

Abstract
Background
Increased numbers and improperly positioned centrosomes, aneuploidy or polyploidy, and chromosomal instability are frequently observed characteristics of cancer cells. While some aspects of these events and the checkpoint mechanisms are well studied, not all players have yet been identified. As the role of proteases other than the proteasome in tumorigenesis is an insufficiently addressed question, we investigated the epigenetic control of the widely conserved protease HTRA1 and the phenotypes of deregulation.
Methods
Mouse embryonal fibroblasts and HCT116 and SW480 cells were used to study the mechanism of epigenetic silencing of HTRA1. In addition, using cell biological and genetic methods, the phenotypes of downregulation of HTRA1 expression were investigated.
Results
HTRA1 is epigenetically silenced in HCT116 colon carcinoma cells via the epigenetic adaptor protein MBD2. On the cellular level, HTRA1 depletion causes multiple phenotypes including acceleration of cell growth, centrosome amplification and polyploidy in SW480 colon adenocarcinoma cells as well as in primary mouse embryonic fibroblasts (MEFs).
Conclusions
Downregulation of HTRA1 causes a number of phenotypes that are hallmarks of cancer cells suggesting that the methylation state of the HtrA1 promoter may be used as a biomarker for tumour cells or cells at risk of transformation.
Electronic supplementary material
The online version of this article (doi:10.1186/s12885-016-2425-8) contains supplementary material, which is available to authorized users.
Keywords: HTRA1, MDB2, serine protease
Background
Mammalian HtrA1 belongs to the widely conserved high-temperature requirement A (HtrA) family of homo-oligomeric serine proteases that are implicated in protein quality control. The ubiquitously expressed HTRA1 is composed of a signal sequence for secretion, a partial insulin like growth factor binding protein-7 domain of unknown function, a serine protease domain resembling chymotrypsin and one C-terminal PDZ domain. HTRA1 has been shown to have at least three cellular locations. The extracytoplasmic pool is involved in the homeostasis of the extracellular matrix as HTRA1 degrades fibronectin, fibromodulin, aggrecan and decorin. In addition, intracellular HTRA1 localizes to microtubules or to the nucleus (for review see [1]).
Human HTRA1 has been implicated in several severe pathologies including cancer, age-related macular degeneration, Alzheimer’s disease, arthritis and familial ischemic cerebral small-vessel disease [1]. In many of these diseases, protein fragments or aggregates are either causative for disease or are disease modifying factors that are produced or degraded by HTRA1. Furthermore, several publications link HTRA1 to tumorigenesis as its gene has been found to be downregulated in many tumours [2], and forcing its re-expression interfered with proliferation of metastatic melanoma cells [3] and cell migration [4], suggesting a tumour suppressor function. In addition, HTRA1 was shown to modulate cisplatin- and paclitaxel-induced cytotoxicity and low levels of HTRA1 correlated with a poor response to drug treatment whilst higher levels of HTRA1 correlated with a higher response rate [5]. Downregulation of the HTRA1 gene in tumour cells has been linked with epigenetic mechanisms [2, 6] and the HTRA1 promoter was identified as a target of the histone deacetylase HDAC1 [7]. Despite these recent advances, the function and mechanism of silencing of intracellular HTRA1 underlying its involvement in cell proliferation, migration and tumorigenesis are currently not well understood.
We show that HTRA1 is epigenetically silenced in HCT116 colon carcinoma cells and during early stages of tumorigenesis in a mouse model of intestinal cancer. Downregulation of HTRA1 causes a multiple phenotypes that are hallmarks of cancer cells including increased proliferation of mouse embryonic fibroblasts (MEF), as well as chromosome and centrosome amplifications.
Methods
Cell lines and drug treatments
This study received ethical approval from Cardiff University’s Animal Welfare and Ethical Review Body (previously known as the ERP), and all animal procedures were conducted in accordance with UK Home Office regulations. HCT116, SW480 cells and MEFs were maintained in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10 % fetal bovine serum, 1 % penicillin and 1 % streptomycin at 37 °C in humidified atmosphere with 5 % CO2. MEFs were isolated from E13.5 and E14.5 embryos derived from four different breedings. Htra1−/− mice were described previously [8]. SW480 and HCT116 cells were obtained from ATCC.
Cells were seeded at a low density for 16 h and were treated with indicated concentrations of 5-Aza-dC (Sigma) or 400 nM TSA (NEB) for 16 h. For drug combination cells were treated with 5-Aza-dC for 48 h followed by TSA for additional 16 h.
Oligonucleotides
All oligonucleotides used are listed in Additional file 1: Table S1.
Lentiviral preparation and viral infection
Hairpin sequences directed against HTRA1 or MBD2 were cloned into the lentiviral pLKO.1puro vector using AgeI and EcoRI. 293 T cells were transfected with lentiviral vectors encoding shRNAs (shHTRA1 D3 and S8) or nonsense RNA (EV ctrl.) and lentiviral packaging vectors pCMVΔR8.2 (gag pol) and pHITG (env). Viruses were collected 48 h after transfection. HCT116 and SW480 cells were infected with the collected viruses twice over 18 h in the presence of polybrene. Infected cells were selected using 1.6 μg/ml puromycin.
Confocal laser microscopy and antibodies
24 h after plating, cells were stained against α-tubulin (Invitrogen), β-tubulin (Molecular Probes), γ-tubulin (Sigma-Aldrich) or Actin (MP Biomedicals). For detection, secondary antibodies conjugated with Alexa-488 or Phalloidin-TRITC (Molecular Probes) were used. Nuclei were stained with DAPI (Molecular Probes). Samples were analysed in a Leica TCS SL (SP5) laser confocal microscope and Leica Confocal Software was used for imaging. Images were taken using an HCX PL APO x 63 oil objective lens.
RNA purification and quantitative real-time-PCR (qRT-PCR) analysis
RNA purification and qRT-PCR were done as described [9]. All mRNA levels were normalized to mRNA levels of the “house-keeping” gene GAPDH for samples from human cell lines or β-actin for samples from murine cell lines to obtain the mean normalized expression. Analysis of data sets was carried out with Q-Gene software [10].
Karyotyping of MEFs and SW480 cells
Exponentially growing SW480 (Parental, EV ctrl. and shHTRA1 D3 and S8) and MEF cultures were incubated in N-deacetyl-N-methylcolchicine (Colcemid; 0.08 μg/ml) for 2 h to arrest mitotic cells in highly condensed metaphase like stages. Monolayers were rinsed and centrifuged for 5 min at 120 g. Cell sediments were hypotonically treated with 5 ml of 75 mM KCl for 10 min. Following centrifugation the swollen cells were gently mixed with 5 ml of fixing solution (methanol/acetic acid; 3/1), centrifuged, and again mixed with fixing solution. Cell suspensions were dropped onto pre-cleaned, wet, ice-cold glass microscope slides to obtain good spreading of the chromosome sets. After air-drying overnight, the preparations were stained in Giemsa-solution (5 %). Intact metaphase cells were counted for their chromosome numbers at 1000 fold magnification (oil-immersion).
Bisulfite modification and bisulfite sequencing PCR (BSP) of genomic DNA
Genomic DNA was prepared from cell lines or murine colon polyp cells using QIAamp DNA Mini Kit (Qiagen). Bisulfite conversion of 2 μg genomic DNA was performed using the EpiTect Bisulfite Kit (Qiagen). Origin of polyps: no. 13 from mouse no. 444, no. 18 from mouse no. 508, no. 22 from mouse no. 509, nos. 97, 98, 99, 101 from mouse no. 1122 and nos. 145 & 147 from mouse no. 495. 3 μl of bisulfite treated genomic DNA were used for PCR amplification. PCR products were purified and cloned into pCR2.1-TOPO using TOPO TA Cloning Kit (Invitrogen). DNA was sequenced and methylation status of the DNA sequences was analysed using BIQAnalyzer [11].
Chromatin immunoprecipitation (ChIP)
Confluent SW480 and HCT116 cells were used for ChIP experiments. For immunoprecipitation, 2 μg of RNApolII (Active Motif, No. 39097), IgG (Active Motif), H3 (Abcam, No. 1791), H3K9ac (Diagenode, pAB-177-050) and 10 μg MBD2a/b (Sigma, M7318) antibodies were used. qRT-PCR was used to determine the enrichment of immunoprecipitated DNA relative to the input material using gene-specific (HTRA1) and control (GAPDH) primer sets (Additional file 1: Table S1). For more details see Additional file 1.
Protein purification
HTRA1 was purified as described [12]. Purified HTRA1 was dialyzed against 50 mM Tris HCl, pH 8.0, 150 mM NaCl and stored at −70 °C. 6His tagged MBD2b, pET28MBD2b were purified using Protino Ni-TED 2000 column (Macherey-Nagel) following manufacturer’s instruction. Subsequently, MBD2b fractions were dialyzed against 50 mM NaH2PO4, pH 8.0 and stored at −70 °C.
EMSA
Electrophoretic Mobility Shift Assay was done in 10 μl of EMSA-buffer (50 mM Tris, 5 mM MgCl2, 10 mM DTT, pH 7.5) for 5 min at RT. Reaction mixtures were loaded on a TBE-gel which was stained with ethidium bromide.
Protease protection assay
Trypsin (Sigma) digests of MBD2b were performed by incubating 5 μg MBD2b with various amounts of trypsin for 20 min at 37 °C in reaction buffer (50 mM Tris, 5 mM MgCl2, pH 7.5). To analyse the effect of DNA-binding, MBD2b was pre-incubated with DNA-oligonucleotides (Additional file 1: Table S1) at equimolar concentration for 10 min at RT before adding trypsin.
Statistical analyses
Statistical analyses were carried out using GraphPad Prism5 software (GraphPad Software). Gaussian distribution of data sets was tested via D’Agostino and Pearson omnibus normality test or (for smaller n) via Komologrov Smirnov normality test with alpha = 0.05. Data sets following a Gaussian distribution were analysed by a two-tailed t-test if variance homogeneity was given. A two-tailed Mann–Whitney U test was used for analysing data sets not following a Gaussian distribution or with significant difference in variances (Levene-test p-value <0.2). For analysis of nominal data sets Fisher’s exact test was performed.
Results
HTRA1 is epigenetically silenced in HCT116 cells and in polyps arising in ApcMin+ mice
As HTRA1 is downregulated in a range of tumours we determined if silencing of HTRA1 occurs in cancer cell lines by analysing methylation of the HTRA1 promoter in two colon carcinoma cell lines (HCT116 and SW480). These data indicate that the HTRA1 promoter is methylated in HCT116 cells but not in SW480 cells and that promoter methylation status correlates with reduced the expression levels of the HTRA1 gene (Fig. 1a, b). To obtain further evidence for epigenetic silencing of the HTRA1 promoter, HCT116 and SW480 cells were treated individually or in combination with the DNA methyltransferase (DNMT) inhibitor 5-aza-deoxycytidine (5-aza-dC) and Trichostatin A (TSA), an inhibitor of histone deacetylases (HDAC). Incorporation of 5-aza-dC into DNA inhibits methylation at CpG islands by DNA methyltransferase whereas Trichostatin A inhibits histone deacetylation by HDAC [13, 14]. Therefore, both inhibitors promote gene expression. Consequently, changes in HTRA1 expression were detected following treatment of HCT116 cells with either 1 μM 5-Aza-dC in combination with 400 nM TSA or 5 μM 5-Aza-dC in combination with 400 nM TSA, leading to 7 and 10 fold higher levels of expression of HTRA1, respectively (Fig. 1c). In contrast, no increases were observed in SW480 cells (Fig. 1c). In this cell line, exposure to TSA even reduced HTRA1 expression, which may be due to the side effect of TSA causing cell cycle arrest in colon carcinoma cells and fibroblasts [15, 16].
Fig. 1.
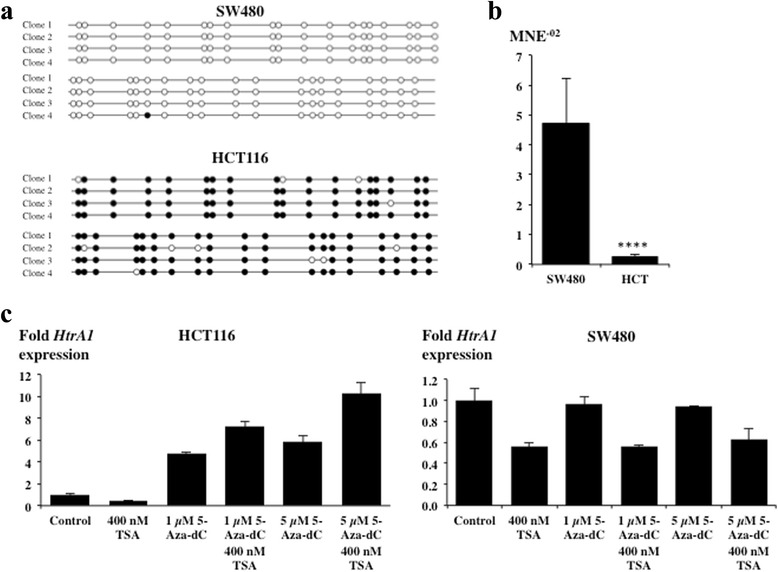
Epigenetic regulation of HTRA1 expression in HCT116 cells. a Methylation of the human HTRA1 promoter (−388 bp to −115 bp) was determined by sequencing 4 independent clones of bisulfite treated DNA of HCT116 and SW480 cells. b Mean normalized expression (MNE) of HTRA1 in SW480 and HCT116 cells determined by qRT-PCR (n = 3 independent cell lines, each done in triplicates, two-tailed Mann–Whitney U test, p-value <0.0001). c HTRA1 mRNA levels in HCT116 and SW480 cells following treatment with 5-Aza-dC and TSA determined by qRT-PCR. Results of one representative experiment are shown. MNE of control was set to 1; standard deviation was calculated from triplicates
To obtain in vivo evidence, we analysed Htra1 expression in the intestines of ApcMin+ mice, a well established model of early events in intestinal tumorigenesis [17], and in polyps of colon adenomas arising in these animals. In the normal intestine samples, Htra1 expression was very low but epigenetic silencing was not involved as none of the samples contained a methylated CpG island (Fig. 2, controls). In contrast, Htra1 expression was differentially regulated during polyp formation and the level of Htra1 expression correlated with the degree of DNA methylation (Fig. 2). Of 9 colonic polyps analysed, the Htra1 promoter was unmethylated in 4 polyps (polyps 13, 22, 145 and 147), methylated in 4 polyps (polyps 18, 98, 99 and 101) and partially methylated in one polyp (polyp 97). These data suggest that silencing of Htra1 is occurring during tumorigenesis in vivo.
Fig. 2.

Htra1 expression and methylation of the Htra1 promoter in polyps. Mean normalized expression (MNE) of Htra1 in polyps isolated from Apc Min+ mice was determined by qRT-PCR. Methylation of the murine Htra1 promoter (−252 bp to −12 bp) was determined by sequencing 3 independent clones of bisulfite treated DNA isolated from the same polyps that were used for measuring mRNA levels by qRT-PCR. Controls are four independent samples taken from large intestine
Methyl Binding Domain protein 2 (MBD2) mediates epigenetic silencing of Htra1
Having established epigenetic silencing of HTRA1 in vivo and in vitro, we wished to obtain a deeper insight by identifying factors that are involved in this process. Since MBD2 has been shown to alter tumorigenesis in ApcMin+ mice [18], we tested whether MBD2 mediates silencing of HTRA1 by stable knockdown of MBD2. HCT116 cells were used for these experiments because the HTRA1 promoter is epigenetically silenced in these cells. Knockdown was done using two independent shRNAs, resulting in an about 5 fold reduction of MBD2 expression which led to a 5 (shMBD2.1) and 10 fold (shMBD2.2) increase in HTRA1 gene expression (Fig. 3a). This effect was specific to MBD2 as levels of MBD1 and MeCP1 were unaffected by the knockdown of MBD2 expression (not shown). These findings expand published data that identified HTRA1 as a target of mouse HDAC1 [7].
Fig. 3.
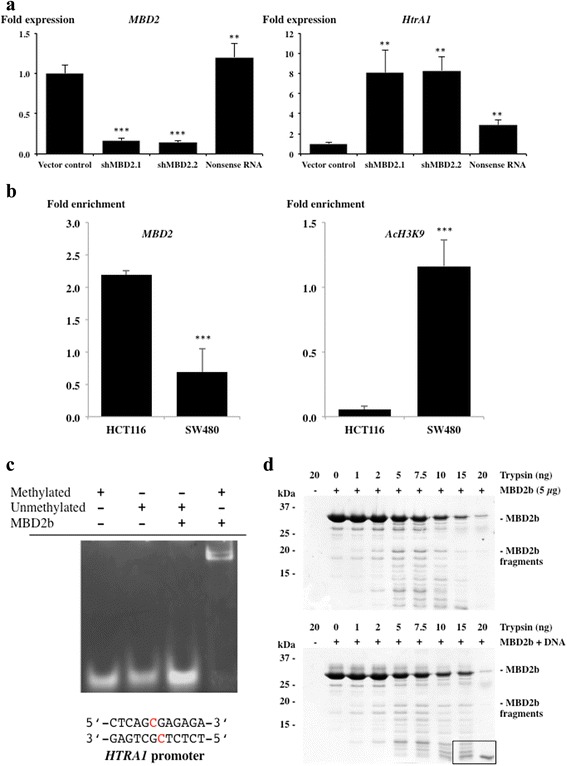
MDB2 mediates silencing of the HTRA1 promoter. a Fold expression of MBD2 and HTRA1 in HCT116 cells stably downregulated by different shRNAs (1 and 2) or random RNA sequence (nonsense RNA) compared to the vector control (set to 1). Standard deviation is indicated (n ≥ 2 independent experiments done in triplicates, two-tailed Mann–Whitney U test, p-values HTRA1 expression: shMBD2.1 = 0.0039, shMBD2.2 = 0.0028, Nonsense RNA = 0.0028; p-values MBD2 expression: shMBD2.1 = 0.0002, shMBD2.2 = 0.0002, Nonsense RNA = 0.0034)). b ChIP analyses of MBD2 binding the human HTRA1 promoter (−453 bp to −336 bp) in HCT116 and SW480 cells. Results are expressed as fold enrichment compared to the reference GAPDH promoter (n = 4, 2 independent chromatin fractions, unpaired two-tailed t-test, p-value <0.001) (left). ChIP analyses of H3K9 acetylation at the human HTRA1 promoter (−453 bp to −336 bp). Results are expressed as fold enrichment compared to the reference histone 3 (n = 3, 2 independent chromatin fractions, unpaired two-tailed t-test, p-value <0.001) (right). c Electric mobility shift assay using methylated or unmethylated 12 bp double stranded oligonucleotide from the CpG island of the HTRA1 promoter and purified MBD2b in equimolar concentrations (70 pmol). MBD2-DNA complex formation was analysed on 12 % agarose gels. d Proteolysis of MBD2b by trypsin. 5 μg of purified MBD2b were incubated with the amounts of trypsin indicated for 20 min at 37 °C. Samples were analysed on SDS PAGE (upper panel). Proteolysis of MBD2b with bound methylated oligonucleotide by trypsin. The protected MBD2b fragment is boxed (lower panel)
To analyse methylation-dependent binding of MBD2 to the HTRA1 promoter we performed ChIP assays in HCT116 and SW480 cells. Primer sets were designed covering the HTRA1 promoter sequence of the methylation analysis shown in Fig. 1a. The specificity of ChIP assays was verified by using nonspecific IgG (binding < 0.02 %). As a positive control, the GAPDH promoter was shown to be equally associated with RNAPolII (data not shown). The occupancy of MBD2 was up to 3-fold higher in HCT116 compared to SW480 cells (Fig. 3b), whereas the acetylation of Lys9 at histone 3, a chromatin marker for transcriptional activity, displayed a reversed pattern (Fig. 3b). Similar results were published in HCT116 cells, where binding of MBD2 to the TFF2 promoter was detected along with reduced histone acetylation levels [19]. To obtain further evidence for MBD2 binding to the HTRA1 promoter we purified recombinant MBD2b, a truncated version of MBD2 lacking the N-terminal 149 residues. This variant mediates silencing of a target promoter in cell culture [20]. To obtain a first indication of the affinity of MBD2 to the HTRA1 promoter we performed electrophoretic mobility shift assays using a specific sequence of 12 nucleotides occurring at position −376 bp to −364 bp in the CpG island of the HTRA1 promoter. These data indicated that the 12mer nucleotide bound in 1:1 stoichiometry to MBD2 in its methylated but not in its unmethylated form (Fig. 3c). These results are in agreement with published data reporting that MBD2 binds to synthetic 34mer nucleotides with high affinity [21]. To obtain independent evidence for binding of MBD2 to HTRA1 promoter DNA, we performed proteolytic digests of MBD2 without or with bound DNA (Fig. 3d). This strategy was chosen as ligand binding commonly causes conformational changes that lead to increased stability of the protein in protease assays. Trypsin digests MBD2 in the presence and absence of DNA. However, in the presence of DNA, several fragments of about 6–10 kDa are protected (Fig. 3d). The size of the prominent band corresponds to the size of the DNA binding domain of MBD2 (residues 24 through 86). Together, these data therefore confirm that MBD2 binds to, and epigenetically regulates, HTRA1 in HCT116 cells.
Loss of HTRA1 causes accelerated proliferation
To investigate whether downregulation of HTRA1 might affect cell growth, we analysed the phenotypes of Htra1+/+ and Htra1−/− MEFs generated from E14.5 mice that expressed either wild type levels of HtrA1 or no HtrA1. In parallel, we also generated a stable knockdown and overproduction of HTRA1 in SW480 cells, derived from a pool of clones, by using two independent shRNA constructs and plasmid p50, respectively. The knockout and knockdown of HTRA1 on mRNA level in MEFs and SW480 cells, respectively, was confirmed by qRT-PCR. While Htra1−/− MEFs showed a 99.6 % depletion of Htra1 mRNA the SW480 shHTRA1 cells had approximately 5 fold less HTRA1 mRNA compared to the empty vector cells (Fig. 4a). Consequently, HTRA1 protein levels were decreased in Htra1−/− MEFs and SW480 cells (Fig. 4a). Note that protein levels of SW480 cells were lower compared to MEFs. Therefore, cell culture supernatants were used for Western blotting to visualize protein levels of SW480 cells because levels of secreted HTRA1 are higher compared to cytoplasmic HTRA1 [1].
Fig. 4.
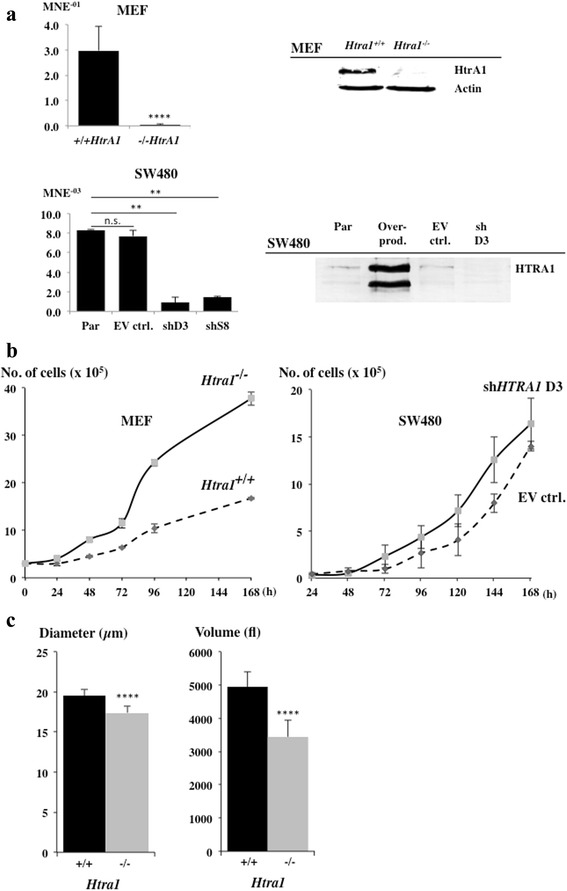
Increased proliferation after HTRA1 depletion. a HTRA1 expression measured by qRT-PCR (n = 2 independent experiments, each assayed in triplicate) and Western blots. Upper panel, Htra1 +/+ and Htra1 −/− MEFs (two-tailed Mann–Whitney U test, p-value <0.0001). Lower panel, supernatants of parental or stably transfected SW480 cells (Par, untransfected SW480; overprod., SW480 stably transfected with plasmid p50HTRA1; EV ctrl., corresponding empty vector control; shD3, downregulated via shHTRA1 D3; shS8, downregulated via shHTRA1 S8; two-tailed Mann–Whitney U test, p-values: EV ctrl. = 0.3776, shD3 = 0.005, shS8 = 0.005). Actin was used as loading control for cell lysates (right). b Growth curves. Left panel, MEFs. 3x105 cells were placed in 6 cm dishes and numbers of cells were counted at the time points indicated. Right panel, SW480 derivatives. 2.5x104 cells were placed into 6-wells and numbers of cells were counted at time points indicated. (n = 2 independent experiments, standard deviations are indicated). c Mean diameter and volume of Htra1 +/+ and Htra1 −/− MEFs (unpaired two-tailed t-test, p-value <0.0001)
To explore the physiologic effects of HTRA1 depletion, we analysed growth curves to measure bulk proliferation rates in MEFs indicating that Htra1−/− MEFs grew significantly faster compared to Htra1+/+ control cells, with a doubling time of 21 h compared to 38 h, respectively (Fig. 4b). In contrast, SW480shHTRA1 cells showed no significant differences in proliferation rates compared to the empty vector control (Fig. 4b), which is probably due to the transformed status of SW480 cancer cells. Since increased proliferation rates are often correlated with a reduction in cell size and volume [22], we determined cell volume and cell diameter in Htra1+/+ and Htra1−/− MEFs. Indeed, the mean cell volume of Htra1−/− MEFs was 2078 fl compared to 3323 fl of Htra1+/+ MEFs and the mean diameter of Htra1−/− MEFs was 17 μm compared to 19 μm of Htra1+/+ MEFs (Fig. 4c). To determine any influence upon senescence, we serially passaged both Htra1+/+ and Htra1−/− MEFs by splitting cells 1:3 twice per week. In 5 independent experiments, Htra1+/+ MEFs reached a senescence plateau after 12 passages (+/− 2), however Htra1−/− MEFs continued to grow until passage 20 (+/− 2).
Reduced HTRA1 expression drives polyploidy and correlates with centrosome amplification
To address the question whether HTRA1 deficiency causes elevated genomic instability, karyotyping was performed to analyse chromosome numbers in Htra1+/+ and Htra1−/− MEFs as well as in SW480 and derivatives of SW480 that were depleted for HTRA1 by using two independent stable shRNA constructs (Fig. 5). These data indicated that both cell lines exhibited polyploidy with the main peaks observed being 4n and 8n instead of 2n and a small peak of 4n in control cells (Fig. 5a, b). Note that SW480 cancer cells are already aneuploid [23].
Fig. 5.
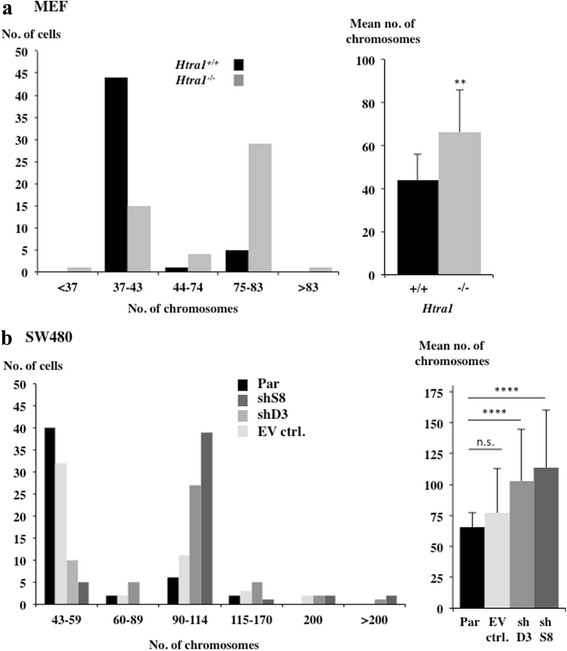
Increased polyploidy in Htra1 −/− MEFs and SW480shHTRA1 cells. a Chromosome numbers of Htra1 −/− and Htra1 +/+ MEFs. 50 mitotic cells were analysed by standard karyotyping. The normal set of chromosomes is 40. Shown is the distribution of chromosome numbers (left panel) as well as the mean number (right panel, two-tailed Mann–Whitney U test, p-value <0.01). b 50 mitotic SW480 cells were analysed by standard karyotyping. The normal set of chromosomes is 56–58. (Par, untransfected SW480; shS8 or D3, SW480 with HTRA1 downregulated via two independent shRNAs; EV ctrl., corresponding to control carrying nonsense shRNA). The distribution of chromosome numbers (left panel) and the mean number (right panel, two-tailed Mann–Whitney U test, p-values: EV ctrl. = 0,7790, shD3 < 0.0001, shS8 < 0.0001) are shown
It has been shown previously that HTRA1 localises to centrosomes and microtubules in PC12 and SKOV3 cells [4, 12]. The cellular location of HTRA1 was confirmed for SW480 cells by analysing HTRA1-mCherry constructs using confocal microscopy (Additional file 1: Figure S1). Centrosomes normally nucleate microtubules to form the spindle that is subsequently used to position the chromosomes during cell division [24]. Since altered microtubule formation can predispose to polyploidy [25], we examined the location and number of centrosomes in mitotic cells by immunofluorescence staining using antibodies against gamma-tubulin in Htra1−/− and Htra1+/+ MEFs. In the absence of HTRA1, an increase in the number of centrosomes (Fig. 6a) and multipolar spindles (Fig. 6b) were detected.
Fig. 6.
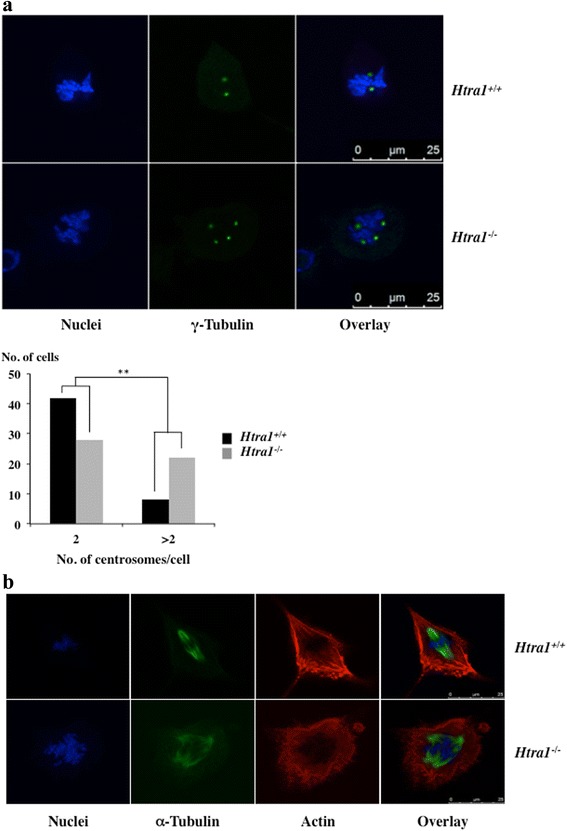
Disturbed spindle formation and centrosome abnormalities in HTRA1 depleted cells. a Representative images of Htra1 +/+ and Htra1 −/− MEFs. MEFs were grown on collagen coated glass slides, synchronised and analysed during mitosis by laser confocal microscopy. Centrosomes were visualised by anti-γ-tubulin antibody using an Alexa488 conjugated secondary antibody. Nuclei were counterstained with DAPI. Scale bar is 25 μm (upper panel). The number of centrosomes were counted in 50 Htra1 +/+ and Htra1 −/− MEFs, respectively (lower panel). For statistical analysis, data were classified into two sets (2 and >2 centrosomes per cell) and analysed via Fisher’s exact test (p-value = 0.0041). b Representative images of immunofluorescence staining with an α-tubulin antibody using an Alexa488 conjugated secondary antibody in primary Htra1 +/+ and Htra1 −/− MEFs (Passage 8). Nuclei were counterstained by DAPI. Scale bar is 25 μm
Discussion
Several proteases such as members of the ADAMTS family, caspase 8 and trypsinogen IV are epigenetically silenced in cancer cells [26, 27]. As the HTRA1 promoter contains a CpG island, we hypothesised that epigenetic mechanisms might be one way of lowering HTRA1 levels in cells. Our data indicate that, indeed, HTRA1 is epigenetically repressed in the human colorectal cell line HCT116, but not in SW480 cells. In HCT116 cells, silencing is mediated by the methyl binding domain protein MBD2. In addition, the Htra1 promoter is methylated and repressed in a proportion of benign polyps that develop in the ApcMin+ mouse model of human familial adenomatous polyposis. These data suggest that Htra1 can be a target for epigenetic repression in intestinal neoplasia and that such targeting might be associated with the development of a subset of tumours. In this regard, it is significant that repression of Htra1 is mediated by MBD2, the deficiency of which we have previously shown to strongly suppress the majority of adenomas in the ApcMin+ mouse [18]. The fact that adenomas do eventually form in this model suggests that MBD2-mediated suppression of target genes such as Htra1 may be relevant to a subset of lesions. An implication of these studies is that Htra1 status will influence adenoma formation in ApcMin+ mice, a hypothesis we are currently testing. Our findings may also have implications in the clinic, as the methylation state of the HtrA1 promoter may act as a convenient biomarker for tumour cells or cells at risk of transformation.
To investigate the mechanistic consequences of HTRA1 repression in tumorigenesis, we have analysed the phenotype of loss of function in both MEFs and SW480 cells. Remarkably, we find multiple phenotypes including increased proliferation, delayed onset of senescence, perturbed centrosome number and positioning, and ultimately polyploidy. Tight control of all aspects of cell division including accurate chromosome replication and partitioning as well as of proliferation rates is clearly critical to both tissue homeostasis and tumour suppression. Mechanistically, the changes we observe implicate a number of different pathways. The delayed onset of senescence suggests a failure in DNA damage checkpoint activation, as senescence can be triggered when telomeres are eroded and generate a DNA damage signal [28].
A second major phenomenon we observe is the deregulation of centrosomes and polyploidy. The perturbation of both centrosome position and number strongly implies a defect in microtubule function in the absence of HTRA1, as centrosomes normally nucleate microtubules to form the spindle, which is subsequently used to position the chromosomes during division. Such altered microtubules can predispose to polyploidisation. Thus, normal cells complete mitosis and enter S phase following activation of cyclin-dependent kinases in G1. However, if microtubule dynamics become perturbed, cells can aberrantly exit mitosis and enter S phase with a >4n DNA content, a process known as endoreduplication, ultimately resulting in polyploidy. This model is supported by the literature showing that HTRA1 is localised to microtubules and is a regulator of microtubule stability [4, 29, 30].
While these models offer initial explanations for some of the detected phenotypes of cells depleted for HTRA1, the underlying molecular mechanisms leading to increased proliferation rates, reduced cell size and the delayed senescence of primary Htra1−/− MEFs remain to be identified. While it is likely that the loss of cell cycle checkpoint functions contributes, it is probably not the only reason. Due to the complexity of the cell cycle mechanism and of cellular transformation it will be interesting to address for example the interconnectivity of different checkpoints, the differential expression of HTRA1 in specific cell cycle phases and its role in regulating centrosome numbers and assembly in future experiments. Another key open question is the regulation of HTRA1 activity in mammalian cells. Previous studies revealed that HtrA proteases reversibly switch from the inactive into the active conformation and that this switch is mediated by specific peptidic ligands [1]. The identification of native modulators of HTRA1s activity (i.e. activators and inhibitors) and the exact circumstances under which these regulators are occurring will provide important insights into how this protease is implicated in the regulation of proliferation.
Conclusions
These data show that MBD2-dependent epigenetic silencing of HTRA1 can occur during tumour development. The phenotypes of reduced HTRA1 expression such as acceleration of cell growth, centrosome amplification and polyploidy provide additional support for the model that proteolytic events are implicated in cancer biology. Moreover, the methylation state of the HTRA1 promoter may be explored as a potential biomarker for tumour cells or cells at risk of transformation.
Abbreviations
5-Aza-Dc, 5-Aza-2′Deoxycytidine; DNMT, DNA methyltransferase; HDAC, histone deacetylase; HtrA, High-temperature requirement A; MBD1, Methyl-CpG-binding domain protein 1; MBD2, Methyl Binding Domain protein 2; Me-CP2, Methyl-CpG binding protein 2; MEF, mouse embryonic fibroblasts; MNE, Mean normalized expression; TSA, Trichostatin A
Acknowledgements
We thank Adrian Bird and Stephan Hahn for providing plasmids, Katharina Günther and Rainer Renkawitz for help with the CHIP assays. M.E. was supported by GRK1431 from Deutsche Forschungsgemeinschaft and A.R.C. by CR UK.
Availability of data and materials
Data and materials are available on request from the corresponding author.
Authors’ contributions
ARC, AB and ME designed the research. NS, II, KR, VL, JN, CJ and MC carried out epigenetic, cell biology and biochemical experiments and analysed data. LP, KG, SA provided essential materials and interpreted data. CO and MK generated the Htra1 mice. ME drafted the manuscript and all authors critically reviewed. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Additional file
contains Supplementary method (Chromatin immunoprecipitation) and Table S1 (oligonucleotides used). Additional figure shows colocalisation of HTRA1 and microtubules in SW480 cells. (ZIP 394 kb)
Contributor Information
Nina Schmidt, Email: schmidt-nina@gmx.net.
Inga Irle, Email: inga@s-irle.de.
Kamilla Ripkens, Email: kamilla.ripkens@uni-due.de.
Vanda Lux, Email: vanda.lux@uni-due.de.
Jasmin Nelles, Email: jasmin.nelles@uni-due.de.
Christian Johannes, Email: christian.johannes@uni-due.de.
Lee Parry, Email: ParryL3@cardiff.ac.uk.
Kirsty Greenow, Email: Greenow@cardiff.ac.uk.
Sarah Amir, Email: amirsarah7@gmail.com.
Mara Campioni, Email: maracampioni@yahoo.it.
Alfonso Baldi, Email: alfonsobaldi@tiscali.it.
Chio Oka, Email: coka@bs.naist.jp.
Masashi Kawaichi, Email: mkawaich@bs.aist-nara.ac.jp.
Alan R. Clarke, Email: ClarkeAR@cardiff.ac.uk
Michael Ehrmann, Email: michael.ehrmann@uni-due.de.
References
- 1.Clausen T, Kaiser M, Huber R, Ehrmann M. HTRA proteases: regulated proteolysis in protein quality control. Nat Rev Mol Cell Biol. 2011;12(3):152–162. doi: 10.1038/nrm3065. [DOI] [PubMed] [Google Scholar]
- 2.Chien J, Campioni M, Shridhar V, Baldi A. HtrA serine proteases as potential therapeutic targets in cancer. Curr Cancer Drug Targets. 2009;9(4):451–468. doi: 10.2174/156800909788486704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Baldi A, De Luca A, Morini M, Battista T, Felsani A, Baldi F, Catricalà C, Amantea A, Noonan DM, Albini A, et al. The HtrA1 serine protease is down-regulated during human melanoma progression and represses growth of metastatic melanoma cells. Oncogene. 2002;21:6684–6688. doi: 10.1038/sj.onc.1205911. [DOI] [PubMed] [Google Scholar]
- 4.Chien J, Ota T, Aletti G, Shridhar R, Boccellino M, Quagliuolo L, Baldi A, Shridhar V. Serine protease HtrA1 associates with microtubules and inhibits cell migration. Mol Cell Biol. 2009;29(15):4177–4187. doi: 10.1128/MCB.00035-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chien J, Aletti G, Baldi A, Catalano V, Muretto P, Keeney GL, Kalli KR, Staub J, Ehrmann M, Cliby WA, et al. Serine protease HtrA1 modulates chemotherapy-induced cytotoxicity. J Clin Invest. 2006;116(7):1994–2004. doi: 10.1172/JCI27698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chien J, Staub J, Hu SI, Erickson-Johnson MR, Couch FJ, Smith DI, Crowl RM, Kaufmann SH, Shridhar V. A candidate tumor suppressor HtrA1 is downregulated in ovarian cancer. Oncogene. 2004;23(8):1636–1644. doi: 10.1038/sj.onc.1207271. [DOI] [PubMed] [Google Scholar]
- 7.Zupkovitz G, Tischler J, Posch M, Sadzak I, Ramsauer K, Egger G, Grausenburger R, Schweifer N, Chiocca S, Decker T, et al. Negative and positive regulation of gene expression by mouse histone deacetylase 1. Mol Cell Biol. 2006;26:7913–7928. doi: 10.1128/MCB.01220-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jones A, Kumar S, Zhang N, Tong Z, Yang JH, Watt C, Anderson J, Amrita, Fillerup H, McCloskey M, et al. Increased expression of multifunctional serine protease, HTRA1, in retinal pigment epithelium induces polypoidal choroidal vasculopathy in mice. Proc Natl Acad Sci U S A. 2011;108(35):14578–14583. doi: 10.1073/pnas.1102853108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Severino A, Campioni M, Straino S, Salloum FN, Schmidt N, Herbrand U, Frede S, Toietta G, Di Rocco G, Bussani R, et al. Identification of protein disulfide isomerase as a cardiomyocyte survival factor in ischemic cardiomyopathy. J Am Coll Cardiol. 2007;50(11):1029–1037. doi: 10.1016/j.jacc.2007.06.006. [DOI] [PubMed] [Google Scholar]
- 10.Simon P. Q-Gene: processing quantitative real-time RT-PCR data. Bioinformatics. 2003;19(11):1439–1440. doi: 10.1093/bioinformatics/btg157. [DOI] [PubMed] [Google Scholar]
- 11.Bock C, Reither S, Mikeska T, Paulsen M, Walter J, Lengauer T. BiQ Analyzer: visualization and quality control for DNA methylation data from bisulfite sequencing. Bioinformatics. 2005;21(21):4067–4068. doi: 10.1093/bioinformatics/bti652. [DOI] [PubMed] [Google Scholar]
- 12.Tennstaedt A, Popsel S, Truebestein L, Hauske P, Brockmann A, Schmidt N, Irle I, Sacca B, Niemeyer CM, Brandt R, et al. Human High Temperature Requirement Serine Protease A1 (HTRA1) Degrades Tau Protein Aggregates. J Biol Chem. 2012;287(25):20931–20941. doi: 10.1074/jbc.M111.316232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Christman JK. 5-Azacytidine and 5-aza-2′-deoxycytidine as inhibitors of DNA methylation: mechanistic studies and their implications for cancer therapy. Oncogene. 2002;21(35):5483–5495. doi: 10.1038/sj.onc.1205699. [DOI] [PubMed] [Google Scholar]
- 14.Yoshida M, Horinouchi S, Beppu T. Trichostatin A and trapoxin: novel chemical probes for the role of histone acetylation in chromatin structure and function. Bioessays. 1995;17(5):423–430. doi: 10.1002/bies.950170510. [DOI] [PubMed] [Google Scholar]
- 15.Flatmark K, Nome RV, Folkvord S, Bratland A, Rasmussen H, Ellefsen MS, Fodstad O, Ree AH. Radiosensitization of colorectal carcinoma cell lines by histone deacetylase inhibition. Radiat Oncol. 2006;1:25. doi: 10.1186/1748-717X-1-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yoshida M, Beppu T. Reversible arrest of proliferation of rat 3Y1 fibroblasts in both the G1 and G2 phases by trichostatin A. Exp Cell Res. 1988;177(1):122–131. doi: 10.1016/0014-4827(88)90030-4. [DOI] [PubMed] [Google Scholar]
- 17.Clarke AR. Wnt signalling in the mouse intestine. Oncogene. 2006;25(57):7512–7521. doi: 10.1038/sj.onc.1210065. [DOI] [PubMed] [Google Scholar]
- 18.Sansom OJ, Berger J, Bishop SM, Hendrich B, Bird A, Clarke AR. Deficiency of Mbd2 suppresses intestinal tumorigenesis. Nat Genet. 2003;34(2):145–147. doi: 10.1038/ng1155. [DOI] [PubMed] [Google Scholar]
- 19.Berger J, Sansom O, Clarke A, Bird A. MBD2 is required for correct spatial gene expression in the gut. Mol Cell Biol. 2007;27(11):4049–4057. doi: 10.1128/MCB.02023-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Boeke J, Ammerpohl O, Kegel S, Moehren U, Renkawitz R. The minimal repression domain of MBD2b overlaps with the methyl-CpG-binding domain and binds directly to Sin3A. J Biol Chem. 2000;275(45):34963–34967. doi: 10.1074/jbc.M005929200. [DOI] [PubMed] [Google Scholar]
- 21.Fodermayr M, Proll J, Zach O, Wechselberger C, Lutz D. In vitro detection of methylated DNA via recombinant protein MBD2b. Mol Biol Rep. 2009;36(7):1859–1862. doi: 10.1007/s11033-008-9391-4. [DOI] [PubMed] [Google Scholar]
- 22.Montagne J. Genetic and molecular mechanisms of cell size control. Mol Cell Biol Res Commun. 2000;4(4):195–202. doi: 10.1006/mcbr.2001.0284. [DOI] [PubMed] [Google Scholar]
- 23.Wang Y, Coffey RJ, Osheroff N, Neufeld KL. Topoisomerase IIalpha binding domains of adenomatous polyposis coli influence cell cycle progression and aneuploidy. PLoS One. 2010;5(4) doi: 10.1371/journal.pone.0009994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nigg EA, Stearns T. The centrosome cycle: Centriole biogenesis, duplication and inherent asymmetries. Nat Cell Biol. 2011;13(10):1154–1160. doi: 10.1038/ncb2345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Thompson SL, Bakhoum SF, Compton DA. Mechanisms of chromosomal instability. Curr Biol. 2010;20(6):R285–R295. doi: 10.1016/j.cub.2010.01.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lopez-Otin C, Matrisian LM. Emerging roles of proteases in tumour suppression. Nat Rev Cancer. 2007;7(10):800–808. doi: 10.1038/nrc2228. [DOI] [PubMed] [Google Scholar]
- 27.Wu Y, Alvarez M, Slamon DJ, Koeffler P, Vadgama JV. Caspase 8 and maspin are downregulated in breast cancer cells due to CpG site promoter methylation. BMC Cancer. 2010;10:32. doi: 10.1186/1471-2407-10-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.d’Adda di Fagagna F, Reaper PM, Clay-Farrace L, Fiegler H, Carr P, Von Zglinicki T, Saretzki G, Carter NP, Jackson SP. A DNA damage checkpoint response in telomere-initiated senescence. Nature. 2003;426(6963):194–198. doi: 10.1038/nature02118. [DOI] [PubMed] [Google Scholar]
- 29.Campioni M, Severino A, Manente L, De Luca A, La Porta R, Vitiello A, Fiore P, Toldo S, Spugnini EP, Paggi MG, et al. Identification of protein-protein interactions of human HtrA1. Front Biosci. 2011;3:1493–1499. doi: 10.2741/e350. [DOI] [PubMed] [Google Scholar]
- 30.Chien J, He X, Shridhar V. Identification of tubulins as substrates of serine protease HtrA1 by mixture-based oriented peptide library screening. J Cell Biochem. 2009;107(2):253–263. doi: 10.1002/jcb.22121. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data and materials are available on request from the corresponding author.