

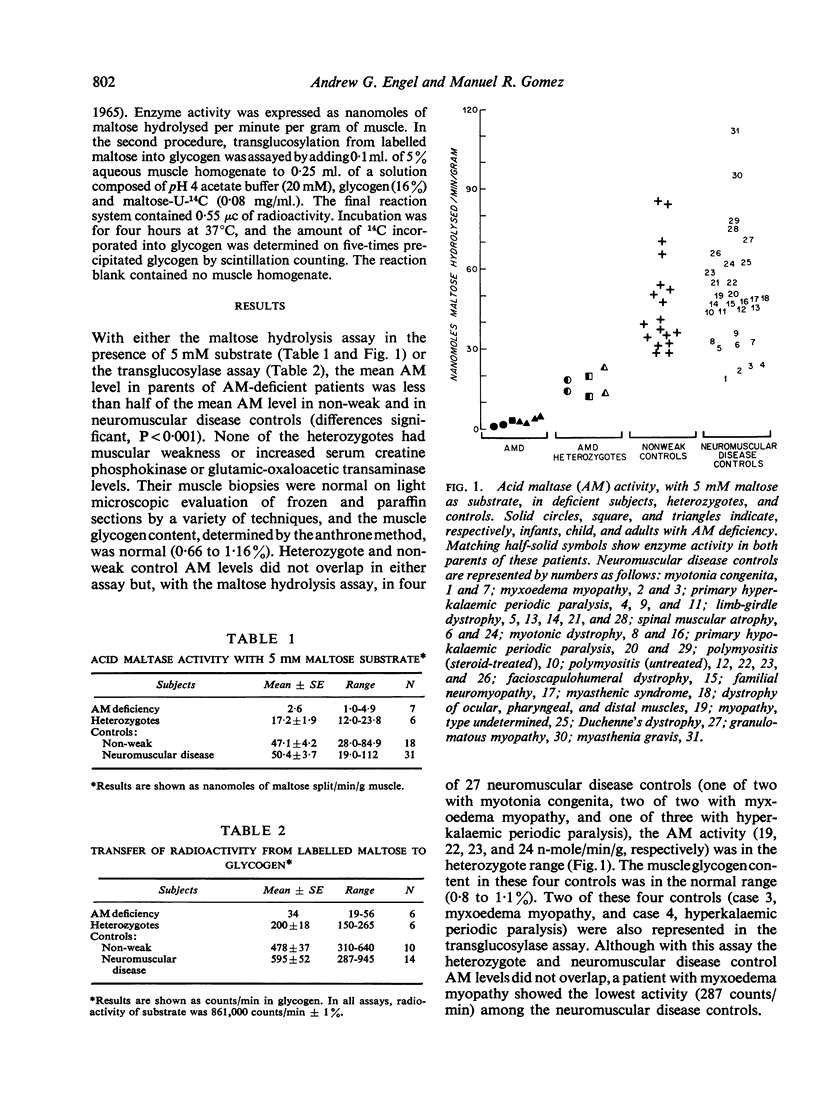
Abstract
Acid maltase (AM) deficiency carriers can be detected by muscle enzyme assay. The assay indicates that, just as in infantile and childhood cases, adult cases of the disease are transmitted by autosomal recessive inheritance. With the maltose hydrolysis assay, in some neuromuscular diseases, muscle AM activity can be as low as in heterozygous AM deficiency. A relatively low muscle AM activity in myxoedema myopathy is confirmed. In human muscle, the Km of the enzyme for maltose hydrolysis is 7·2 to 9 × 10−3M. A modification of the enzyme assay based on this fact is recommended.
Full text
PDF



Selected References
These references are in PubMed. This may not be the complete list of references from this article.
- Auricchio F., Bruni C. B., Sica V. Further purification and characterization of the acid alpha-glucosidase. Biochem J. 1968 Jun;108(2):161–167. doi: 10.1042/bj1080161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- COOPER A. C., MILLDR J. R. Progressive muscular dystrophy: a review. Rev Can Biol. 1962 Sep-Dec;21:337–351. [PubMed] [Google Scholar]
- Di Mauro S., Angelini C., Catani C. Enzymes of the glycogen cycle and glycolysis in various human neuromuscular disorders. J Neurol Neurosurg Psychiatry. 1967 Oct;30(5):411–415. doi: 10.1136/jnnp.30.5.411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engel A. G. Acid maltase deficiency in adults: studies in four cases of a syndrome which may mimic muscular dystrophy or other myopathies. Brain. 1970;93(3):599–616. doi: 10.1093/brain/93.3.599. [DOI] [PubMed] [Google Scholar]
- Engel A. G., Dale A. J. Autophagic glycogenosis of late onset with mitochondrial abnormalities: light and electron microscopic observations. Mayo Clin Proc. 1968 Apr;43(4):233–279. [PubMed] [Google Scholar]
- Hirschhorn K., Nadler H. L., Waithe W. I., Brown B. I., Hirschhorn R. Pompe's disease: detection of heterozygotes by lymphocyte stimulation. Science. 1969 Dec 26;166(3913):1632–1633. doi: 10.1126/science.166.3913.1632. [DOI] [PubMed] [Google Scholar]
- Hudgson P., Gardner-Medwin D., Worsfold M., Pennington R. J., Walton J. N. Adult myopathy from glycogen storage disease due to acid maltase deficiency. Brain. 1968 Sep;91(3):435–462. doi: 10.1093/brain/91.3.435. [DOI] [PubMed] [Google Scholar]
- Hurwitz L. J., McCormick D., Allen I. V. Reduced muscle alpha-glucosidase (acid-maltase) activity in hypothyroid myopathy. Lancet. 1970 Jan 10;1(7637):67–69. doi: 10.1016/s0140-6736(70)91849-0. [DOI] [PubMed] [Google Scholar]
- Krause E. G., Wollenberger A. Abhängigkeit der Aktivität einiger Enzyme des Glykogenstoffwechsels des Skeletmuskels der Ratte vom Schilddrüsenstatus. Acta Biol Med Ger. 1968;21(5):615–624. [PubMed] [Google Scholar]
- Nitowsky H. M., Grunfeld A. Lysosomal alpha-glucosidase in type II glycogenosis; activity in leukocytes and cell cultures in relation to genotype. J Lab Clin Med. 1967 Mar;69(3):472–484. [PubMed] [Google Scholar]