cascade. It is produced in the brain either immediately after cerebral hemorrhage or after the blood-brain barrier (BBB) breakdown that occurs following brain injuries1. It has been demonstrated that the role of thrombin acting on its receptors in ischemic, hemorrhagic and traumatic brain injury is concentration-dependent. Increasing evidence indicates that high concentrations (more than 500 nM) of thrombin within the brain parenchyma can be deleterious2,3. For example, thrombin can induce brain edema, thrombosis and inflammation by directly acting on its receptors, protease activated receptors (PARs)4,5. PARs are a subfamily of G protein-coupled receptors (GPCRs) with four members, namely, PAR1, PAR2, PAR3 and PAR4. PAR1 is highly expressed in many different cell types and transmits signals by coupling with Gαq/11, Gαi/O and Gα12/13 proteins6,7. Under physiological condition, PAR1 is detected in the mammalian brain cortex, basal ganglia, and striatum and nucleus accumbens neurons. PAR1 plays an important role in astrocyte proliferation, stimulus induced long-term potentiation (LTP) and nerve growth factor (NGF) secretion8,9,10. Activation of PAR1 by thrombin at high concentrations contributes to the transient focal cerebral ischemia injury11. In addition, PAR1 deficient mice had a 3.1-fold reduction in infarct volume after transient focal cerebral ischemia. Similarly, intracerebro-ventricular injection of PAR1 antagonist BMS-200261 reduced infarct volume by 2.7-fold11. These observations suggest that PAR1 activation exacerbates neuronal damage in many brain injury models in a manner that is dependent on thrombin overproduction12.
The N-methyl-D-aspartic acid receptor (NMDAR) is a ligand-gated ion channel receptor composed of two types of subunits, GluN1 and GluN2 subunits13. NMDAR plays a critical role in many pathological processes of brain injuries and has been implicated in neurological diseases such as stroke, traumatic brain injury, dementia and schizophrenia14. Under ischemia and reperfusion (I/R) condition, excessive glutamate release causes influx of Na+ and Ca2+ through the NMDAR15,16. Activation of Src kinases by influx of Ca2+ mediates GluN2A and GluN2B tyrosine phosphorylation17, up-regulates NMDAR activity, feedback-increases the influx of Ca2+ and aggravates the brain damage18,19. Previously we reported that cerebral I/R could induce Src autophosphorylation and activation, resulting in tyrosine phosphorylation and activation of NMDA receptor, and that postsynaptic density protein 95 (PSD95) participated in the formation of Src-PSD95-GluN2A signaling modules19,20. Although Src is known to bind to PSD95 and mediate phosphorylation of NMDA receptors21, how Src participates in NMDAR activation in ICH model is unclear yet.
It has been reported that PAR1 activation led to cell depolarization and potentiation of synaptically activated NMDAR function in dentate granule neurons of hippocampus12. Indeed, Shigetomi et al. found that the NMDAR was involved in PAR1 mediated Ca2+ elevation via PAR1-NMDA interplay in astrocytes22. Numerous studies suggest that PAR1 can enhance neuronal excitability associated with NMDAR mediated neuronal damage and NMDAR activation is necessary for thrombin/PAR1-induced neurodegenerative effects under pathological conditions, such as cerebral ischemia and hemorrhage22,23,24,25. The route from PAR1 activation to NMDA receptor current potentiation has been described in ischemia by the Traynelis group23, however, the underlying mechanism by which PAR1 affects NMDAR and induces brain injuries under ICH condition remains to be addressed.
In this study, we tested our hypothesis that activation of PAR1 by thrombin during ICH enhanced interaction of Src-PSD95-GluN2A signaling modules and increased the phosphorylation of Src and NMDAR, which eventually led to brain injuries. To determine the mechanism of PAR1 in the regulation of NMDAR during ICH, we used PAR1 agonist SFLLR and antagonist RLLFS, antisense oligonucleotide against PSD95 AS-ODNs, Src antagonist PP2 or NMDAR antagonist MK-801. We expect that elucidation of PAR1 signaling pathway in ICH would provide potential drug targets and strategies for treatment of hemorrhagic brain injuries.
Experimental Procedures
Antibodies, peptides and reagents
GluN2A (AB1555, 1:1000) was purchased from Millipore (Billerica, MA, USA). Goat anti-rabbit IgG-AP and goat anti-mouse IgG-AP antibodies were purchased from Sigma-Aldrich Corp. (St. Louis, MO, USA) while antibodies for Src (ab7950, 1:500), phosphotyrosine (ab9319, 1:500) and anti-β-actin antibody (ab32572) were purchased from Abcam Biotechnology (Cambridge, UK). Anti-Phospho-Src Family (Tyr416) (2101S, 1:1000), anti-PSD95 (2507S, 1:1000) were obtained from Cell Signaling Biotechnology (Beverly, MA, USA). Peptides including SFLLR (SF), RLLFS (RL), AS and MS- PSD95 were obtained from Shanghai Biotechnology Company (Shanghai, China). The sequences of antisense and missense ODNs against PSD95 are as follows: AS, 5′-TGTGATCTCCTCATACTC-3′; and MS, 5′-AAGCCCTTGTTCCCATTT-3′.
Experimental cerebral hemorrhagic model
One hundred and fifty adult male Sprague-Dawley (SD) rats weighing 200–250 g were obtained from Shanghai Experimental Animal Center (Shanghai, China). The experimental procedures and protocols were approved by the Animal Ethics Committee of Xuzhou Medical College (Approval ID: SCXK (SU) 2010-0003). All experiments were conducted in compliance with institutional guidelines. The animals were randomly divided into control group, in which rats did not receive any treatment; ICH group, in which rats were subjected to ICH; dissolvent group, in which rats were intraperitoneally or intracerebro-ventricularly given corresponding solvent, H2O, 0.9% NaCl, 10 mM TE buffer or 1% DMSO; drug treatment group, in which rats subjected to ICH were treated with SF, RL, SF + RL, MS, AS, SF + AS, PP2, SF + PP2, MK801 respectively. ICH model was developed as previously described26. Briefly, rats were intraperitoneal injected with 10% chloral hydrate (300 mg/kg) for anesthesia. After anesthesia induction, rats were fixed at prone position on the stereotaxic apparatus. Subsequently, 50 μl of autologous whole blood was injected into the right caudate nucleus (striatum coordinates: 1.0 mm anterior, 3.0 mm lateral and 5.0 mm ventral, with respect to Bregma). Rats were allowed for free access to food and water when the incision was sutured. To confirm the success of the model, nerve functional scores tests were conducted according to Zea Longa et al.27. The nerve function was scored on a five-point scale: a score of 0 indicated no neurologic deficit, a score of 1 (failure to extend left forepaw fully) a mild focal neurologic deficit, a score of 2 (circling to the left) a moderate focal neurologic deficit, and a score of 3 (falling to the left) a severe focal deficit; rats with a score of 4 did not walk spontaneously and had a depressed level of consciousness. Level 2 to 4 were considered as successful model.
SFLLR (SF, 300 μM) was administered 30 min before hemorrhage and RLLFS (100 μM) was administered 30 min after hemorrhage (at the dose of 10 μl) in rats via intracerebroventricularly injection. One hundred micrograms of specific antisense ODNs against PSD95 (AS-PSD95) in 10 μl TE buffer (10 mM Tris-HCl, pH 8.0, 1 mM EDTA) were administrated to the rats every 24 hr for 3 days through cerebral ventricular injection before ICH (IV, anteroposterior, 0.8 mm; lateral, 1.5 mm; depth, 3.5 mm from bregma). The same dose of missense ODNs (MS-PSD95) and vehicle were used as control. MK-801 (3.0 mg/kg) was given twice injection 30 min and 60 min before hemorrhage by IP. Ten microliters of PP2 (15 μg dissolved in DMSO to make 10% solution) was injected 30 min before hemorrhage by IV. In parallel experiments, solvent was used as control.
Sample preparation
After 3 h of ICH, the rats were decapitated and the brain caudate putamen was obtained. The brain tissues were added to 1.0 ml of homogenization buffer and homogenized using Teflon homogenizer (10 s × 6 times). Following the addition of 1.0 ml homogenization buffer, the samples were centrifuged at 2000 g at 4 °C for 20 min, and the supernatant was collected (mainly the caudate putamen cytoplasmic portion). Protein concentration was determined by the Lowry method.
Immunoprecipitation and immunoblotting
For immunoprecipitation, tissue homogenates (400 μg of protein) were diluted four-fold with HEPES buffer containing 50 mM HEPES (pH 7.4), 150 mM NaCl, 10% glycerol, 1% Triton X-100, and 1 mM each of EGTA, EDTA, PMSF and Na3VO4. Samples (from ICH models) were pre-incubated for 1 h with 20 μl protein A/G and then centrifuged to remove any adhered protein. The supernatants were incubated with 2–5 μg proper antibody for 4 h at 4 °C. After the addition of proteinA/G-sepharose, the mixture was incubated at 4 °C for 2 h. After being washed with HEPES buffer, samples were eluted by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) loading buffer and boiled at 100 °C for 5 min. Supernatants were used for western blot analysis.
Western blot analysis was carried out on 10% SDS-PAGE gel. Proteins (100 μg) were electro-transferred onto nitrocellulose filter (NC, pore size, 0.45 μm). After blocking for 3 h in phosphate buffer (PBS) with 0.1% Tween 20 (PBST) and 3% BSA, the membranes were incubated overnight with primary antibody in PBST containing 3% BSA. Detection was carried out by using proper alkaline phosphatase conjugated IgG (1:20,000) and developed with NBT/BCIP assay kit (Promega, Madison, USA).
Primary neuronal cell culture
Primary cultured hippocampal neurons were prepared from 18-day-old SD rat embryos. In brief, hippocampi were isolated in ice-cold high-glucose Dulbecco’s modified Eagle medium (Gibco, Grand Island, NY). Hippocampal cells were dissociated by trypsinization [0.25% (wt/vol) trypsin and 0.02% (wt/vol) EDTA in Ca2+ and Mg2+ -free Hank’s balanced salt solution] at 37 °C for 15 minutes, followed by gentle triturating in plating medium (high-glucose Dulbecco’s modified Eagle medium supplemented with 10% fetal bovine serum and 10% horse serum; Gibco). Cells were seeded onto poly-L-lysine (Sigma)-coated wells or cover slips at a density of 0.5 × 105 cells/cm2 and incubated at 37 °C in 5% CO2 atmosphere. After 24 hours, culture medium was replaced with neurobasal medium supplemented with B-27 (Gibco) and 0.5 mM glutamine. Medium was half-replaced twice every week.
Patch Clamp Recording
All experiments were carried out at room temperature (22 ~ 25 °C). Electrophysiological recording was performed in the conventional whole-cell patch-clamp recording configuration under voltage-clamp conditions as previous reported28. Briefly, glass pipettes were pulled from 1.5 mm glass capillaries on a two-stage puller (PP-830, Narishige). The resistance between the recording electrode filled with pipette solution (in mM: CsCl 140, TEA 20, Hepes 10, EGTA 5, MgCl2 1, Mg-ATP 4, pH7.2) and the reference electrode was 3–5 MΩ in standard extracellular solution (SS, in mM: NaCl 140, glucose 10, Hepes 10, KCl 5, CaCl2 2, MgCl2 1, pH7.4). Membrane currents were detected with AxonPatch 700B (Axon Instruments, Foster City, CA, USA), filtered at 2 kHz, sampled and analyzed using a DigiData 1440A interface and a computer with the pClamp 10 system (Axon Instruments). The series resistance compensation was automated. The membrane potential was held at −60 mV throughout the experiment. NMDA receptor-mediated whole-cell currents (INMDA) was detected by application of 100 μmol/L NMDA dissolved in Mg2+ -free SS following 5 s application of Mg2+ -free SS. Src kinase inhibitor PP2 and its non-active analog PP3 were administered in the pipette solution. The INMDA were detected at least 5 min after the cells were patched to allow the diffusion of PP2 and PP3. The chemical reagents used in patch-clamp recording were obtained from Sigma-Aldrich except otherwise indicated.
COS7 Cell Culture, transfection and treatments
COS7 Cells derived from monkey kidney were grown in Dulbecco’s modified Eagle medium (DMEM; Gibco BRL, Grand Island, NY) supplemented with 10% fetal calf serum (37 °C, 5% CO2) in 5 cm × 5 cm or 5 cm × 7 cm culture flasks in a humidified incubator. Cells were transfected with the indicated plasmid vectors (PSD95, 6 μg; 6 μg GluN2A; or 3 μg Src) at the time of 90–95% confluent by using Lipofectamine™ 2000. COS7 cells were incubated for forty eight hours and were stimulated with thrombin (5 U/ml) for 1 hour to mimic the ICH model followed by immediate harvest. PP2 (10 μmol/L) or control buffer was added to cells 30 minute before thrombin treatment, while SF (1 μmol/L) was added to cells 60 minute before thrombin treatment.
Statistical Analysis Statistical
Statistical analysis was carried out by using one-way ANOVA followed by Newman-Keuls test. P < 0.05 was considered as statistically significant. Data are expressed as Mean ± S.D.
Results
Activated PAR1 up-regulated NMDA receptor ion channel activity via Src kinase in primary cell cultures
To investigate the interactions of PAR1 with NMDA receptors, NMDA receptor-mediated whole-cell currents (INMDA) were recorded by patch-clamp recording on 12- to 16-day cultured rat hippocampal neurons. As shown in Fig. 1A–F, 5 min incubation of PAR1 agonist SF (20 μmol/L) could lead to an increase in INMDA, while incubation of PAR1 antagonist RL led to a decrease of INMDA. To further investigate the signaling mechanism of the upregulation effects of SF on INMDA, 2 μmol/L Src family kinase inhibitor PP2 and its non-active analog PP3 were used through intracellular administration. INMDA was detected at least 5 min after the cells were patched. As shown in Fig. 1C,D, intra-pipette administration of PP2 or PP3 did not affect the basal INMDA. However, the upregulation effects of SF on INMDA were blocked by PP2.
Figure 1. PAR1 up-regulated NMDA receptor ion channel activity via Src kinase.

(A,B) PAR1 agonist SF and antagonist RL affected NMDA receptor-mediated whole-cell currents (INMDA) on 12- to 16 day cultured rat hippocampal neurons. (C) Statistics for INMDA after stimulation for 5 min by SF or RL (aP < 0.05 vs. group of pre-Control, bP < 0.05 vs group of SF, n = 6 from 3 cultures). (D,E) The effects of Src family kinase inhibitor PP2 and its non-active analog PP3 on INMDA. (F) Statistic results of effects of PP2 and PP3 on SF upregulation effects on INMDA (aP < 0.05 vs. group of SS + PP2, bP < 0.05 vs. group of SF + Vehicle, n = 6 from 3 cultures).
PAR1 activated Src-mediated phosphorylation of NMDAR after ICH
To study PAR1 induced phosphorylation of NMDAR via Src in ICH model, rats were treated with the PAR1 agonist SFRRL, antagonist RLLSF or Src inhibitor PP2. Results showed that phosphorylation of Src and GluN2A were significantly increased in the hemorrhagic brain compared with the control rats. RLLSF reduced the p-Src and p-GluN2A compared with the ICH, SF and H2O groups (H2O, water, the solvent of SF or RL). To further prove PAR1 regulation of the NMDAR function, co-application of RL and SF, it was found that RL + SF neutralized the effect of SF on p-Src and p-GluN2A (Fig. 2A,C). These results indicated the important role of PAR1 in the regulation of Src and NMDAR phosphorylation after ICH. To further demonstrate that PAR1 regulates the NMDAR function though Src, we treated the rats with Src inhibitor PP2. Results showed that PP2 treatment significantly reduced the phosphorylation of Src and GluN2A when compared with either ICH or SF or DMSO (DMSO, the solvent of PP2) groups. Additionally, we also found that when Src inhibitor PP2 and PAR1 agonist SF were combined, the inhibitory role of PP2 was abolished and phosphorylation of Src and NR2A was enhanced (Fig. 2B,C). The total protein of GluN2A and Src remained unchanged.
Figure 2. PAR1 activated Src-mediated phosphorylation of NMDAR after rat ICH in caudate putamen (CPu).

(A) p-Src, c-Src, GluN2A were determined by western blot analysis and p-GluN2A was examined by immunoprecipitation with anti-p-Y antibody followed by immunoblotting with antibody against NR2A in control, ICH, H2O, RL, SF + RL and SF groups. β-actin was used as a loading control. (B) Similar to A, but in control, ICH, SF, PP2, SF + RL and DMSO groups. (C) Statistic the results of p-Src and p-GluN2A in each experimental group. The results are shown as mean ± S.D (n = 5). aP < 0.05 vs. Control group, bP < 0.05 vs. ICH 3 h treatment group, cP < 0.05 vs. RL-treatment group.
In vitro, treatment of COS7 cells with thrombin (5 U/ml) enhanced the phosphorylation of Src and GluN2A when compared with the control group. PP2 treatment significantly reduced the p-Src and p-GluN2A when compared with thrombin or DMSO (the solvent of PP2) groups. When Src inhibitor PP2 and PAR1 agonist SF were combined, the inhibitory role of PP2 was reversed. p-Src and p-GluN2A were enhanced in PP2 + SF groups when compared with the PP2 group. The total protein of GluN2A and Src remained unchanged (Fig. 3A,B). Both in vivo and in vitro results suggested that PAR1 mediated phosphorylation of GluN2A via Src.
Figure 3. Thrombin enhanced the interaction of PSD95 and GluN2A, as well as phosphorylation of Src and GluN2A in COS7 cells.
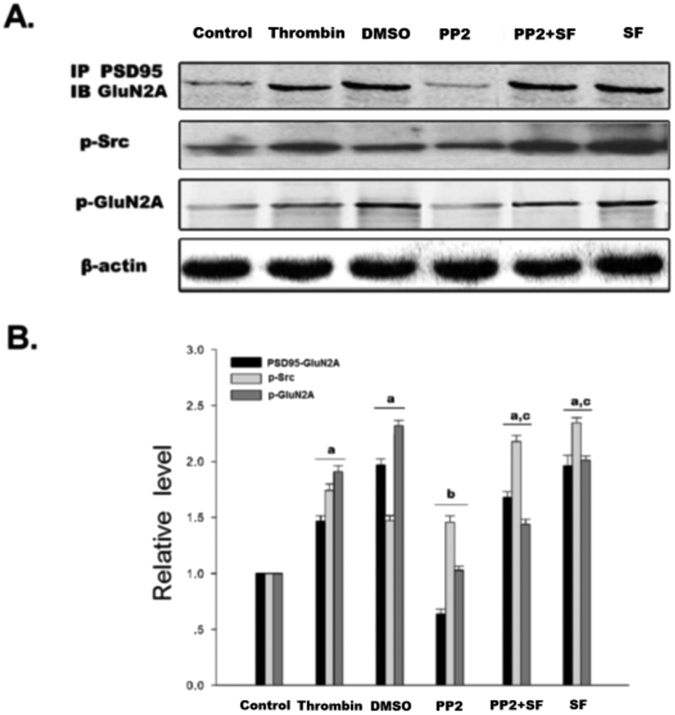
(A) The interaction of PSD95 and GluN2A was examined by immunoprecipitation (IP) with PSD95 antibody followed by immunoblotting with anti-GluN2A antibody. The expression level p-Src was detected by immunoblotting with anti- p-Src antibody. Tyrosine phosphorylation of GluN2A was detected by immunoprecipitation of p-Y and immunoblotting of GluN2A. β-actin was used as a loading control. (B) Statistics for the interaction of PSD95 and GluN2A, p-Src and p-GluN2A in control, thrombin, DMSO, PP2, PP2 + SF and SF groups. The results are shown as mean ± S.D (n = 5). aP < 0.05 vs. Control, bP < 0.05 vs. Thrombin treatment group, cP < 0.05 vs. PP2- treatment group.
ICH enhanced the assembly of the Src-PSD95-GluN2A signaling modules and promoted the phosphorylation of Src and GluN2A
To further investigate the interactions among components of the Src-PSD95-GluN2A module, three kinds of protein immunoprecipitation (IP) and Immunoblotting (IB) were conducted. Previously we found that interactions among Src, PSD95 and GluN2A reached maximum after 3 h of ICH. Among the six time points after ICH including 0.5 h, 1 h, 3 h, 6 h, 12 h, 24 h, 3 h and 6 h time points showed significant differences (data not here). Present results showed that antisense oligonucleotide against PSD95 (AS-PSD95) inhibited the expression of PSD95 and reduced the interactions between Src-PSD95, Src-GluN2A, and PSD95-GluN2A, while the MS-PSD95 could not do so (Fig. 4A,B). In addition, the phosphorylation of Src and GluN2A was significantly increased when compared with the control rats, while AS-PSD95 or PP2 reduced p-Src or p-GluN2A in rats after 3 h of ICH induction (Figs 5A,B and 2B,C).
Figure 4. ICH enhanced the interactions among Src, PSD95 and GluNR2A in rat caudate putamen (CPu).

(A) Reciprocal co-IP analysis of interactions of GluNR2A and PSD95 with Src. Sample proteins were IP with anti-GluNR2A or anti-PSD95 or anti- Src antibodies and then IB with anti-GluNR2A or anti-PSD95 or anti-Src antibody in control, ICH, TE, MS and AS groups. β-actin was used as a loading control. (B) Statistics for the interactions in each group. The results are shown as mean ± S.D (n = 5). aP < 0.05 vs. Control, bP < 0.05 vs. ICH 3 h treatment group.
Figure 5. AS-PSD95 and Src antagonist PP2 reduced the tyrosine phosphorylation of Src, GluNR2A after rat ICH in CPu.
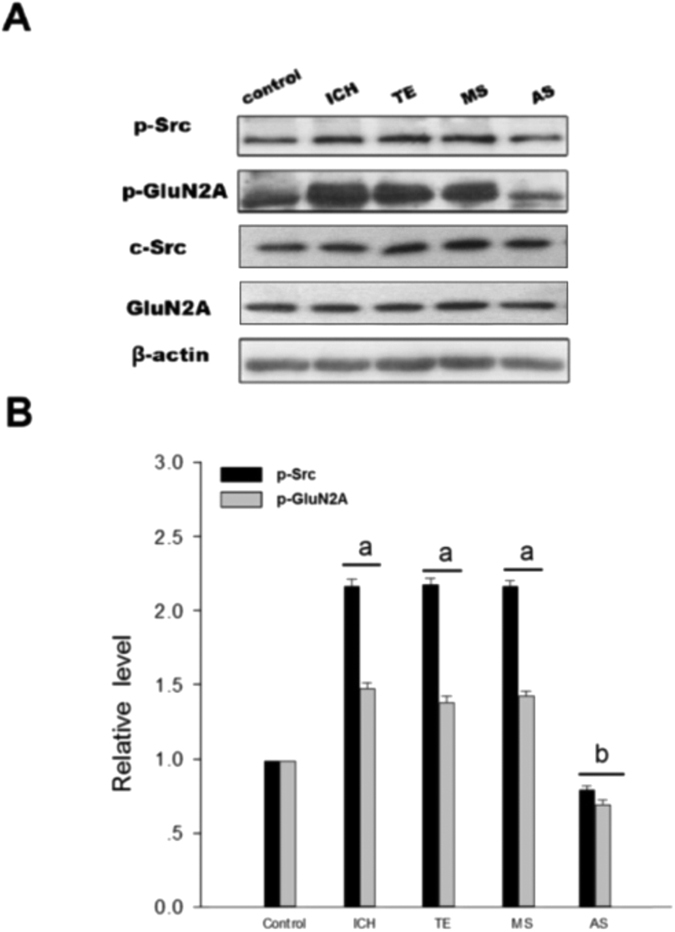
(A) The protein levels of Src, p-Src and GluNR2A were determined by western blot analyses. p-GluNR2A was examined by immunoprecipitation with anti-p-Y antibody followed by immunoblotting with anti-GluNR2A antibody. (B) Statistic for p-Src and p-GluN2A in control, ICH, TE, MS and AS groups. The results are shown as mean ± S.D (n = 5). aP < 0.05 vs. Control group, bP < 0.05 vs. ICH group.
In COS7 cells, treatment of thrombin (5 U/ml, to mimic the rats ICH model) enhanced the interaction between PSD95 and GluN2A when compared with the control cells; treatment with PP2 reduced PSD95-GluN2A interaction, while co-application of PP2 and SFLLR eliminated the inhibitory effect of PP2 (Fig. 3A,B). Both in vivo and in vitro results suggested that ICH enhanced the assembly of the Src-PSD95-GluN2A signaling modules and promoted the phosphorylation of Src and GluN2A.
Enhanced interaction of Src-PSD95-GluN2A and phosphorylation of Src and GluN2A after ICH were PAR1 dependent
Here we showed that treatment of the rats with the PAR1 agonist SF could increase the interaction among Src-PSD95, Src-GluN2A, and PSD95-GluN2A, while treatment of PAR1 antagonist RL significantly reduced the interaction compared with SF group (Fig. 6A,B). To further illustrate whether enhanced interaction of Src-PSD95-GluN2A after ICH were dependent on PAR1, co-application of SF and AS-PSD95 was adopted, and the results showed that AS-PDS95 could counteract the effects of SF. The results indicate that PAR1 mediated the interaction of Src-PSD95-GluN2A molecules after rat ICH. Similar results were also obtained in thrombin (5 U/ml) treated COS7 cells when co-application of SF and PP2 were used (Fig. 3A,B). Collectively, these results indicate that assembly of Src-PSD95-GluN2A signaling molecules after ICH was closely related to PAR1 activation after rat ICH.
Figure 6. PAR1 enhanced interactions among GluNR2A, PSD95 and Src after rat ICH in CPu.

(A) Reciprocal co-IP analysis of interactions of GluNR2A and PSD95 with Src. Sample proteins were immunoprecipitated with anti-GluNR2A or anti-PSD95 or anti-Src antibodies and then blotted with anti-GluNR2A or anti-PSD95 or anti-Src antibody. (B) Statistics for the interactions in control, ICH, SF, RL, AS + SF and TE groups. The results are given as mean ± S.D (n = 5). aP < 0.05 vs. Control, bP < 0.05 vs. ICH 3 h treatment group, cP < 0.05 vs. RL-treatment group.
In accordance, treatment of AS-PSD95 or MK801 in SD rats reduced GluN2A and Src phosphorylation, while co-application of SF and AS-PSD95 could abolish the effect of SF (Fig. 7A–C). These results indicated that PAR1 mediated GluN2A and Src phosphorylation after ICH.
Figure 7. PAR1 increased phosphorylation of Src and GluN2A after rat ICH in CPu.

(A,B) Western blot analysis for Src, p-Src and GluNR2A protein levels after SF, AS-PSD95, SF + AS-PSD95 and MK-801 treatment. p-GluNR2A was examined by immunoprecipitation with anti-p-Y antibody followed by immunoblotting with anti-GluNR2A antibody. (C) Statistics for p-Src, p-GluNR2A, Src and GluNR2A in control, ICH, SF, AS + SF, TE, MK801 and saline groups. The results are shown as mean ± S.D (n = 5). aP < 0.05 vs. Control, bP < 0.05 vs. ICH 3 h treatment group, cP < 0.05 vs. AS group.
Discussion
The major finding of this study is that PAR1 enhanced Src-PSD95-GluN2A signaling molecules interaction through phosphorylation of Src, thereby leading to the activation of NMDA receptor under ICH condition.
ICH enhanced thrombin release and mediated PAR1 activation. Hematoma mass effect is a major cause of cerebral ischemic and edema in ICH11,12. The level of thrombin generated after ICH was positively correlated with the degree of edema surrounding the hematoma tissue, suggesting that the release of thrombin could be an important factor for brain injuries after hemorrhage29,30. As a first identified receptor of thrombin, PAR1 is one of the most important PARs involved in neurotoxicity and neuronal apoptosis31,32. Studies showed that excess thrombin could induce neurotoxic effects and neuronal apoptosis via PAR1 activation33,34,35,36. Based on these lines of evidence, we speculated that thrombin directly activated PAR1 in the course of ICH, leading to the activation of a series of downstream signaling pathways that mediate ICH injuries. For induction of ICH, we injected the 50 μl tail blood directly into the rat caudate putamen so that prothrombin could be transformed into thrombin during the process of the coagulation.
Src family is a class of non-receptor tyrosine kinase. Src was not only a cytoplasmic effector enzyme of G protein-coupled receptor PAR137,38, but also a functional enzyme regulating ion channel NMDAR18,19,20. Therefore, Src might be an important bridge linking G protein-coupled receptors (PAR1) and NMDAR. In the central nerve systems, NMDAR was the first identified ionotropic glutamate receptor regulated by Src family kinase20. Electrophysiological analysis showed that phosphorylation/dephosphorylation of tyrosine controlled the electronic current of NMDAR. NMDAR is co-regulated by two classes of enzymes: PTK and PTP39. Inhibiting PTP or inducing PTK activation could increase the electronic current of NMDAR40. Src belongs to PTK, which can enhance the function of NMDAR18 and affect the current flux through native NMDAR channels41. We previously showed that excessive release of glutamate during cerebral ischemia/reperfusion injury caused influx of Na+ and Ca2+ through NMDAR, and activated Src kinases mediated GluN2A and 2B tyrosine phosphorylation, subsequently increased the activity of NMDAR19,42,43. Herein, we hypothesized that thrombin could activate Src through G protein-coupled receptor PAR1, subsequently Src leads to activation of NMDAR. In this study, we found that PAR1 activator SF significantly increased NMDAR-mediated fEPSP, while antagonist RL reduced f EPSP (Fig. 1A–C). To further investigate PAR1 up-regulated NMDAR activity via Src kinase, co-application of SF and Src inhibitor PP2, the electrophysiological analysis showed that Src inhibitor PP2 significantly abolished the effects of SF on INMDA (Fig. 1D,E). Such results that SF + PP2 abolished the effects of SF were further confirmed in vivo by determining the activation of GluN2A phosphorylation with immunoblotting (Fig. 2A,B) as well as in COS7 cells (Fig. 3A,B). Those results indicate that regulatory effects of PAR1 on NMDAR activity are mediated by Src kinase during ICH. Although a broad spectrum inhibitor PP2 of the Src kinase (not the specific Src inhibitor) was used in this study, GluN2A might be tyr-phosphorylated by Fyn and there may be other src-family tyrosine kinases such as Lck/Fyn in the IPed (immunoprecipitated) complex that are inhibited by PP244, Src kinase should be an important occurring18,19,20. It is possible that PAR1 could regulate the Src-mediated activation of NMDAR, and therefore increased glutamate-mediated neuronal death45,46.
In the glutamatergic excitatory post-synaptic density (PSD), receptor proteins, cytoskeletal proteins, and various signaling complex molecules (including protein kinase, phosphatase) could directly or indirectly bind to NMDAR reversibly, forming a dynamic signaling complex with NMDAR as the core47,48. PSD95 contained three N-terminal PDZ domains (PDZ1, PDZ2, PDZ3), one SH3 domain and one GK domain at the C-terminus. PSD95 could bind to GluN2 subunits (GluN2A and GluN2B) of NMDAR via PDZ249. PSD95 bond SH2 of SrcPTKs through its PDZ3 domain50. During cerebral ischemic injury, SrcPTKs and NMDAR substrate GluN2A were recruited to PSD, forming Src-PSD95-GluN2A signaling modules19. Formation of this signaling module could firmly anchor SrcPTKs to the NMDAR, resulting in Src close GluN2A and enhance GluN2A tyrosine phosphorylation51,52. Up-regulation of NMDAR functions thereby aggravates the injuries in excitatory neurons53,54. In the present study, our results showed that the assembly of the Src-PSD95-GluN2A signaling module, and phosphorylation of Src and GluN2A were enhanced under ICH in vivo (Figs 2A,B and 4A,B). AS-PSD95, Src inhibitor PP2, NMDA receptor antagonist MK801 or the PAR1 antagonist peptide RL inhibited the interaction Src-PSD95, Src- GluN2A, and PSD95-GluN2A (Figs 2A,B and 3A,B) and p-Src, p-GluN2A (Figs 2B,C, 3A,B and 7A,B), PAR1 activator SFLLR enhanced such interaction and p-Src, p- GluN2A, while co-application of SF + AS-PSD95, SF + PP2 neutralized the increase of SF (Figs 2A, 3A,B and 6A,B), indicating that GluN2A-PSD95-Src signaling cascades were regulated by PAR1 (Fig. 6). All these results support the hypothesis that PAR1 induces Src activation. Src is a cytoplasmic effector enzyme of G protein-coupled receptor PAR1 which could enhance the interactions among the Src-PSD95-GluN2A signaling modules and enhance the anchor of SrcPTKs to the NMDAR, subsequently inducing tyrosine phosphorylation of GluN2A (Fig. 8). High amount of Ca2+ may enter the postsynaptic pyramidal cells through the NMDA receptors, which further activates Src18. This positive feedback mechanism may result in the delayed neuronal death after ICH.
Figure 8. Possible mechanisms for PAR1modulating NMDA receptor.
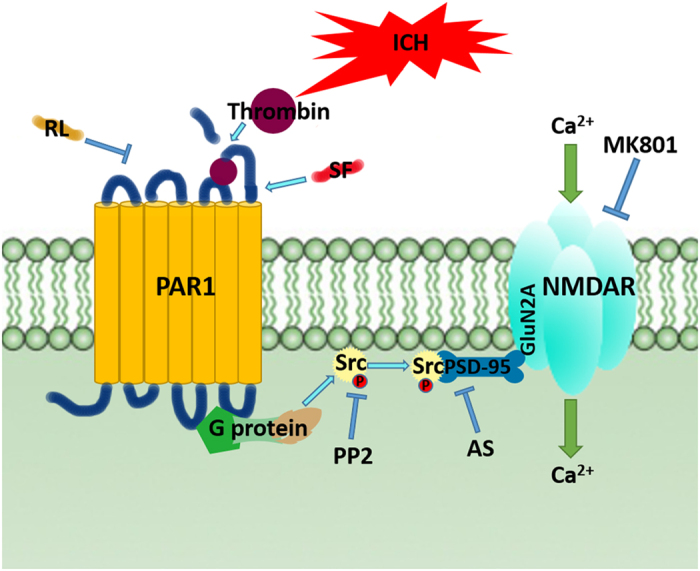
Diagram summarizing our findings that during ICH, activated PAR1 increased the Src activation and subsequently enhanced the interactions among the Src-PSD95-NR2A signaling molecules, which promoted phosphorylation of NMDAR and activation.
In summary, our results demonstrated that during ICH, activated G protein-coupled receptor PAR1 mediated activation of its effector enzyme Src. Activated Src enhanced the interactions among the Src-PSD95-GluN2A signaling molecules and promoted phosphorylation of NMDAR (Fig. 8). By studying this signaling pathway (thrombin → PAR1 → Src-PSD95-GluN2A signal modules → GluN2A), we expect to further explore the role of Src kinase and NMDA receptors in ICH brain injuries, and provide new strategies and drug targets for the treatment of hemorrhagic brain injury.
Additional Information
How to cite this article: Duan, Z.-Z. et al. Protease activated receptor 1 (PAR1) enhances Src-mediated tyrosine phosphorylation of NMDA receptor in intracerebral hemorrhage (ICH). Sci. Rep. 6, 29246; doi: 10.1038/srep29246 (2016).
Acknowledgments
The authors wish to thank Professor Jian Hong Luo from Zhejiang University for providing Src plasmid and Professor Sheng M for providing PSD95 and GluN2A plasmids for this study. This work was supported by Grants from the National Natural Science Foundation of China (No. 81171075), Natural Science Foundation of the Jiangsu Higher Education Institutions of China (No. 13KJA310005), the Priority Academic Program Development of Jiangsu Higher Education Student Research Train Program (SRTP) (No. 201510313014Z). And the 333 project for the cultivation of high-level talents of Jiangsu (To Suhua Qi and Yong Liu).
Footnotes
Author Contributions All (Z.-Z.D., F.Z., F.-Y.L., Y.-F.L., P.G., Y.-H.L., Y.L. and S.-H.Q.) the authors contributed significantly to this study. Z.-Z.D. finished the most parts of experiments (about 70%), F.Z. about 10%, F.-Y.L. about 6%, Y.-F.L. finished the about 5%, P.G. and Y.-H.L. finished the about 9%. F.Z. drafted the proposal of this study and wrote the first draft of the manuscript. Y.L. discussed about the proposal and critically revised the draft. S.-H.Q. is the leader of this study. She came up with the original idea, designed and revised the proposal. S.-H.Q. also revised the draft critically and finally approved the manuscript to be submitted.
References
- Lee K. R., Betz A. L., Kim S., Keep R. F. & Hoff J. T. The role of the coagulation cascade in brain edema formation after intracerebral hemorrhage. Acta Neurochir. 138, 396–400, discussion 400–391 (1996). [DOI] [PubMed] [Google Scholar]
- Lee K. R. et al. Edema from intracerebral hemorrhage: the role of thrombin. J. Neurosurg. 84, 91–96 (1996). [DOI] [PubMed] [Google Scholar]
- Striggow F. et al. The protease thrombin is an endogenous mediator of hippocampal neuroprotection against ischemia at low concentrations but causes degeneration at high concentrations. P. Natl. Acad. Sci. USA 97, 2264–2269 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao D. et al. Effects of focal mild hypothermia on thrombin-induced brain edema formation and the expression of protease activated receptor-1, matrix metalloproteinase-9 and aquaporin 4 in rats. Mol. Med. Rep. 11, 3009–3014 (2015). [DOI] [PubMed] [Google Scholar]
- Coughlin S. R. Protease-activated receptors in hemostasis, thrombosis and vascular biology. J. Thromb. Haemost. 3, 1800–1814 (2005). [DOI] [PubMed] [Google Scholar]
- Ender M., Andreoni F., Zinkernagel A. S. & Schuepbach R. A. Streptococcal SpeB cleaved PAR1 suppresses ERK phosphorylation and blunts thrombin-induced platelet aggregation. PloS one 8, e81298 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramachandran R. & Hollenberg M. D. Proteinases and signalling: pathophysiological and therapeutic implications via PARs and more. Brit. J. Pharmacol. 153 Suppl 1, S263–S282 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noorbakhsh F., Vergnolle N., Hollenberg M. D. & Power C. Proteinase-activated receptors in the nervous system. Nat. Rev. Neurosci. 4, 981–990 (2003). [DOI] [PubMed] [Google Scholar]
- Junge C. E. et al. Protease-activated receptor-1 in human brain: localization and functional expression in astrocytes. Exp. Neurol. 188, 94–103 (2004). [DOI] [PubMed] [Google Scholar]
- Junge C. E. et al. The contribution of protease-activated receptor 1 to neuronal damage caused by transient focal cerebral ischemia. P. Natl. Acad. Sci. USA 100, 13019–13024 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein E. S. et al. Thrombin induces ischemic LTP (iLTP): implications for synaptic plasticity in the acute phase of ischemic stroke. Sci. Rep. 5, 7912 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han K. S. et al. Activation of protease activated receptor 1 increases the excitability of the dentate granule neurons of hippocampus. Mol. Brain 4, 32 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatterton J. E. et al. Excitatory glycine receptors containing the NR3 family of NMDA receptor subunits. Nature 415, 793–798 (2002). [DOI] [PubMed] [Google Scholar]
- Hollmann M., O’Shea-Greenfield A., Rogers S. W. & Heinemann S. Cloning by functional expression of a member of the glutamate receptor family. Nature 342, 643–648 (1989). [DOI] [PubMed] [Google Scholar]
- Takagi N. et al. Transient ischemia differentially increases tyrosine phosphorylation of NMDA receptor subunits 2A and 2B. J. Neurochem. 69, 1060–1065 (1997). [DOI] [PubMed] [Google Scholar]
- Lee J. M., Zipfel G. J. & Choi D. W. The changing landscape of ischaemic brain injury mechanisms. Nature 399, A7–14 (1999). [DOI] [PubMed] [Google Scholar]
- Ali D. W. & Salter M. W. NMDA receptor regulation by Src kinase signalling in excitatory synaptic transmission and plasticity. Curr. Opin. Neurobiol. 11, 336–342 (2001). [DOI] [PubMed] [Google Scholar]
- Yu X. M., Askalan R., Keil G. J. 2nd & Salter M. W. NMDA channel regulation by channel-associated protein tyrosine kinase Src. Science 275, 674–678 (1997). [DOI] [PubMed] [Google Scholar]
- Xu J., Liu Y. & Zhang G. Y. Neuroprotection of GluR5-containing kainate receptor activation against ischemic brain injury through decreasing tyrosine phosphorylation of N-methyl-D-aspartate receptors mediated by Src kinase. J. Biol. Chem. 283, 29355–29366 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y., Zhang G., Gao C. & Hou X. NMDA receptor activation results in tyrosine phosphorylation of NMDA receptor subunit 2A(NR2A) and interaction of Pyk2 and Src with NR2A after transient cerebral ischemia and reperfusion. Brain Res. 909, 51–58 (2001). [DOI] [PubMed] [Google Scholar]
- Kalia L. V., Pitcher G. M., Pelkey K. A. & Salter M. W. PSD-95 is a negative regulator of the tyrosine kinase Src in the NMDA receptor complex. EMBO. J. 25, 4971–4982 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shigetomi E., Bowser D. N., Sofroniew M. V. & Khakh B. S. Two forms of astrocyte calcium excitability have distinct effects on NMDA receptor-mediated slow inward currents in pyramidal neurons. J. Neurosci. 28, 6659–6663 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mannaioni G. et al. Plasmin potentiates synaptic N-methyl-D-aspartate receptor function in hippocampal neurons through activation of protease-activated receptor-1. J. Biol. Chem. 283, 20600–20611 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamill C. E. et al. Special lecture: glial reactivity after damage: implications for scar formation and neuronal recovery. Clin. Neurosurg. 52, 29–44 (2005). [PubMed] [Google Scholar]
- Hamill C. E., Mannaioni G., Lyuboslavsky P., Sastre A. A. & Traynelis S. F. Protease-activated receptor 1-dependent neuronal damage involves NMDA receptor function. Exp. Neurol. 217, 136–146 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan Z. Z. et al. Protection of Momordica charantia polysaccharide against intracerebral hemorrhage-induced brain injury through JNK3 signaling pathway. J. Recept. Signal. Transduct. Res. 35, 1–7 (2015). [DOI] [PubMed] [Google Scholar]
- Longa E. Z., Weinstein P. R., Carlson S. & Cummins R. Reversible middle cerebral artery occlusion without craniectomy in rats. Stroke 20, 84–91 (1989) [DOI] [PubMed] [Google Scholar]
- Qi S. H. et al. Neuroprotection of ethanol against ischemia/reperfusion-induced brain injury through decreasing c-Jun N-terminal kinase 3 (JNK3) activation by enhancing GABA release. Neuroscience 167, 1125–1137 (2010). [DOI] [PubMed] [Google Scholar]
- Lerma J., Morales M., Vicente M. A. & Herreras O. Glutamate receptors of the kainate type and synaptic transmission. Trends. Neurosci. 20, 9–12 (1997). [DOI] [PubMed] [Google Scholar]
- Gingrich M. B., Junge C. E., Lyuboslavsky P. & Traynelis S. F. Potentiation of NMDA receptor function by the serine protease thrombin. J. Neurosci. 20, 4582–4595 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bae J. S., Kim I. S. & Rezaie A. R. Thrombin down-regulates the TGF-beta-mediated synthesis of collagen and fibronectin by human proximal tubule epithelial cells through the EPCR-dependent activation of PAR-1. J. Cell Physiol. 225, 233–239 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zigler M., Kamiya T., Brantley E. C., Villares G. J. & Bar-Eli M. PAR-1 and thrombin: the ties that bind the microenvironment to melanoma metastasis. Cancer Res. 71, 6561–6566 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volbers B. et al. Intraventricular fibrinolysis does not increase perihemorrhagic edema after intracerebral hemorrhage. Stroke 44, 362–366 (2013). [DOI] [PubMed] [Google Scholar]
- Sansing L. H., Messe S. R., Cucchiara B. L., Lyden P. D. & Kasner S. E. Anti-adrenergic medications and edema development after intracerebral hemorrhage. Neurocrit. Care 14, 395–400 (2011). [DOI] [PubMed] [Google Scholar]
- Donovan F. M., Pike C. J., Cotman C. W. & Cunningham D. D. Thrombin induces apoptosis in cultured neurons and astrocytes via a pathway requiring tyrosine kinase and RhoA activities. J. Neurosci. 17, 5316–5326 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Q. B. et al. Baicalin attenuates brain edema in a rat model of intracerebral hemorrhage. Inflammation 37, 107–115 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harper M. T. & Sage S. O. PAR-1-dependent pp60src activation is dependent on protein kinase C and increased [Ca2+]: evidence that pp60src does not regulate PAR-1-dependent Ca2+ entry in human platelets. J. Thromb. Haemost. 4, 2695–2703 (2006). [DOI] [PubMed] [Google Scholar]
- Chiu Y. C. et al. Thrombin-induced IL-6 production in human synovial fibroblasts is mediated by PAR1, phospholipase C, protein kinase C alpha, c-Src, NF-kappa B and p300 pathway. Mol. Immunol. 45, 1587–1599 (2008). [DOI] [PubMed] [Google Scholar]
- Wang Y. T., Yu X. M. & Salter M. W. Ca(2+)-independent reduction of N-methyl-D-aspartate channel activity by protein tyrosine phosphatase. P. Natl. Acad. Sci. USA 93, 1721–1725 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roche S., Koegl M., Barone M. V., Roussel M. F. & Courtneidge S. A. DNA synthesis induced by some but not all growth factors requires Src family protein tyrosine kinases. Mol. Cell Biol. 15, 1102–1109 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohr G. & Seeburg P. H. Subtype-specific regulation of recombinant NMDA receptor-channels by protein tyrosine kinases of the src family. J. Physiol. 492 (Pt 2), 445–452 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang F. et al. Activation of GABA receptors attenuates neuronal apoptosis through inhibiting the tyrosine phosphorylation of NR2A by Src after cerebral ischemia and reperfusion. Neuroscience 150, 938–949 (2007). [DOI] [PubMed] [Google Scholar]
- Ma J., Zhang G. Y., Liu Y., Yan J. Z. & Hao Z. B. Lithium suppressed Tyr-402 phosphorylation of proline-rich tyrosine kinase (Pyk2) and interactions of Pyk2 and PSD-95 with NR2A in rat hippocampus following cerebral ischemia. Neurosci. Res. 49, 357–362 (2004). [DOI] [PubMed] [Google Scholar]
- Knox R. et al. Enhanced NMDA receptor tyrosine phosphorylation and increased brain injury following neonatal hypoxia-ischemia in mice with neuronal Fyn overexpression. Neurobiol. Dis. 51, 113–119 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai S. H. et al. Activation of mGluR5 attenuates NMDA-induced neurotoxicity through disruption of the NMDAR-PSD-95 complex and preservation of mitochondrial function in differentiated PC12 cells. Int. J. Mol. Sci. 15, 10892–10907 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimmyo Y., Kihara T., Akaike A., Niidome T. & Sugimoto H. Three distinct neuroprotective functions of myricetin against glutamate-induced neuronal cell death: involvement of direct inhibition of caspase-3. J. Neurosci. Res. 86, 1836–1845 (2008). [DOI] [PubMed] [Google Scholar]
- Sheng M. & Kim M. J. Postsynaptic signaling and plasticity mechanisms. Science 298, 776–780 (2002). [DOI] [PubMed] [Google Scholar]
- Husi H., Ward M. A., Choudhary J. S., Blackstock W. P. & Grant S. G. Proteomic analysis of NMDA receptor-adhesion protein signaling complexes. Nat. Neurosci. 3, 661–669 (2000). [DOI] [PubMed] [Google Scholar]
- Gardoni F., Polli F., Cattabeni F. & Di Luca M. Calcium-calmodulin-dependent protein kinase II phosphorylation modulates PSD-95 binding to NMDA receptors. Eur. J. Neurosci. 24, 2694–2704 (2006). [DOI] [PubMed] [Google Scholar]
- Tezuka T., Umemori H., Akiyama T., Nakanishi S. & Yamamoto T. PSD-95 promotes Fyn-mediated tyrosine phosphorylation of the N-methyl-D-aspartate receptor subunit NR2A. P. Natl. Acad. Sci. USA 96, 435–440 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sala C. & Sheng M. The fyn art of N-methyl-D-aspartate receptor phosphorylation. P. Natl. Acad. Sci. USA 96, 335–337 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lei G. et al. Gain control of N-methyl-D-aspartate receptor activity by receptor-like protein tyrosine phosphatase alpha. EMBO. J. 21, 2977–2989 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- He Y., Wan S., Hua Y., Keep R. F. & Xi G. Autophagy after experimental intracerebral hemorrhage. J. Cereb. Blood Flow Metab. 28, 897–905 (2008). [DOI] [PubMed] [Google Scholar]
- Bimpis A. et al. Modulation of crucial adenosinetriphosphatase activities due to U-74389G administration in a porcine model of intracerebral hemorrhage. Metab. Brain Dis. 28, 439–446 (2013). [DOI] [PubMed] [Google Scholar]